
Improved Titer in Late-Stage Mammalian Cell Culture Manufacturing by Re-Cloning

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Media, Re-Clone and Seed Expansion
2.2. N-1 Seed Cultures
2.3. Fed-Batch Production Cultures
2.4. In-Process Cell Culture and Quality Attribute Assays
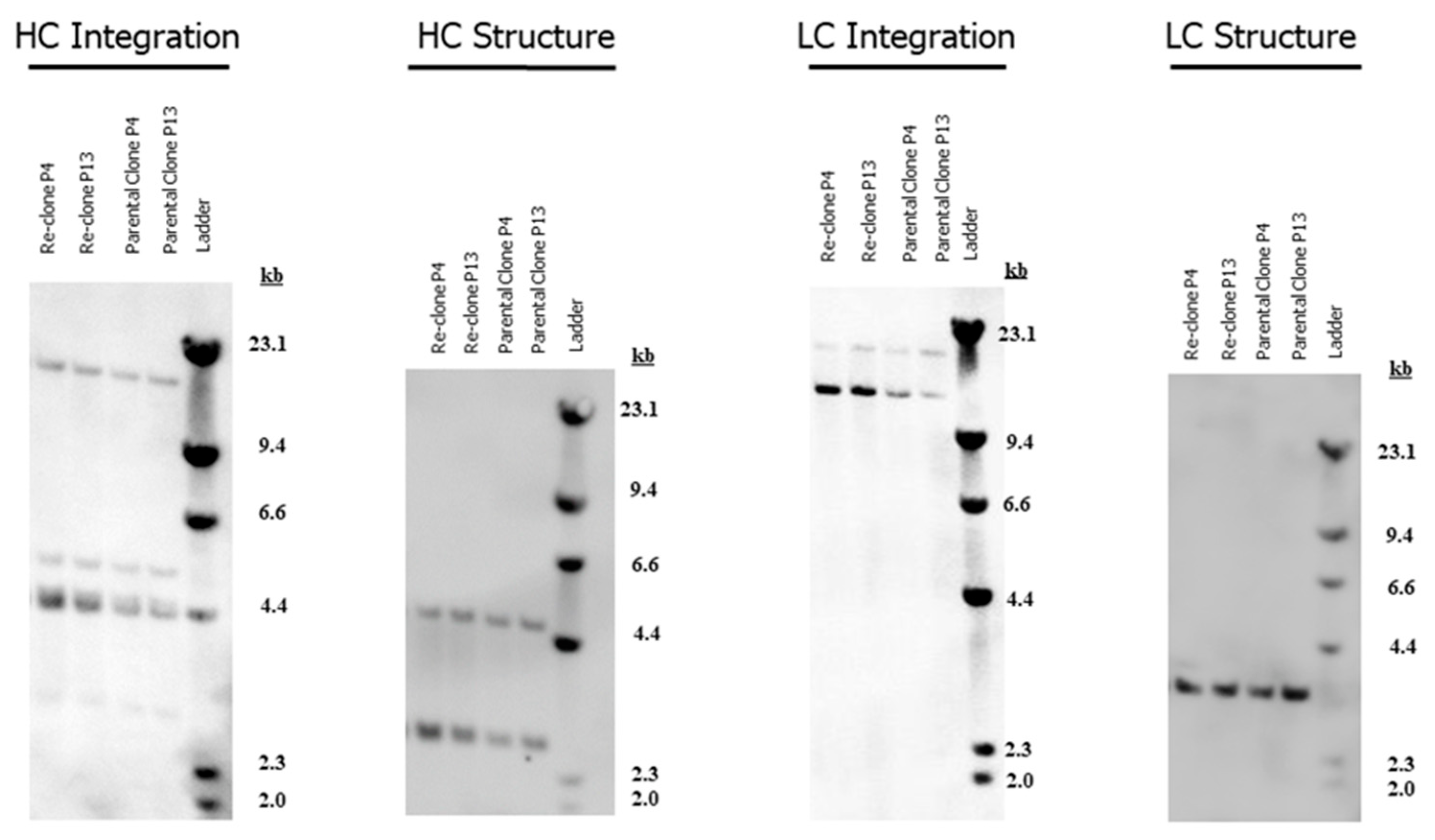
2.5. Copy Number Analysis
2.6. Southern Blot Analysis
3. Results
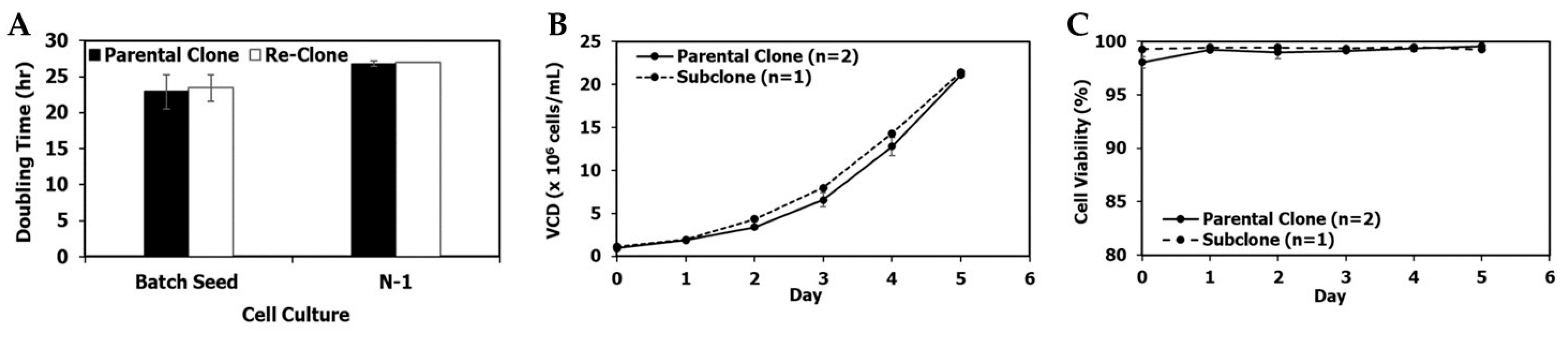
