The Needle in the Haystack—Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Ethics Statement
2.2. Whole Genome Sequencing (WGS)
2.3. Whole Genome Bisulfite Sequencing (WGBS)
2.4. Filtering for Disease-Relevant Genetic and Epigenetic Alterations
2.5. Statistics
3. Results
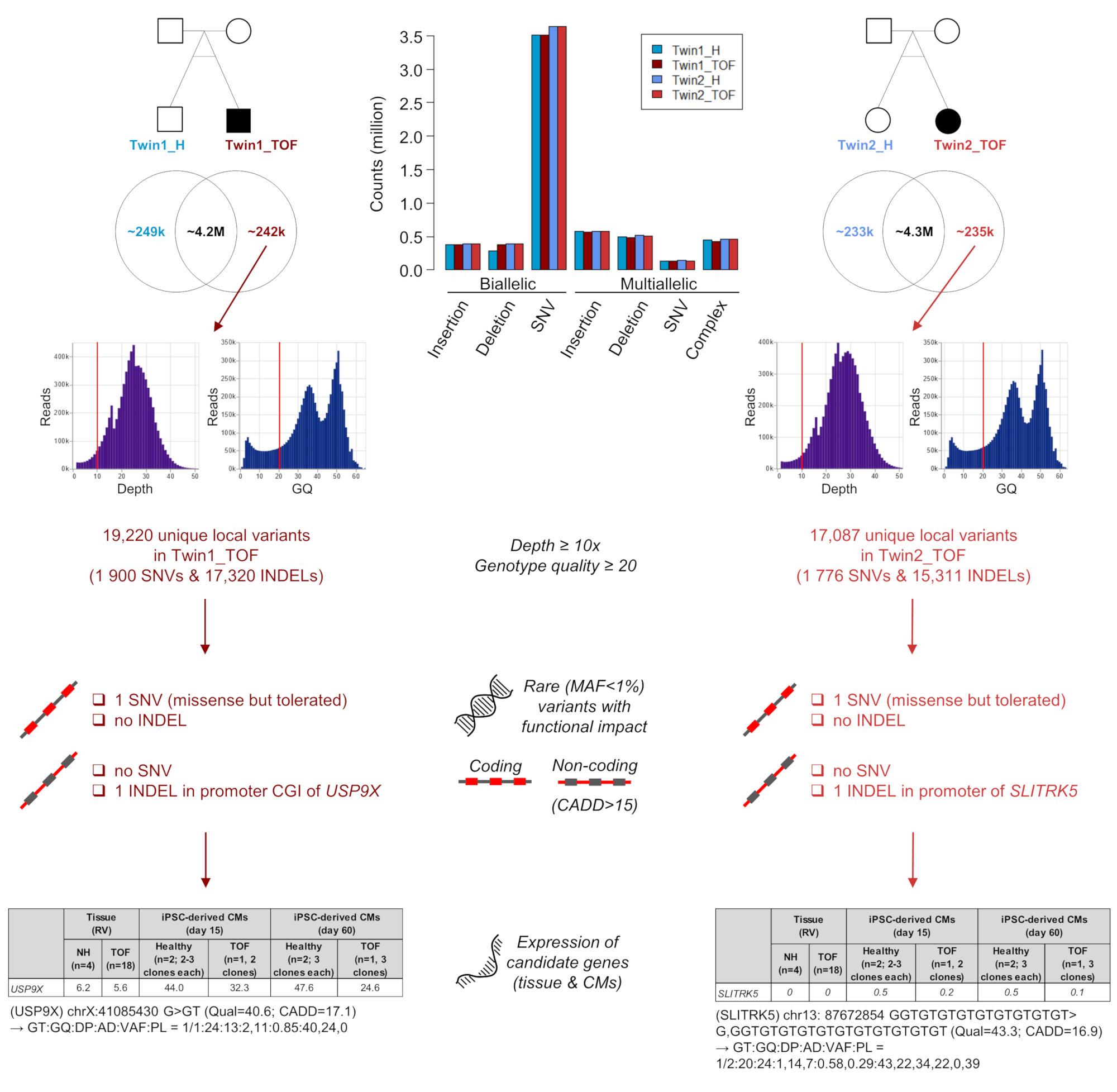
3.1. Genomic Variations in Affected Twins
3.2. Structural Genomic Variations in Affected Twins
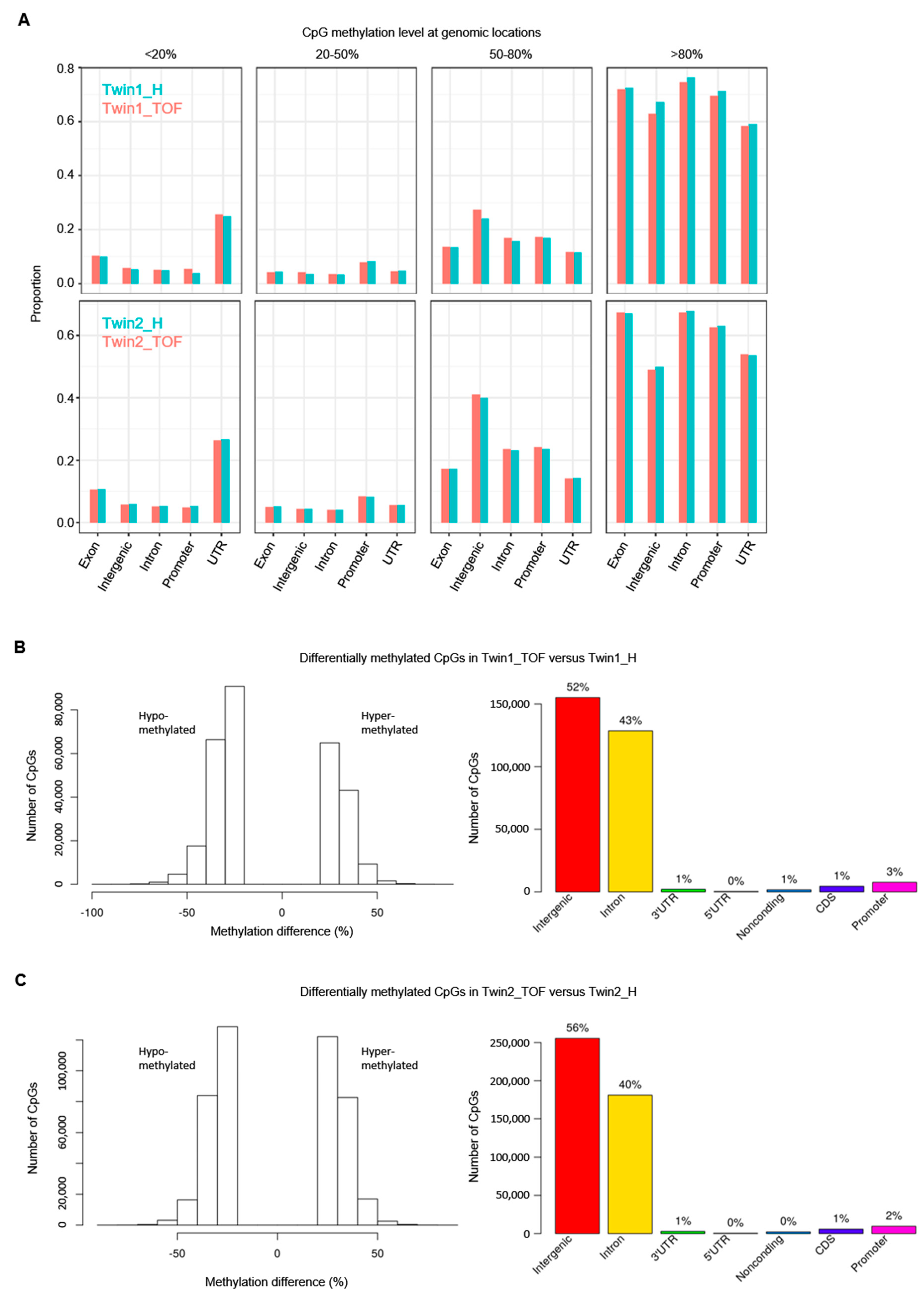
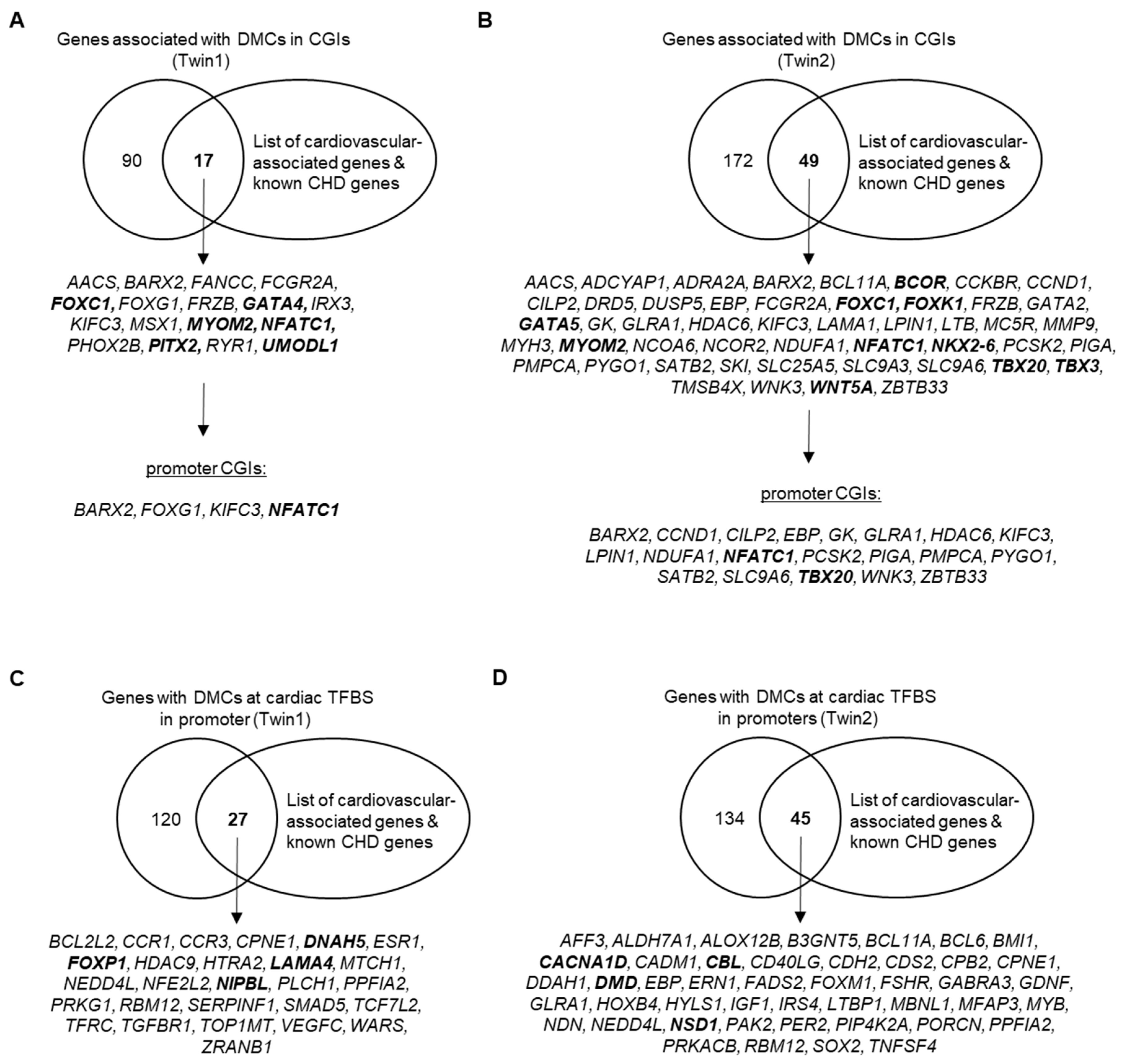
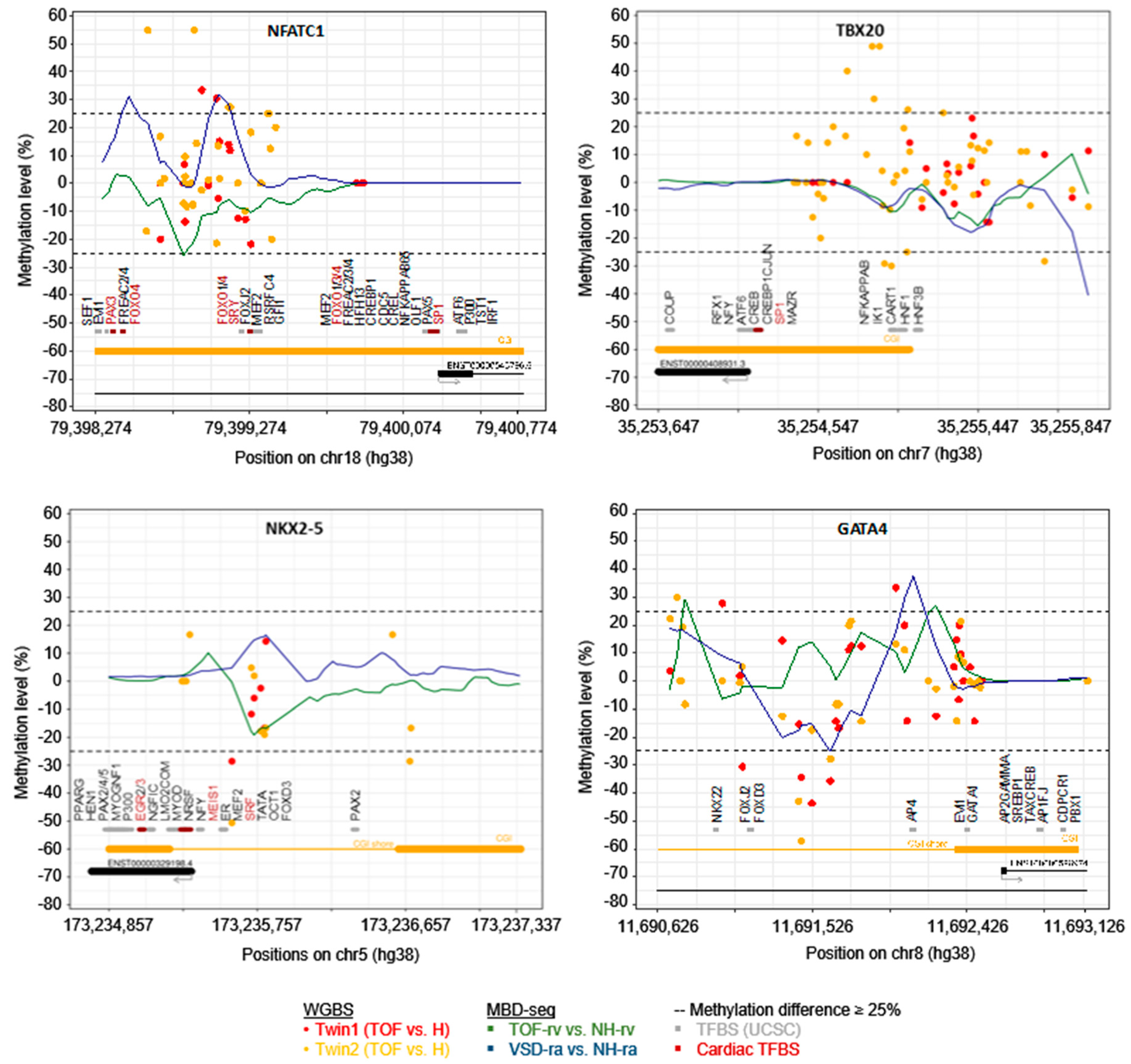
3.3. DNA Methylation Differences between Discordant Twins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [Green Version]
