
Effective Label-Free Sorting of Multipotent Mesenchymal Stem Cells from Clinical Bone Marrow Samples

 , , ,
, , ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Harvesting and Preparation
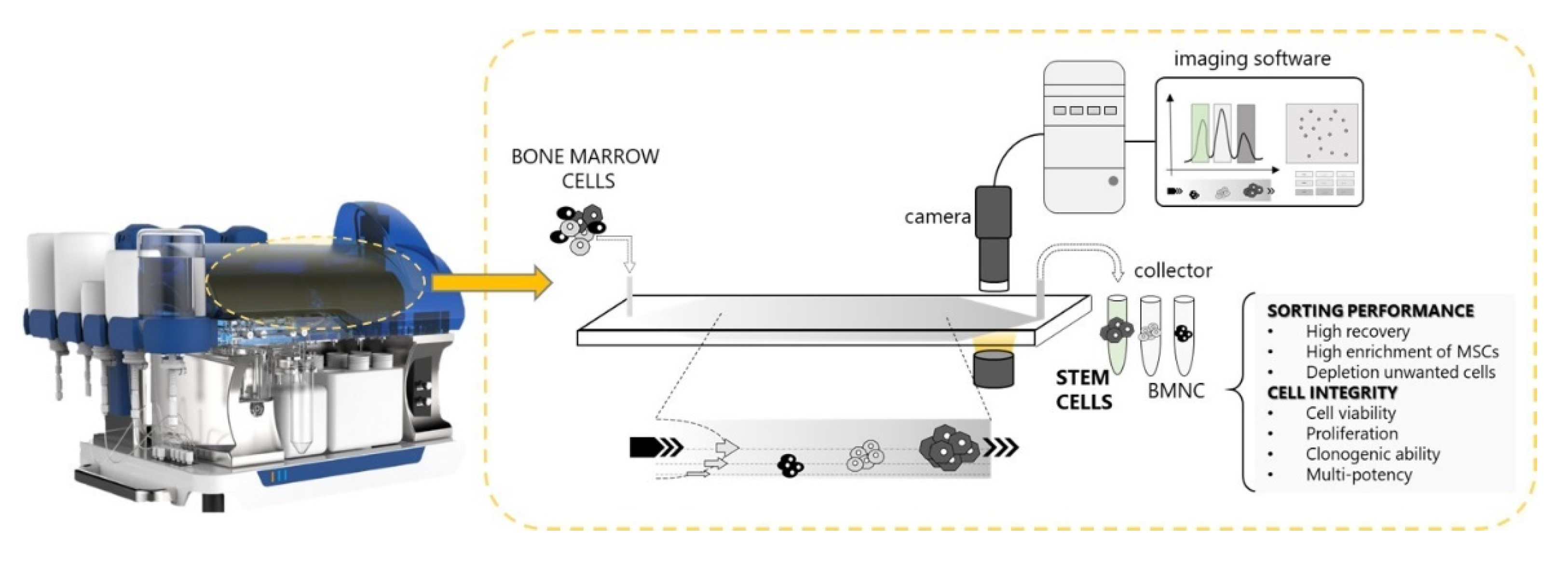
2.2. Celector® Instrument
2.3. Fractionation Principle and Procedure
2.4. Optical Analysis
2.5. Analysis and Cell Collection
2.6. Physical Characteristics
2.7. Colony-Forming Units-Fibroblast Assay (CFU-F)
2.8. Phenotype Characterization
2.9. Differentiation Capacity
2.10. Statistical Analysis
3. Results
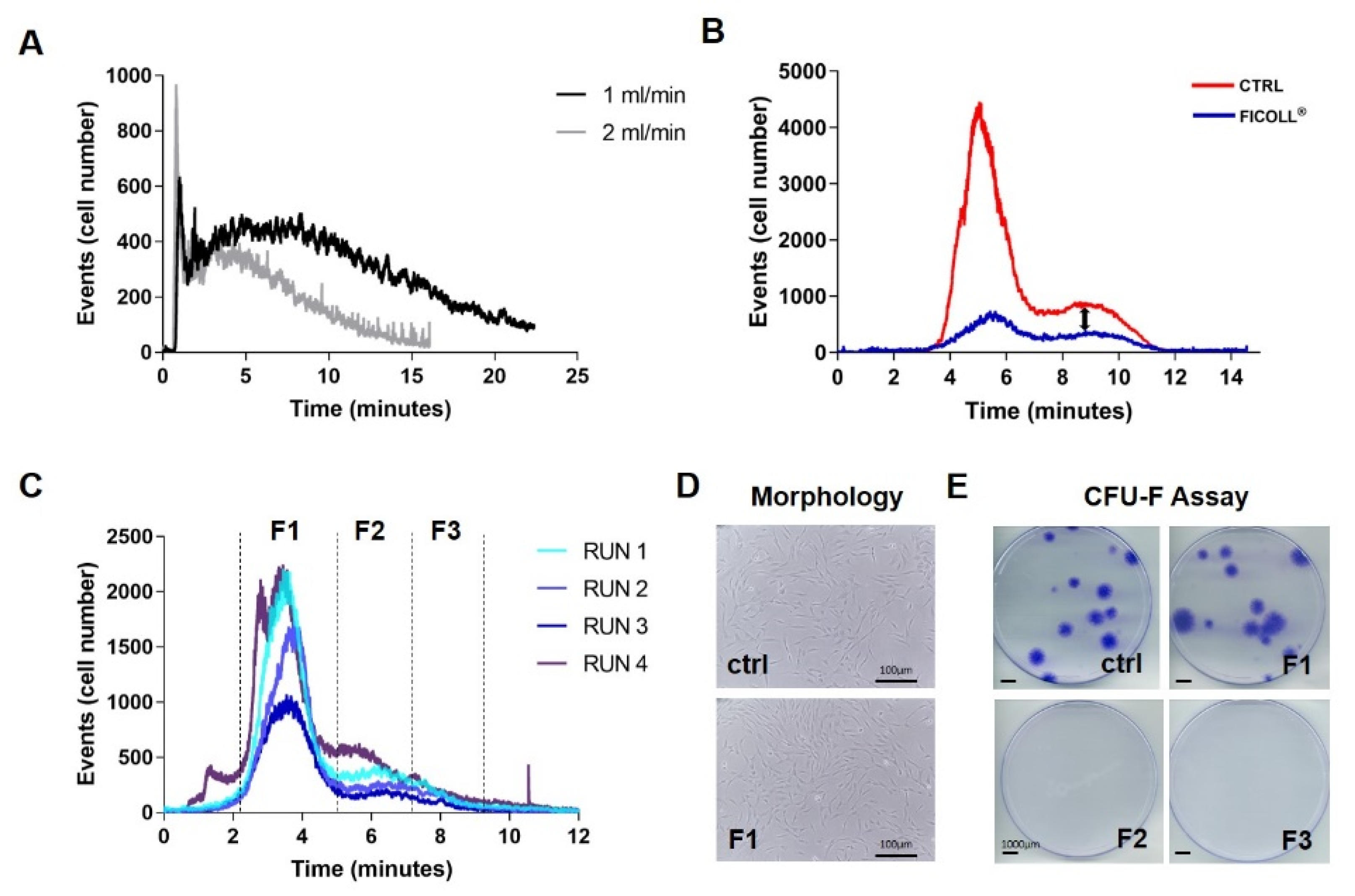
3.1. Fractionation Method Development
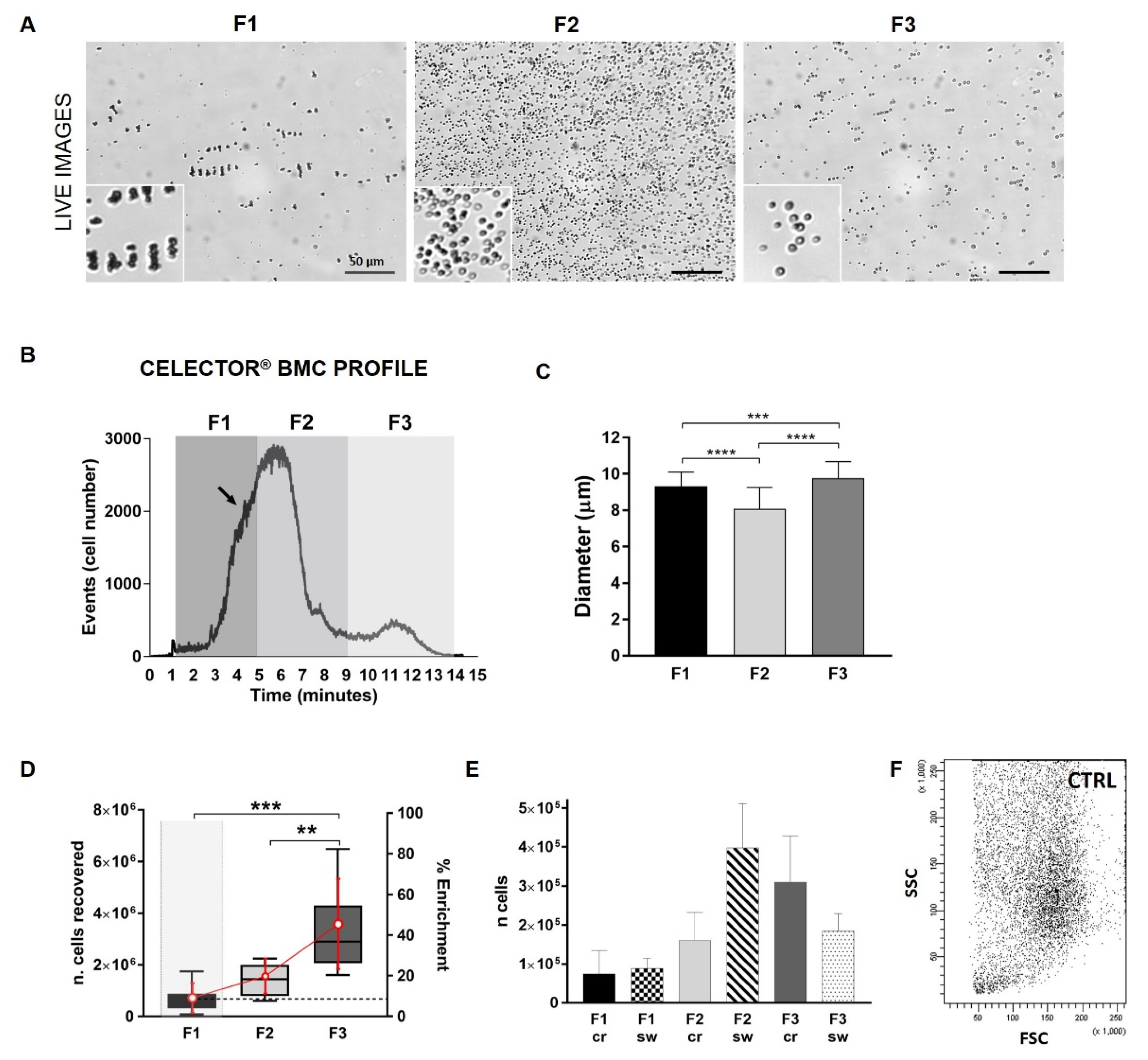
3.2. Celector® Bone Marrow Concentrate Profile
3.3. Representation of Cells Physical Parameters
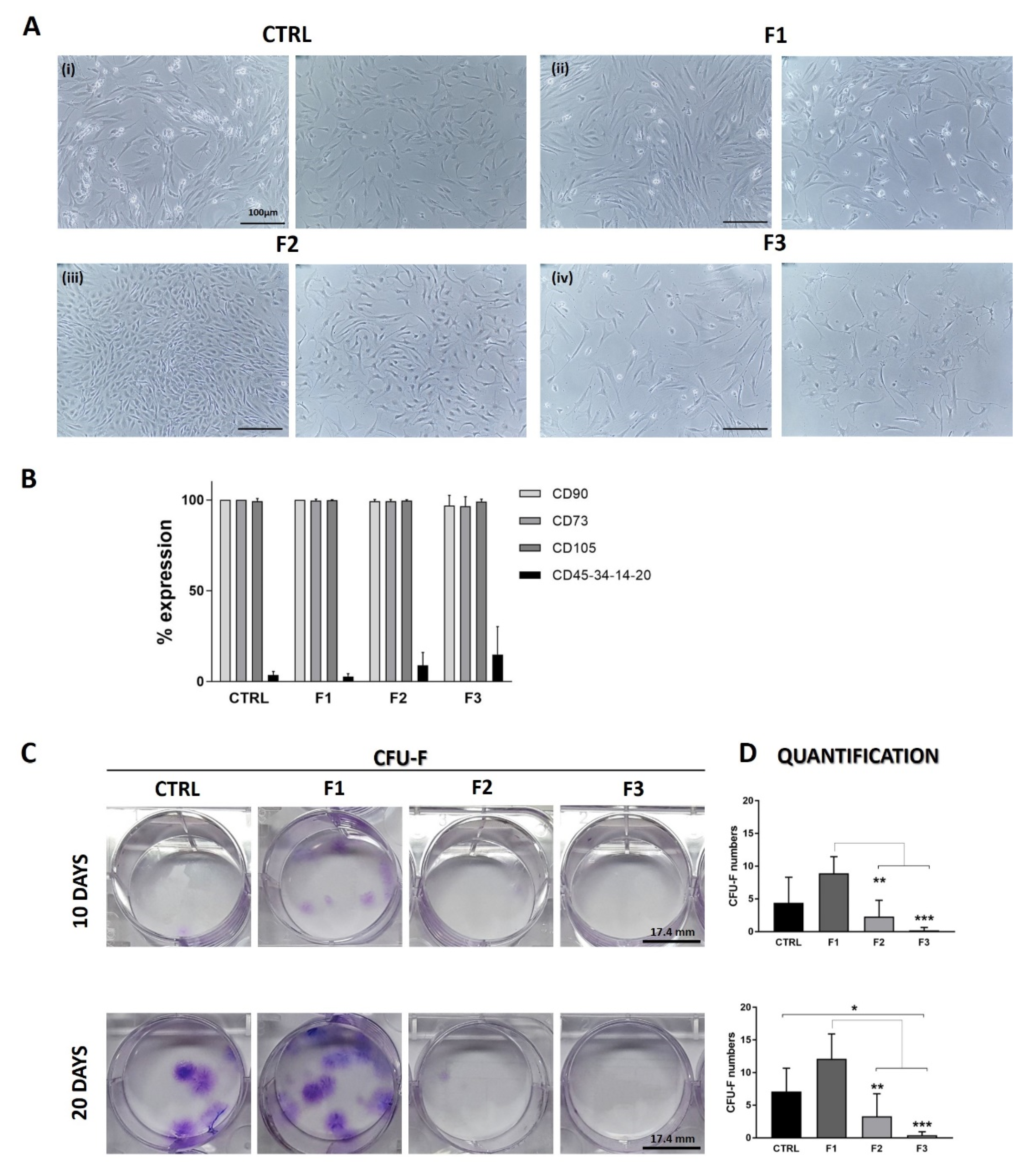
3.4. Morphological and Phenotypical Analysis
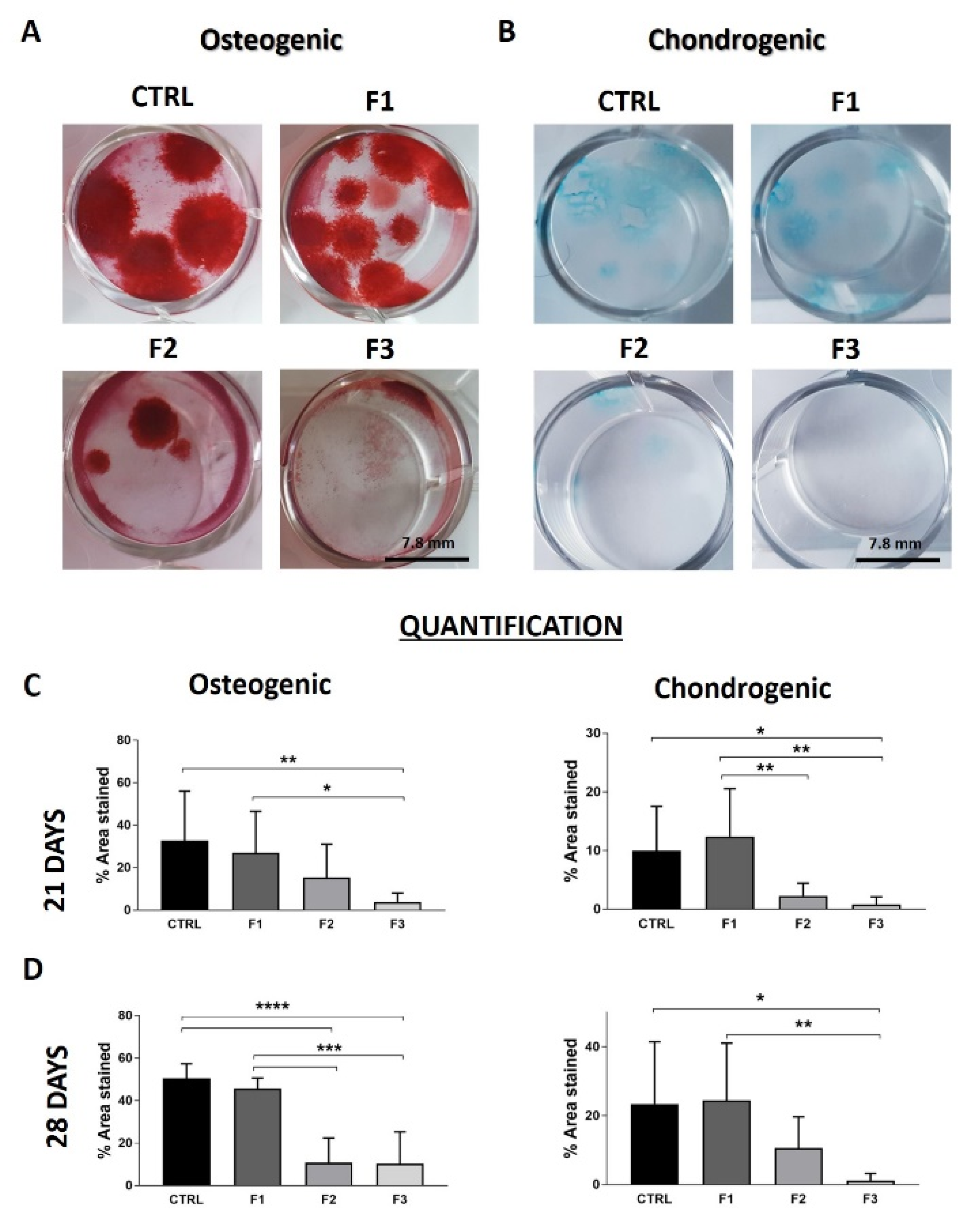
3.5. Clonogenic Activity
3.6. Differentiation Capacity
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trento, C.; Bernardo, M.E.; Nagler, A.; Kuci, S.; Bornhäuser, M.; Köhl, U.; Strunk, D.; Galleu, A.; Sanchez-Guijo, F.; Gaipa, G.; et al. Manufacturing Mesenchymal Stromal Cells for the Treatment of Graft-versus-Host Disease: A Survey among Centers Affiliated with the European Society for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Madry, H.; Gao, L.; Eichler, H.; Orth, P.; Cucchiarini, M. Bone Marrow Aspirate Concentrate-Enhanced Marrow Stimulation of Chondral Defects. Stem Cells Int. 2017, 2017, 1609685. [Google Scholar] [CrossRef] [PubMed]