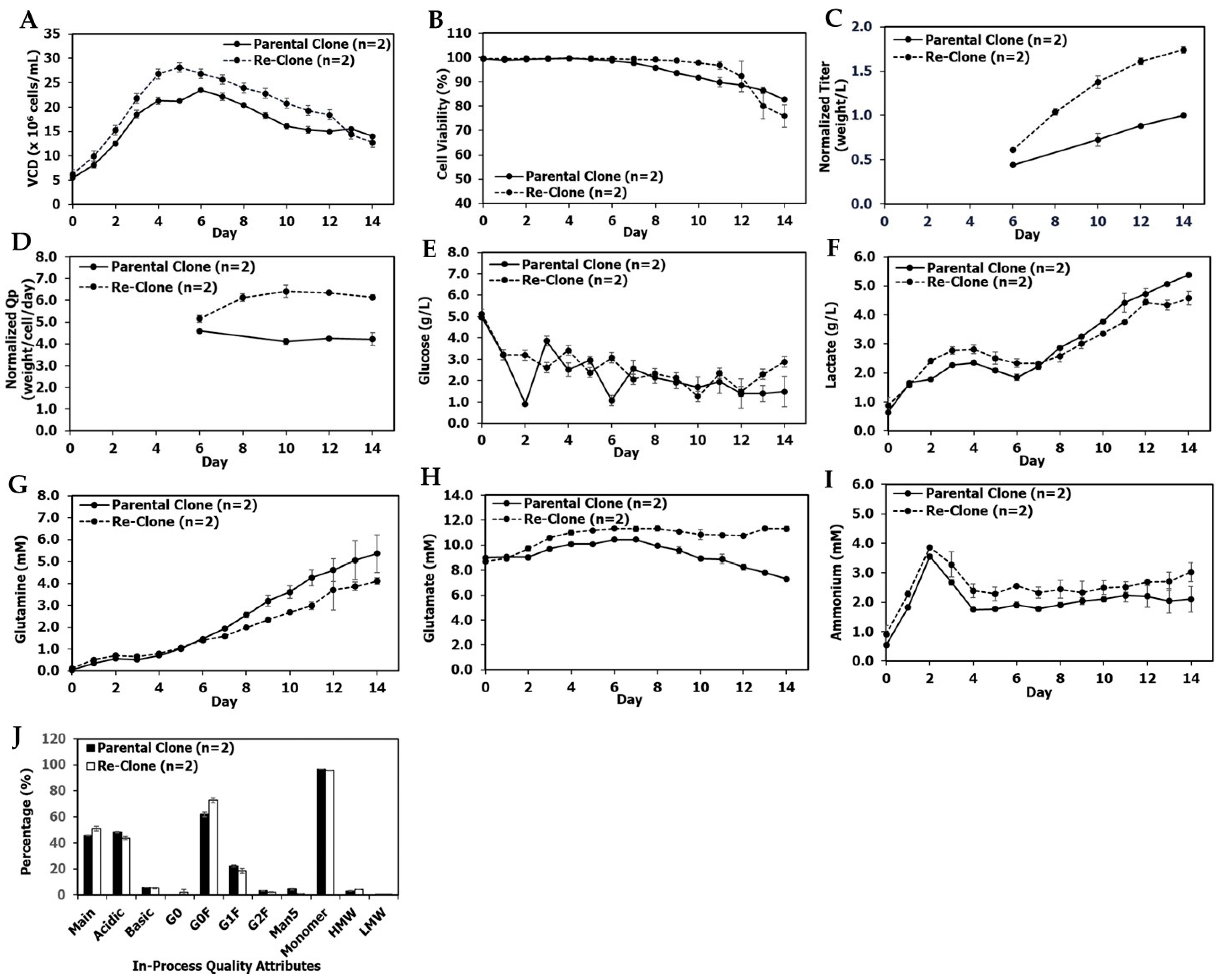
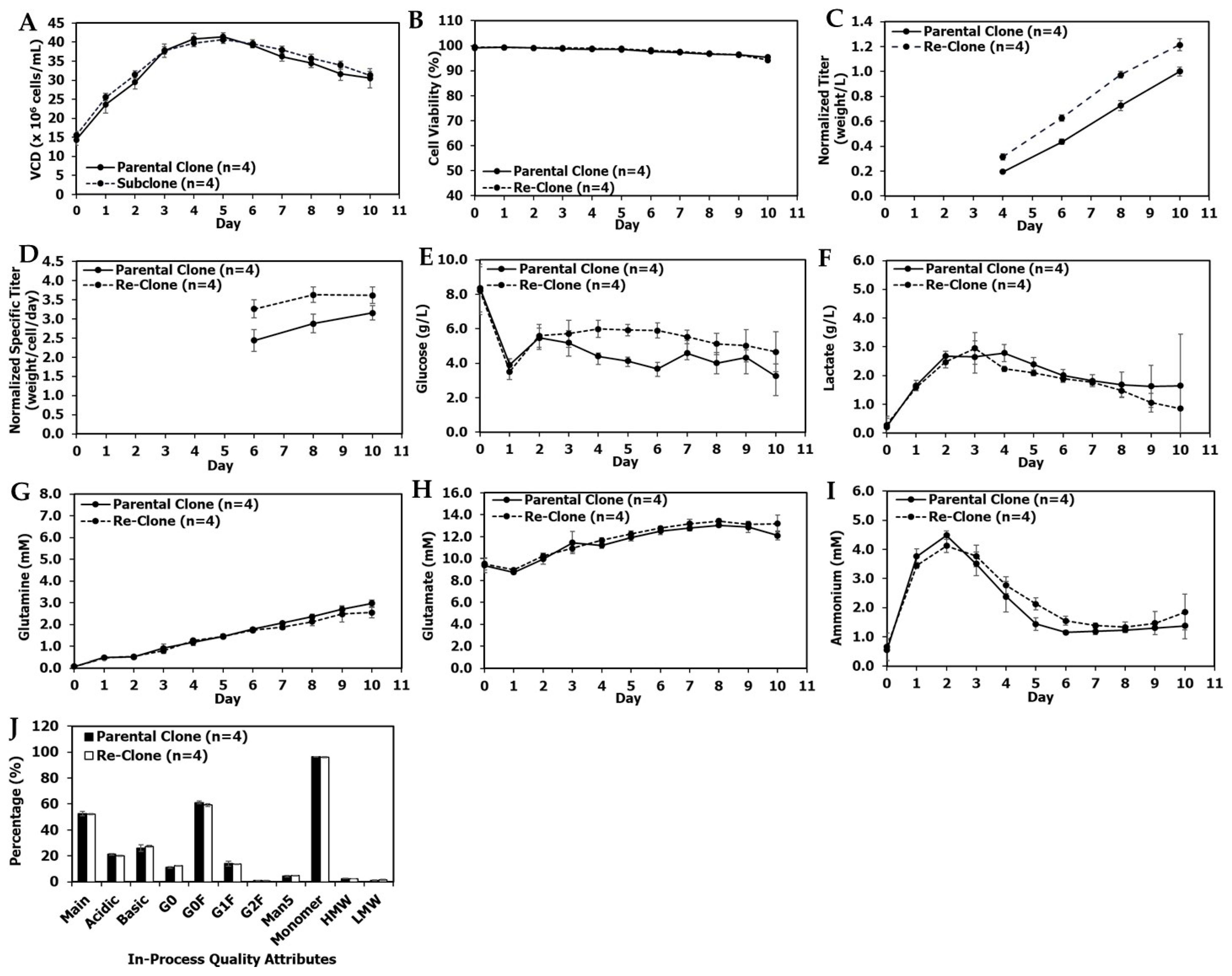
3.1. Re-Clone Process Development for mAb1 Production
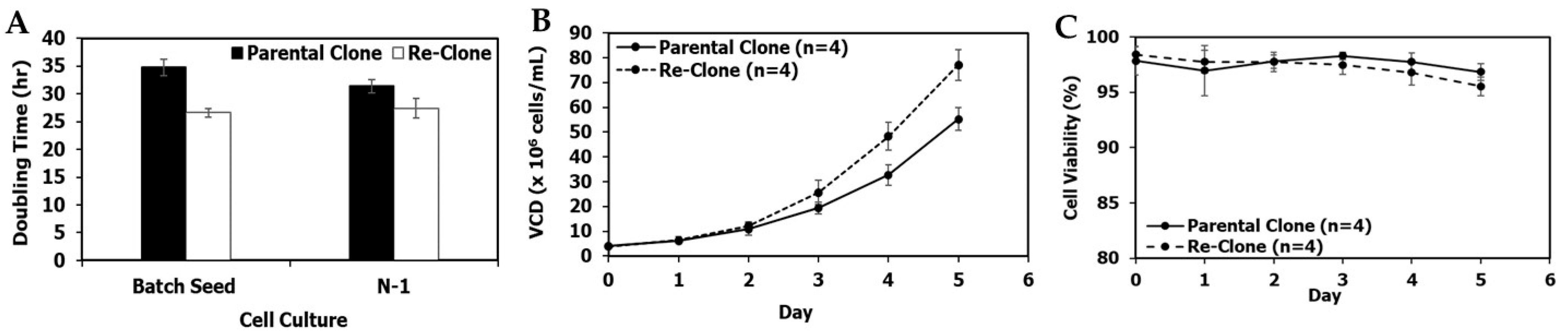
3.2. Re-Clone Process Development for mAb2 Production
3.3. Re-Clone Process Development for mAb3 Production
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reports and Data. Biologics Market to Reach USD 625.6 Million by 2026. GlobeNewswire, 10 October 2019.