- Nora, J.J. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. The genetic-environmental interaction. Circulation 1968, 38, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickert-Sperling, S.; Kelly, R.G.; Driscoll, D.J. (Eds.) Congenital Heart Diseases: The Broken Heart; Clinical Features, Human Genetics and Molecular Pathways; Springer: Wien, Austria, 2016; ISBN 978-3-7091-1883-2. [Google Scholar]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, T.A.; Troelsen, K.d.L.L.; Larsen, L.A. Of mice and men: Molecular genetics of congenital heart disease. Cell. Mol. Life Sci. 2014, 71, 1327–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.A.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Dorn, C.; Schueler, M.; Dunkel, I.; Schlesinger, J.; Mebus, S.; Alexi-Meskishvili, V.; Perrot, A.; Wassilew, K.; Timmermann, B.; et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum. Mol. Genet. 2014, 23, 3115–3128. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Dorn, C.; Cui, H.; Dunkel, I.; Schulz, K.; Schoenhals, S.; Sun, W.; Berger, F.; Chen, W.; Sperling, S.R. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc. Res. 2016, 112, 464–477. [Google Scholar] [CrossRef]
- Yuan, W.; Xia, Y.; Bell, C.G.; Yet, I.; Ferreira, T.; Ward, K.J.; Gao, F.; Loomis, A.K.; Hyde, C.L.; Wu, H.; et al. An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat. Commun. 2014, 5, 5719. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.; Reichenberg, A.; Willfors, C.; Austin, C.; Gennings, C.; Berggren, S.; Lichtenstein, P.; Anckarsäter, H.; Tammimies, K.; Bölte, S. Fetal and postnatal metal dysregulation in autism. Nat. Commun. 2017, 8, 15493. [Google Scholar] [CrossRef]
- Castillo-Fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome Med. 2014, 6, 60. [Google Scholar] [CrossRef]
- Lyu, G.; Zhang, C.; Ling, T.; Liu, R.; Zong, L.; Guan, Y.; Huang, X.; Sun, L.; Zhang, L.; Li, C.; et al. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genom. 2018, 19, 428. [Google Scholar] [CrossRef]
- Andrews, S.R. FastQC. Available online: https://github.com/s-andrews/FastQC (accessed on 1 August 2020).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 1 August 2020).