- Pösel, C.; Möller, K.; Fröhlich, W.; Schulz, I.; Boltze, J.; Wagner, D.-C. Density Gradient Centrifugation Compromises Bone Marrow Mononuclear Cell Yield. PLoS ONE 2012, 7, e50293. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.R.; Jiang, Y.; Leary, E.; Yavanian, G.; Eminli, S.; O’Neill, D.W.; Marasco, W.A. Identification and Isolation of Small CD44-Negative Mesenchymal Stem/Progenitor Cells From Human Bone Marrow Using Elutriation and Polychromatic Flow Cytometry. STEM CELLS Transl. Med. 2013, 2, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Harichandan, A.; Bühring, H.-J. Prospective isolation of human MSC. Best Pract. Res. Clin. Haematol. 2011, 24, 25–36. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Sivasubramaniyan, K.; Lehnen, D.; Ghazanfari, R.; Sobiesiak, M.; Harichandan, A.; Mortha, E.; Petkova, N.; Grimm, S.; Cerabona, F.; De Zwart, P.; et al. Phenotypic and functional heterogeneity of human bone marrow- and amnion-derived MSC subsets. Ann. N. Y. Acad. Sci. 2012, 1266, 94–106. [Google Scholar] [CrossRef]
- Mabuchi, Y.; Okawara, C.; Mendez-Ferrer, S.; Akazawa, C. Cellular Heterogeneity of Mesenchymal Stem/Stromal Cells in the Bone Marrow. Front. Cell Dev. Biol. 2021, 9, 689366. