
PDGFRα: Expression and Function during Mitral Valve Morphogenesis

 , ,
, ,  , , ,
, , ,  and
and 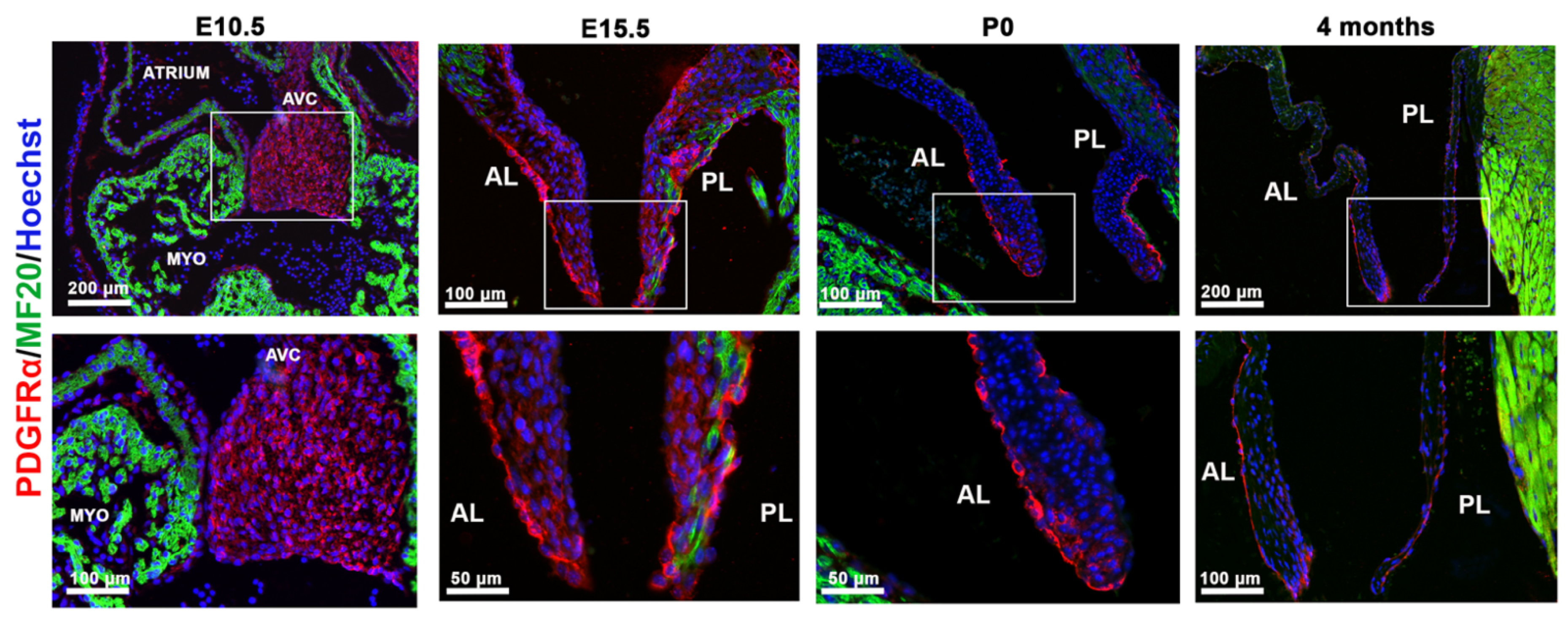{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Husbandry and Genotyping
2.2. Immunohistochemistry Staining and Imaging
2.3. Three-dimensional Valve Reconstructions