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Devon, Ryan MethylDackel. Available online: https://github.com/dpryan79/MethylDackel (accessed on 1 August 2020).
- Park, Y.; Figueroa, M.E.; Rozek, L.S.; Sartor, M.A. MethylSig: A whole genome DNA methylation analysis pipeline. Bioinformatics 2014, 30, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- May, D.; Blow, M.J.; Kaplan, T.; McCulley, D.J.; Jensen, B.C.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2011, 44, 89–93. [Google Scholar] [CrossRef]
- Kaynak, B.; von Heydebreck, A.; Mebus, S.; Seelow, D.; Hennig, S.; Vogel, J.; Sperling, H.-P.; Pregla, R.; Alexi-Meskishvili, V.; Hetzer, R.; et al. Genome-wide array analysis of normal and malformed human hearts. Circulation 2003, 107, 2467–2474. [Google Scholar] [CrossRef]
- Toenjes, M.; Schueler, M.; Hammer, S.; Pape, U.J.; Fischer, J.J.; Berger, F.; Vingron, M.; Sperling, S. Prediction of cardiac transcription networks based on molecular data and complex clinical phenotypes. Mol. Biosyst. 2008, 4, 589–598. [Google Scholar] [CrossRef]
- Ricci, M.; Xu, Y.; Hammond, H.L.; Willoughby, D.A.; Nathanson, L.; Rodriguez, M.M.; Vatta, M.; Lipshultz, S.E.; Lincoln, J. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLoS ONE 2012, 7, e29784. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, J.; Tönjes, M.; Schueler, M.; Zhang, Q.; Dunkel, I.; Sperling, S.R. Evaluation of the LightCycler 1536 Instrument for high-throughput quantitative real-time PCR. Methods 2010, 50, S19–S22. [Google Scholar] [CrossRef]
- Rodemoyer, A.; Kibiryeva, N.; Bair, A.; Marshall, J.; O’Brien, J.E.; Bittel, D.C. A tissue-specific gene expression template portrays heart development and pathology. Hum. Genom. 2014, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Wang, H.; Ma, X.; Qian, Y.; Zhang, P.; Wu, Y.; Zheng, F.; Chen, L.; Huang, G.; Ma, D. LINE-1 methylation status and its association with tetralogy of fallot in infants. BMC Med. Genom. 2012, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Qian, Y.; Zhang, P.; Wu, Y.; Wang, H.; Ma, X.; Chen, L.; Ma, D.; Huang, G. Association of promoter methylation statuses of congenital heart defect candidate genes with Tetralogy of Fallot. J. Transl. Med. 2014, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Erickson, S.W.; MacLeod, S.L.; Cleves, M.A.; Hu, P.; Karim, M.A.; Hobbs, C.A. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS ONE 2011, 6, e16506. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Qian, Y.; Wang, H.; Ma, X.; Zhang, P.; Diao, L.; An, Q.; Chen, L.; Ma, D.; Huang, G. DNA methylation status of NKX2-5, GATA4 and HAND1 in patients with tetralogy of fallot. BMC Med. Genom. 2013, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Bansal, V.; Dorn, C.; Grunert, M.; Klaassen, S.; Hetzer, R.; Berger, F.; Sperling, S.R. Outlier-based identification of copy number variations using targeted resequencing in a small cohort of patients with Tetralogy of Fallot. PLoS ONE 2014, 9, e85375. [Google Scholar] [CrossRef]
- Warburton, D.; Ronemus, M.; Kline, J.; Jobanputra, V.; Williams, I.; Anyane-Yeboa, K.; Chung, W.; Yu, L.; Wong, N.; Awad, D.; et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 2014, 133, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Greenway, S.C.; Pereira, A.C.; Lin, J.C.; DePalma, S.R.; Israel, S.J.; Mesquita, S.M.; Ergul, E.; Conta, J.H.; Korn, J.M.; McCarroll, S.A.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Tomita-Mitchell, A.; Mahnke, D.K.; Struble, C.A.; Tuffnell, M.E.; Stamm, K.D.; Hidestrand, M.; Harris, S.E.; Goetsch, M.A.; Simpson, P.M.; Bick, D.P.; et al. Human gene copy number spectra analysis in congenital heart malformations. Physiol. Genom. 2012, 44, 518–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, A.R.; Chang, S.-W.; Koenig, S.N.; Zinn, A.R.; Garg, V. Submicroscopic chromosomal copy number variations identified in children with hypoplastic left heart syndrome. Pediatr. Cardiol. 2012, 33, 757–763. [Google Scholar] [CrossRef]