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Rui, T.; Zhang, S.; Ding, Z. Heterogeneity of MSC: Origin, Molecular Identities, and Functionality. Stem Cells Int. 2019, 2019, 9281520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, B.; Lanzoni, G.; Alviano, F.; Zattoni, A.; Costa, R.; Di Carlo, A.; Marchionni, C.; Franchina, M.; Ricci, F.; Tazzari, P.L.; et al. A Novel Stem Cell Tag-Less Sorting Method. Stem Cell Rev. Rep. 2009, 5, 420–427. [Google Scholar] [CrossRef]
- Roda, B.; Reschiglian, P.; Zattoni, A.; Alviano, F.; Lanzoni, G.; Costa, R.; Di Carlo, A.; Marchionni, C.; Franchina, M.; Bonsi, L.; et al. A tag-less method of sorting stem cells from clinical specimens and separating mesenchymal from epithelial progenitor cells. Cytom. Part B Clin. Cytom. 2009, 76, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Roda, B.; Zia, S.; Vigliotta, M.I.; Zannini, C.; Alviano, F.; Bonsi, L.; Zattoni, A.; Reschiglian, P.; Gennai, A. Characterization of the Tissue and Stromal Cell Components of Micro-Superficial Enhanced Fluid Fat Injection (Micro-SEFFI) for Facial Aging Treatment. Aesthetic Surg. J. 2018, 40, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, C.; Desando, G.; Cattini, L.; Cavallo, M.; Buda, R.; Giannini, S.; Facchini, A.; Grigolo, B. Bone marrow concentrated cell transplantation: Rationale for its use in the treatment of human osteochondral lesions. J. Boil. Regul. Homeost. Agents 2013, 27, 165–175. [Google Scholar]
- Tong, X.; Caldwell, K.D. Separation and Characterization of Red Blood Cells with Different Membrane Deformability Using Steric Field-Flow Fractionation. J. Chromatogr. B Biomed. Sci. Appl. 1995, 674, 39–47. [Google Scholar] [CrossRef]