- van Beers, M.M.; Bardor, M. Minimizing Immunogenicity of Biopharmaceuticals by Controlling Critical Quality Attributes of Proteins. Biotechnol. J. 2012, 7, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hu, Z.; Pacis, E.; Vijayasankaran, N.; Shen, A.; Li, F. Understanding the Intracellular Effect of Enhanced Nutrient Feeding toward High Titer Antibody Production Process. Biotechnol. Bioeng. 2011, 108, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Rehmann, M.S.; Xu, M.; Zheng, S.; Hill, C.; He, Q.; Michael CBorys, M.C.; Li, Z.J. Development of an Intensified Fed-Batch Production Platform with Doubled Titers Using N-1 Perfusion Seed for Cell Culture Manufacturing. Bioresour. Bioprocess. 2020, 7, 17. [Google Scholar] [CrossRef]
- Lin, P.C.; Chan, K.F.; Kiess, I.A.; Tan, J.; Shahreel, W.; Wong, S.Y.; Song, Z. Attenuated Glutamine Synthetase as a Selection Marker in Cho Cells to Efficiently Isolate Highly Productive Stable Cells for the Production of Antibodies and Other Biologics. mAbs 2019, 11, 965–976. [Google Scholar] [CrossRef]
- Wurm, F.M. Cho Quasispecies—Implications for Manufacturing Processes. Processes 2013, 1, 296–311. [Google Scholar] [CrossRef]
- Ha, T.K.; Lee, J.S.; Lee, G.M. Chapter 1—Platform Technology for Therapeutic Protein Production. Cell Cult. Eng. 2019, 1–22. [Google Scholar] [CrossRef]
- Galbraith, S.C.; Bhatia, H.; Liu, H.; Yoon, S. Media Formulation Optimization: Current and Future Opportunities. Curr. Opin. Chem. Eng. 2018, 22, 42–47. [Google Scholar] [CrossRef]
- Huang, Y.M.; Hu, W.; Rustandi, E.; Chang, K.; Yusuf-Makagiansar, H.; Ryll, T. Maximizing Productivity of CHO Cell-Based Fed-Batch Culture Using Chemically Defined Media Conditions and Typical Manufacturing Equipment. Biotechnol. Prog. 2010, 26, 1400–1410. [Google Scholar] [CrossRef]
- Xu, J.; Rehmann, M.S.; Xu, X.; Huang, C.; Tian, J.; Qian, N.X.; Li, Z.J. Improving Titer While Maintaining Quality of Final Formulated Drug Substance Via Optimization of CHO Cell Culture Conditions in Low-Iron Chemically Defined Media. mAbs 2018, 10, 488–499. [Google Scholar] [CrossRef]
- Xu, J.; Rehmann, M.S.; Tian, J.; He, Q.; Chen, J.; Lee, J.; Borys, M.C.; Li, Z.J. Rosmarinic Acid, a New Raw Material, Doubled Monoclonal Antibody Titer in Cell Culture Manufacturing. Biochem. Eng. J. 2020, 160, 107637. [Google Scholar] [CrossRef]
- Fan, L.; Frye, C.; Racher, A. The Use of Glutamine Synthetase as a Selection Marker: Recent Advances in Chinese Hamster Ovary Cell Line Generation Processes. Pharm. Bioprocess. 2013, 1, 487–502. [Google Scholar] [CrossRef]
- Chu, L.; Robinson, D.K. Industrial Choices for Protein Production by Large-Scale Cell Culture. Curr. Opin. Biotechnol. 2001, 12, 180–187. [Google Scholar] [CrossRef]
- Butler, M.; Spearman, M. The Choice of Mammalian Cell Host and Possibilities for Glycosylation Engineering. Curr. Opin. Biotechnol. 2014, 30, 107–112. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of Recombinant Protein Therapeutics in Cultivated Mammalian Cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Rita Costa, A.; Elisa Rodrigues, M.; Henriques, M.; Azeredo, J.; Oliveira, R. Guidelines to Cell Engineering for Monoclonal Antibody Production. Eur. J. Pharm. Biopharm. 2010, 74, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Chusainow, J.; Yang, Y.S.; Yeo, J.H.; Toh, P.C.; Asvadi, P.; Wong, N.S.; Yap, M.G. A Study of Monoclonal Antibody-Producing CHO Cell Lines: What Makes a Stable High Producer? Biotechnol. Bioeng. 2009, 102, 1182–1196. [Google Scholar] [CrossRef]