- de Souza, K.R.; Mergener, R.; Huber, J.; Campos Pellanda, L.; Riegel, M. Cytogenomic Evaluation of Subjects with Syndromic and Nonsyndromic Conotruncal Heart Defects. Biomed. Res. Int. 2015, 2015, 401941. [Google Scholar] [CrossRef]
- Geng, J.; Picker, J.; Zheng, Z.; Zhang, X.; Wang, J.; Hisama, F.; Brown, D.W.; Mullen, M.P.; Harris, D.; Stoler, J.; et al. Chromosome microarray testing for patients with congenital heart defects reveals novel disease causing loci and high diagnostic yield. BMC Genom. 2014, 15, 1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittel, D.C.; Zhou, X.-G.; Kibiryeva, N.; Fiedler, S.; O’Brien, J.E.; Marshall, J.; Yu, S.; Liu, H.-Y. Ultra high-resolution gene centric genomic structural analysis of a non-syndromic congenital heart defect, Tetralogy of Fallot. PLoS ONE 2014, 9, e87472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo-Gómez, A.; Arteaga-Vázquez, J.; Svyryd, Y.; Calderón-Colmenero, J.; Zamora-González, C.; Vargas-Alarcón, G.; Mutchinick, O.M. Identification of Copy Number Variations in Isolated Tetralogy of Fallot. Pediatr. Cardiol. 2015, 36, 1642–1646. [Google Scholar] [CrossRef]
- Hitz, M.-P.; Lemieux-Perreault, L.-P.; Marshall, C.; Feroz-Zada, Y.; Davies, R.; Yang, S.W.; Lionel, A.C.; D’Amours, G.; Lemyre, E.; Cullum, R.; et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012, 8, e1002903. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, F.; Larsen, L.A.; Zhang, L.; Tümer, Z.; Tommerup, N.; Chen, W.; Jacobsen, J.R.; Schubert, M.; Jurkatis, J.; Tzschach, A.; et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J. Med. Genet. 2008, 45, 704–709. [Google Scholar] [CrossRef]
- Soemedi, R.; Wilson, I.J.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.D.; Shen, P.; Fung, E.; Karakikes, I.; Zhang, A.; InanlooRahatloo, K.; Odegaard, J.; Sallam, K.; Davis, R.W.; Lui, G.K.; et al. A rapid, high-quality, cost-effective, comprehensive and expandable targeted next-generation sequencing assay for inherited heart diseases. Circ. Res. 2015, 117, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Silversides, C.K.; Lionel, A.C.; Costain, G.; Merico, D.; Migita, O.; Liu, B.; Yuen, T.; Rickaby, J.; Thiruvahindrapuram, B.; Marshall, C.R.; et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012, 8, e1002843. [Google Scholar] [CrossRef]
- Richards, A.A.; Santos, L.J.; Nichols, H.A.; Crider, B.P.; Elder, F.F.; Hauser, N.S.; Zinn, A.R.; Garg, V. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr. Res. 2008, 64, 358–363. [Google Scholar] [CrossRef]
- O’Brien, J.E.; Kibiryeva, N.; Zhou, X.-G.; Marshall, J.A.; Lofland, G.K.; Artman, M.; Chen, J.; Bittel, D.C. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012, 5, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA expression in hypoplastic left heart syndrome. J. Card. Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Appelt, S.; Dunkel, I.; Berger, F.; Sperling, S.R. Altered microRNA and target gene expression related to Tetralogy of Fallot. Sci. Rep. 2019, 9, 19063. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Appelt, S.; Schönhals, S.; Mika, K.; Cui, H.; Cooper, A.; Cyganek, L.; Guan, K.; Sperling, S.R. Induced pluripotent stem cells of patients with Tetralogy of Fallot reveal transcriptional alterations in cardiomyocyte differentiation. Sci. Rep. 2020, 10, 10921. [Google Scholar] [CrossRef]