- Roda, B.; Reschiglian, P.; Alviano, F.; Lanzoni, G.; Bagnara, G.P.; Ricci, F.; Buzzi, M.; Tazzari, P.L.; Pagliaro, P.; Michelini, E.; et al. Gravitational field-flow fractionation of human hemopoietic stem cells. J. Chromatogr. A 2009, 1216, 9081–9087. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, G.; Tazzari, P.; Ricci, F.; Vaselli, C.; Buzzi, M.; Conte, R.; Orrico, C.; Foroni, L.; Stella, A.; Alviano, F.; et al. Ultrastructural Characteristics of Human Mesenchymal Stromal (Stem) Cells Derived from Bone Marrow and Term Placenta. Ultrastruct. Pathol. 2007, 31, 23–31. [Google Scholar] [CrossRef]
- Ahmed, N.; Vogel, B.; Rohde, E.; Strunk, D.; Grifka, J.; Schulz, M.B.; Grässel, S. CD45-positive cells of haematopoietic origin enhance chondrogenic marker gene expression in rat marrow stromal cells. Int. J. Mol. Med. 2006, 18, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Siegel, G.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schäfer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Muraglia, A.; Cancedda, R.; Quarto, R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J. Cell Sci. 2000, 113, 1161–1166. [Google Scholar] [CrossRef]
- Jones, E.A.; English, A.; Kinsey, S.E.; Straszynski, L.; Emery, P.; Ponchel, F.; McGonagle, D. Optimization of a flow cytometry-based protocol for detection and phenotypic characterization of multipotent mesenchymal stromal cells from human bone marrow. Cytom. Part B Clin. Cytom. 2006, 70, 391–399. [Google Scholar] [CrossRef]