- Brown, M.E.; Renner, G.; Field, R.P.; Hassell, T. Process Development for the Production of Recombinant Antibodies Using the Glutamine Synthetase (GS) System. Cytotechnology 1992, 9, 231–236. [Google Scholar] [CrossRef]
- Noh, S.M.; Shin, S.; Lee, G.M. Comprehensive Characterization of Glutamine Synthetase-Mediated Selection for the Establishment of Recombinant CHO Cells Producing Monoclonal Antibodies. Sci. Rep. 2018, 8, 5361. [Google Scholar] [CrossRef]
- Ogata, N.; Nishimura, A.; Matsuda, T.; Kubota, M.; Omasa, T. Single-Cell Transcriptome Analyses Reveal Heterogeneity in Suspension Cultures and Clonal Markers of Cho-K1 Cells. Biotechnol. Bioeng. 2021, 118, 944–951. [Google Scholar] [CrossRef]
- Lewis, N.E.; Liu, X.; Li, Y.; Nagarajan, H.; Yerganian, G.; O’Brien, E.; Bordbar, A.; Roth, A.M.; Rosenbloom, J.; Bian, C.; et al. Genomic Landscapes of Chinese Hamster Ovary Cell Lines as Revealed by the Cricetulus Griseus Draft Genome. Nat. Biotechnol. 2013, 31, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; O’Callaghan, P.M.; Droms, K.A.; James, D.C. A Mechanistic Understanding of Production Instability in CHO Cell Lines Expressing Recombinant Monoclonal Antibodies. Biotechnol. Bioeng. 2011, 108, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, T.; Barkhordarian, H.; Tejeda, N.; Daris, K.; Yaghmour, S.; Yam, P.; Lu, F.; Goudar, C.; Munro, T.; Stevens, J. Characterization of Phenotypic and Genotypic Diversity in Subclones Derived from a Clonal Cell Line. Biotechnol. Prog. 2018, 34, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Park, J.H.; Ha, T.K.; Samoudi, M.; Lewis, N.E.; Palsson, B.O.; Kildegaard, H.F.; Lee, G.M. Revealing Key Determinants of Clonal Variation in Transgene Expression in Recombinant CHO Cells Using Targeted Genome Editing. ACS Synth. Biol. 2018, 7, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Vcelar, S.; VJadhav, V.; Melcher, M.; Auer, N.; Hrdina, A.; Sagmeister, R.; Heffner, K.; Puklowski, A.; Betenbaugh, M.; Wenger, T.; et al. Karyotype Variation of CHO Host Cell Lines over Time in Culture Characterized by Chromosome Counting and Chromosome Painting. Biotechnol. Bioeng. 2018, 115, 165–173. [Google Scholar] [CrossRef]
- Derouazi, M.; Martinet, D.; Schmutz, N.B.; Flaction, R.; Wicht, M.; Bertschinger, M.; Hacker, D.L.; Beckmann, J.S.; Wurm, F.M. Genetic Characterization of CHO Production Host DG44 and Derivative Recombinant Cell Lines. Biochem. Biophys. Res. Commun. 2006, 340, 1069–1077. [Google Scholar] [CrossRef]
- Feichtinger, J.; Hernández, I.; Fischer, C.; Hanscho, M.; Auer, N.; Hackl, M.; Jadhav, V.; Baumann, M.; Krempl, P.M.; Schmidl, C.; et al. Comprehensive Genome and Epigenome Characterization of CHO Cells in Response to Evolutionary Pressures and over Time. Biotechnol. Bioeng. 2016, 113, 2241–2253. [Google Scholar] [CrossRef] [Green Version]
- Wippermann, A.; Noll, T. DNA Methylation in CHO Cells. J. Biotechnol. 2017, 258, 206–210. [Google Scholar] [CrossRef]
- Ko, P.; Misaghi, S.; Hu, Z.; Zhan, D.; Tsukuda, J.; Yim, M.; Sanford, M.; Shaw, D.; Shiratori, M.; Snedecor, M. Probing the Importance of Clonality: Single Cell Subcloning of Clonally Derived CHO Cell Lines Yields Widely Diverse Clones Differing in Growth, Productivity, and Product Quality. Biotechnol. Prog. 2018, 34, 624–634. [Google Scholar] [CrossRef]
- Davies, S.L.; Lovelady, C.S.; Grainger, R.K.; Racher, A.J.; Young, R.J.; James, D.C. Functional Heterogeneity and Heritability in CHO Cell Populations. Biotechnol. Bioeng. 2013, 110, 260–274. [Google Scholar] [CrossRef]
- FDA. Q5D Quality of Biotechnological/Biological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products; Section 2.1.3; ICH: Geneva, Switzerland, 1998. [Google Scholar]