- Wu, B.; Baldwin, H.S.; Zhou, B. Nfatc1 directs the endocardial progenitor cells to make heart valve primordium. Trends Cardiovasc. Med. 2013, 23, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Kirk, E.P.; Sunde, M.; Costa, M.W.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Dorn, C.; Rickert-Sperling, S. Cardiac Transcription Factors and Regulatory Networks. In Congenital Heart Diseases: The Broken Heart; Rickert-Sperling, S., Kelly, R.G., Driscoll, D.J., Eds.; Springer: Vienna, Austria, 2016; pp. 139–152. ISBN 978-3-7091-1882-5. [Google Scholar]
- Boccuni, P.; MacGrogan, D.; Scandura, J.M.; Nimer, S.D. The human L(3)MBT polycomb group protein is a transcriptional repressor and interacts physically and functionally with TEL (ETV6). J. Biol. Chem. 2003, 278, 15412–15420. [Google Scholar] [CrossRef] [Green Version]
- Trojer, P.; Li, G.; Sims, R.J.; Vaquero, A.; Kalakonda, N.; Boccuni, P.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Nimer, S.D.; et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 2007, 129, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Feng, S.; Joo, J.W.J.; Jacobsen, S.E.; Pellegrini, M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011, 12, R62. [Google Scholar] [CrossRef] [Green Version]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Yehya, A.; Souki, R.; Bitar, F.; Nemer, G. Differential duplication of an intronic region in the NFATC1 gene in patients with congenital heart disease. Genome 2006, 49, 1092–1098. [Google Scholar] [CrossRef]
- Abdul-Sater, Z.; Yehya, A.; Beresian, J.; Salem, E.; Kamar, A.; Baydoun, S.; Shibbani, K.; Soubra, A.; Bitar, F.; Nemer, G. Two heterozygous mutations in NFATC1 in a patient with Tricuspid Atresia. PLoS ONE 2012, 7, e49532. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, J.K.; Mileikovskaia, M.; Koshiba-Takeuchi, K.; Heidt, A.B.; Mori, A.D.; Arruda, E.P.; Gertsenstein, M.; Georges, R.; Davidson, L.; Mo, R.; et al. Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 2005, 132, 2463–2474. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, T.; Pu, T.; Cao, R.; Long, F.; Chen, S.; Sun, K.; Xu, R. Copy Number Variants and Exome Sequencing Analysis in Six Pairs of Chinese Monozygotic Twins Discordant for Congenital Heart Disease. Twin Res. Hum. Genet. 2017, 20, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Breckpot, J.; Thienpont, B.; Gewillig, M.; Allegaert, K.; Vermeesch, J.R.; Devriendt, K. Differences in Copy Number Variation between Discordant Monozygotic Twins as a Model for Exploring Chromosomal Mosaicism in Congenital Heart Defects. Mol. Syndromol. 2012, 2, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Chaiyasap, P.; Kulawonganunchai, S.; Srichomthong, C.; Tongsima, S.; Suphapeetiporn, K.; Shotelersuk, V. Whole genome and exome sequencing of monozygotic twins with trisomy 21, discordant for a congenital heart defect and epilepsy. PLoS ONE 2014, 9, e100191. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.S.; Bonow, R.O.; Stolker, J.M.; Braddock, S.R.; Lee, R. Discordant Aortic Valve Morphology in Monozygotic Twins: A Clinical Case Series. JAMA Cardiol. 2016, 1, 1043–1047. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Thiele, H.; Bartmann, P.; Hilger, A.C.; Berg, C.; Herberg, U.; Klingmüller, D.; Nürnberg, P.; Ludwig, M.; Reutter, H. Whole-Exome Sequencing in Nine Monozygotic Discordant Twins. Twin Res. Hum. Genet. 2016, 19, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Erlich, Y. Blood ties: Chimerism can mask twin discordance in high-throughput sequencing. Twin Res. Hum. Genet. 2011, 14, 137–143. [Google Scholar] [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Lindinger, A.; Schwedler, G.; Hense, H.-W. Prevalence of congenital heart defects in newborns in Germany: Results of the first registration year of the PAN Study (July 2006 to June 2007). Klin. Padiatr. 2010, 222, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Cosselman, K.E.; Navas-Acien, A.; Kaufman, J.D. Environmental factors in cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 627–642. [Google Scholar] [CrossRef]