- Chan, N.; Bourdeau, A.; Vera, S.; Abdalla, S.; Gross, M.; Wong, J.; Cymerman, U.; Paterson, A.; Mullen, B.; Letarte, M. Umbilical Vein and Placental Vessels from Newborns with Hereditary Haemorrhagic Telangiectasia Type 1 Genotype are Normal despite Reduced Expression of Endoglin. Placenta 2004, 25, 208–217. [Google Scholar] [CrossRef]
- Narravula, S.; Lennon, P.F.; Mueller, B.U.; Colgan, S.P. Regulation of Endothelial CD73 by Adenosine: Paracrine Pathway for Enhanced Endothelial Barrier Function. J. Immunol. 2000, 165, 5262–5268. [Google Scholar] [CrossRef] [Green Version]
- Haasters, F.; Prall, W.C.; Anz, D.; Bourquin, C.; Pautke, C.; Endres, S.; Mutschler, W.; Docheva, D.; Schieker, M. Morphological and immunocytochemical characteristics indicate the yield of early progenitors and represent a quality control for human mesenchymal stem cell culturing. J. Anat. 2009, 214, 759–767. [Google Scholar] [CrossRef]
- Ahmadbeigi, N.; Soleimani, M.; Babaeijandaghi, F.; Mortazavi, Y.; Gheisari, Y.; Vasei, M.; Azadmanesh, K.; Rostami, S.; Shafiee, A.; Nardi, N.B. The aggregate nature of human mesenchymal stromal cells in native bone marrow. Cytotherapy 2012, 14, 917–924. [Google Scholar] [CrossRef]
- Blazsek, I.; Chagraoui, J.; Péault, B. Ontogenic emergence of the hematon, a morphogenetic stromal unit that supports multipotential hematopoietic progenitors in mouse bone marrow. Blood 2000, 96, 3763–3771. [Google Scholar] [CrossRef]