- Frye, C.; Deshpande, R.; Estes, S.; Francissen, K.; Joly, J.; Lubiniecki, A.; Munro, T.; Russell, R.; Wang, T.; Anderson, K. Industry View on the Relative Importance of “Clonality” of Biopharmaceutical-Producing Cell Lines. Biologicals 2016, 44, 117–122. [Google Scholar] [CrossRef]
- Yongky, A.; Xu, J.; Tian, J.; Oliveira, C.; Zhao, J.; McFarland, K.; Borys, M.C.; Li, Z.J. Process Intensification in Fed-Batch Production Bioreactors Using Non-Perfusion Seed Cultures. mAbs 2019, 11, 1502–1514. [Google Scholar] [CrossRef]
- Tian, J.; He, Q.; Oliveira, C.; Qian, Y.; Egan, S.; Xu, J.; Qian, N.X.; Langsdorf, E.; Warrack, B.; Aranibar, N.; et al. Increased Msx Level Improves Biological Productivity and Production Stability in Multiple Recombinant GS CHO Cell Lines. Eng. Life Sci. 2020, 20, 112–125. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Huang, C.; Angelo, J.; Oliveira, C.; Xu, M.; Xu, X.; Temel, D.; Ding, J.; Ghose, S.; et al. Biomanufacturing evolution from conventional to intensified processes for productivity improvement: A case study. mAbs 2020, 12, 1770669. [Google Scholar] [CrossRef]
- Huhn, S.; Chang, M.; Kumar, A.; Liu, R.; Jiang, B.; Betenbaugh, M.; Lin, H.; Nyberg, G.; Du, Z. Chromosomal instability drives convergent and divergent evolution toward advantageous inherited traits in mammalian CHO bioproduction lineages. iScience 2022, 25, 104074. [Google Scholar] [CrossRef]
- O’Callaghan, P.M.; Berthelot, M.E.; Young, R.J.; Graham, J.W.A.; Racher, A.J.; Aldana, D. Diversity in Host Clone Performance within a Chinese Hamster Ovary Cell Line. Biotechnol. Prog. 2015, 31, 1187–1200. [Google Scholar] [CrossRef]
- Wurm, M.J.; Wurm, F.M. Naming CHO cells for bio-manufacturing: Genome plasticity and variant phenotypes of cell populations in bioreactors question the relevance of old names. Biotechnol. J. 2021, 16, e2100165. [Google Scholar] [CrossRef]
- Weinguny, M.; Klanert, G.; Eisenhut, P.; Lee, I.; Timp, W.; Borth, N. Subcloning Induces Changes in the DNA-Methylation Pattern of Outgrowing Chinese Hamster Ovary Cell Colonies. Biotechnol. J. 2021, 16, 2000350. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | HC GCN | LC GCN | |
|---|---|---|---|
| mAb1 | Parental clone | 94.9 ± 18.6 | 148.9 ± 23.7 |
| Re-clone | 84.7 ± 10.7 | 131.0 ± 8.7 | |
| mAb2 | Parental clone | 40.5 ± 9.1 | 37.2 ± 6.8 |
| Re-clone | 39.0 ± 1.0 | 35.7 ± 0.8 | |
| mAb3 | Parental clone | 49.4 ± 1.1 | 49.7 ± 0.4 |
| Re-clone | 49.1 ± 1.7 | 49.6 ± 0.9 |
| Cell Line | Clone | Normalized Titer |
|---|---|---|
| mAb1 | Parental clone | 1.00 ± 0.02 |
| Re-clone | 1.62 ± 0.09 | |
| mAb2 | Parental clone | 1.00 ± 0.01 |
| Re-clone | 1.25 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Q.; Rehmann, M.S.; Tian, J.; Xu, J.; Sabino, L.; Vandermark, E.; Basson, Z.; Po, I.; Bierilo, K.; Tremml, G.; et al. Improved Titer in Late-Stage Mammalian Cell Culture Manufacturing by Re-Cloning. Bioengineering 2022, 9, 173. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9040173
He Q, Rehmann MS, Tian J, Xu J, Sabino L, Vandermark E, Basson Z, Po I, Bierilo K, Tremml G, et al. Improved Titer in Late-Stage Mammalian Cell Culture Manufacturing by Re-Cloning. Bioengineering. 2022; 9(4):173. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9040173
Chicago/Turabian StyleHe, Qin, Matthew S. Rehmann, Jun Tian, Jianlin Xu, Luzmary Sabino, Erik Vandermark, Ziev Basson, Iris Po, Kathleen Bierilo, Gabi Tremml, and et al. 2022. "Improved Titer in Late-Stage Mammalian Cell Culture Manufacturing by Re-Cloning" Bioengineering 9, no. 4: 173. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering9040173