- Porter, G.A. Environmental Signals. In Congenital Heart Diseases: The Broken Heart; Rickert-Sperling, S., Kelly, R.G., Driscoll, D.J., Eds.; Springer: Vienna, Austria, 2016; pp. 223–235. ISBN 978-3-7091-1882-5. [Google Scholar]
- Abu-Halima, M.; Weidinger, J.; Poryo, M.; Henn, D.; Keller, A.; Meese, E.; Abdul-Khaliq, H. Micro-RNA signatures in monozygotic twins discordant for congenital heart defects. PLoS ONE 2019, 14, e0226164. [Google Scholar] [CrossRef] [Green Version]
- Czyz, W.; Morahan, J.M.; Ebers, G.C.; Ramagopalan, S.V. Genetic, environmental and stochastic factors in monozygotic twin discordance with a focus on epigenetic differences. BMC Med. 2012, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020. [Google Scholar] [CrossRef]
- Karatza, A.A.; Wolfenden, J.L.; Taylor, M.J.O.; Wee, L.; Fisk, N.M.; Gardiner, H.M. Influence of twin-twin transfusion syndrome on fetal cardiovascular structure and function: Prospective case-control study of 136 monochorionic twin pregnancies. Heart 2002, 88, 271–277. [Google Scholar] [CrossRef]
- Marceau, K.; McMaster, M.T.B.; Smith, T.F.; Daams, J.G.; van Beijsterveldt, C.E.M.; Boomsma, D.I.; Knopik, V.S. The Prenatal Environment in Twin Studies: A Review on Chorionicity. Behav. Genet. 2016, 46, 286–303. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Kidd, A.; Zuccollo, J.; Parker, S.; Richardson, V.; Tait, J.; Pringle, K.C. A case of amyoplasia in a monochorionic twin pregnancy: A sequela from twin-twin transfusion syndrome? Fetal Diagn. Ther. 2009, 25, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.W.; Wang, C.; Yao, J.; Walsh, S.A.; Sawatzke, A.B.; Hu, S.; Sunderland, J.J.; Segar, J.L.; Ponto, L.L.B. Effect of insulin and dexamethasone on fetal assimilation of maternal glucose. Endocrinology 2011, 152, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Mahle, W.T. What we can learn from twins: Congenital heart disease in the danish twin registry. Circulation 2013, 128, 1173–1174. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Twin1_TOF vs. Twin1_H | Twin2_TOF vs. Twin2_H | |
|---|---|---|
| Differentially methylated CpGs (DMCs) with ≥25% methylation difference | 299,643 | 457,108 |
| DMCs in promoters | 18,052 | 22,537 |
| DMCs in CpG islands (CGIs) | 397 | 713 |
| DMCs in promoter CGIs | 131 | 429 |
| DMCs in CGI shores | 17,337 | 21,455 |
| DMCs at TFBS | 23,716 | 31,789 |
| DMCs at cardiac TFBS | 2066 | 2751 |
| DMCs at TFBS in promoters | 2125 | 2929 |
| DMCs at cardiac TFBS in promoters | 215 | 264 |
| DMCs in cardiac enhancers (p300) | 2042 | 2518 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grunert, M.; Appelt, S.; Grossfeld, P.; Sperling, S.R. The Needle in the Haystack—Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot. J. Cardiovasc. Dev. Dis. 2020, 7, 55. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7040055
Grunert M, Appelt S, Grossfeld P, Sperling SR. The Needle in the Haystack—Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot. Journal of Cardiovascular Development and Disease. 2020; 7(4):55. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7040055
Chicago/Turabian StyleGrunert, Marcel, Sandra Appelt, Paul Grossfeld, and Silke R. Sperling. 2020. "The Needle in the Haystack—Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot" Journal of Cardiovascular Development and Disease 7, no. 4: 55. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7040055