- Janel, A.; Berger, J.; Bourgne, C.; Lemal, R.; Boiret-Dupré, N.; Dubois-Galopin, F.; Déchelotte, P.; Bothorel, C.; Hermet, E.; Chabi, S.; et al. Bone marrow hematons: An access point to the human hematopoietic niche. Am. J. Hematol. 2017, 92, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Di Girolamo, C.M.; Stokes, D.; Colter, D.; Phinney, D.; Class, R.; Prockop, D.J. Propagation and senescence of human marrow stromal cells in culture: A simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br. J. Haematol. 1999, 107, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Pochampally, R.; Perry, A.; Hsu, S.; Prockop, D.J. Isolation of a Highly Clonogenic and Multipotential Subfraction of Adult Stem Cells from Bone Marrow Stroma. STEM CELLS 2004, 22, 823–831. [Google Scholar] [CrossRef]
- Colter, D.C.; Sekiya, I.; Prockop, D.J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7841–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assmus, B.; Tonn, T.; Seeger, F.H.; Yoon, C.-H.; Leistner, D.; Klotsche, J.; Schächinger, V.; Seifried, E.; Zeiher, A.M.; Dimmeler, S. Red Blood Cell Contamination of the Final Cell Product Impairs the Efficacy of Autologous Bone Marrow Mononuclear Cell Therapy. J. Am. Coll. Cardiol. 2010, 55, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Beitzel, K.; McCarthy, M.B.; Coté, M.; Russell, R.P.; Arciero, R.A.; Mazzocca, A.D. Comparison of Mesenchymal Stem Cells (Osteoprogenitors) Harvested From Proximal Humerus and Distal Femur During Arthroscopic Surgery. Arthrosc. J. Arthrosc. Relat. Surg. 2013, 29, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Narbona-Carceles, J.; Vaquero, J.; Suárez-Sancho, S.B.S.; Forriol, F.; Santos, M.E.F. Bone marrow mesenchymal stem cell aspirates from alternative sources Is the knee as good as the iliac crest? Injury 2014, 45, S42–S47. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zia, S.; Cavallo, C.; Vigliotta, I.; Parisi, V.; Grigolo, B.; Buda, R.; Marrazzo, P.; Alviano, F.; Bonsi, L.; Zattoni, A.; et al. Effective Label-Free Sorting of Multipotent Mesenchymal Stem Cells from Clinical Bone Marrow Samples. Bioengineering 2022, 9, 49. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9020049
Zia S, Cavallo C, Vigliotta I, Parisi V, Grigolo B, Buda R, Marrazzo P, Alviano F, Bonsi L, Zattoni A, et al. Effective Label-Free Sorting of Multipotent Mesenchymal Stem Cells from Clinical Bone Marrow Samples. Bioengineering. 2022; 9(2):49. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9020049
Chicago/Turabian StyleZia, Silvia, Carola Cavallo, Ilaria Vigliotta, Valentina Parisi, Brunella Grigolo, Roberto Buda, Pasquale Marrazzo, Francesco Alviano, Laura Bonsi, Andrea Zattoni, and et al. 2022. "Effective Label-Free Sorting of Multipotent Mesenchymal Stem Cells from Clinical Bone Marrow Samples" Bioengineering 9, no. 2: 49. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9020049