2.4. Image Analyses
2.5. Human Valve Endothelial Cell Culture and Treatments
2.6. Western Blotting
2.7. In Situ Hybridization
2.8. Statistical Analysis
3. Results
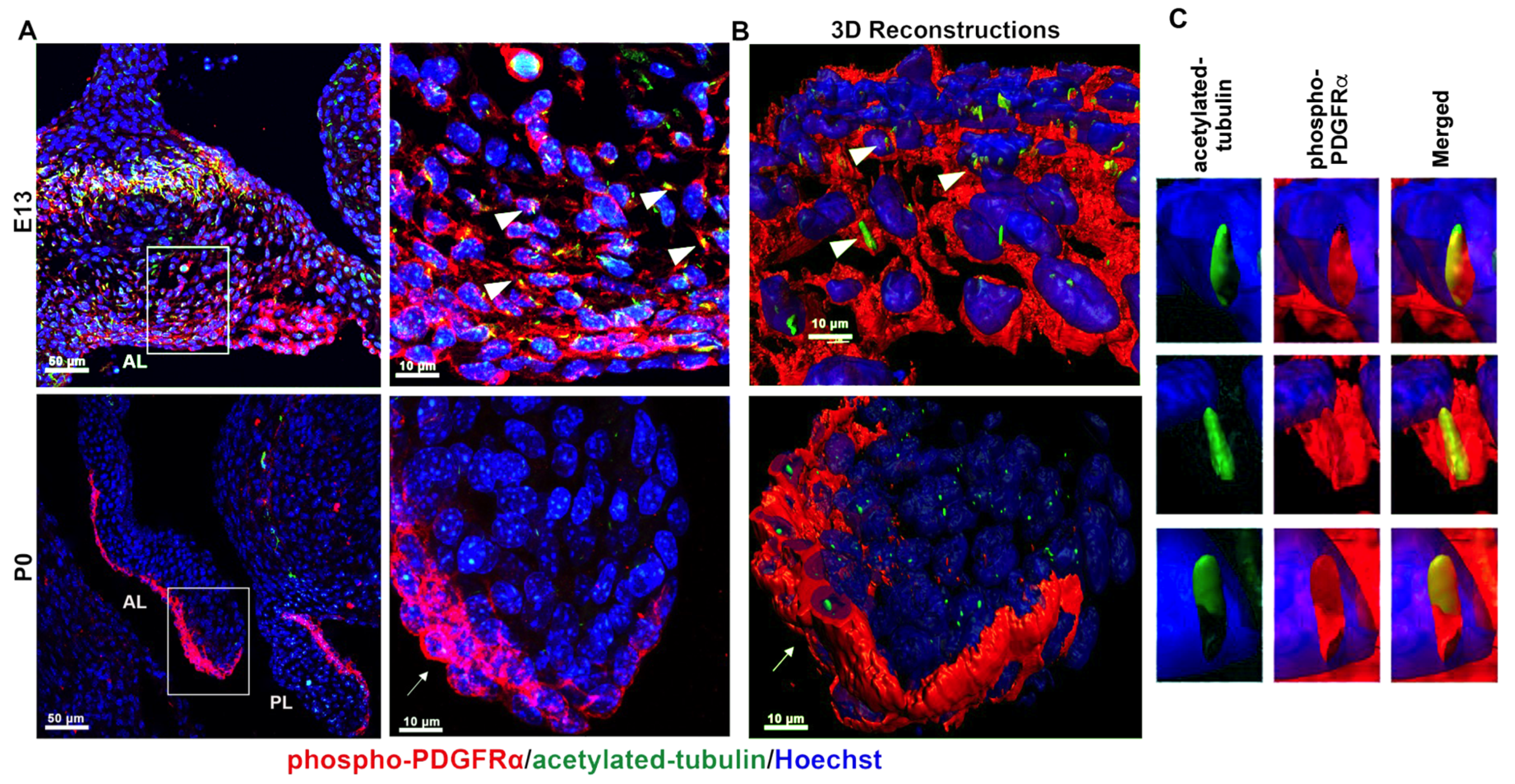
3.1. PDGFRα Is Spatially and Temporally Regulated in Developing Mitral Valves
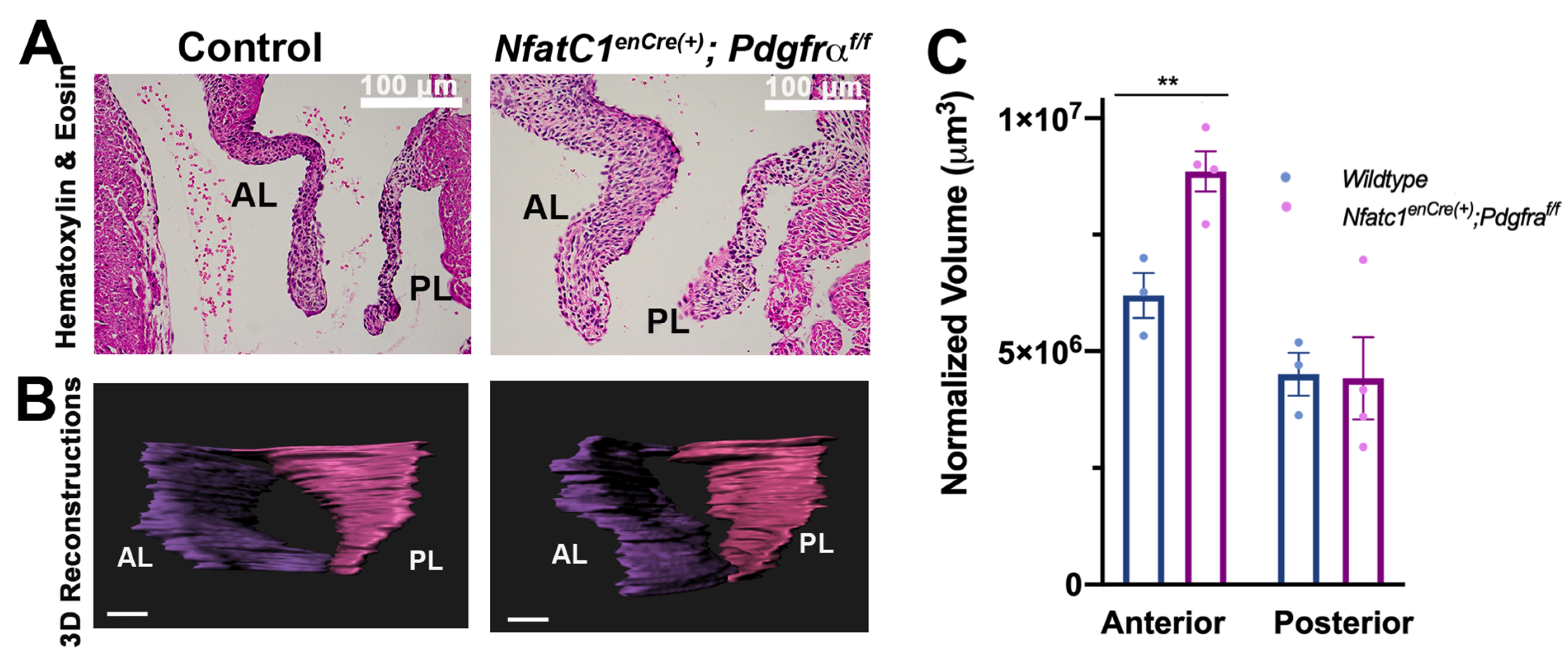
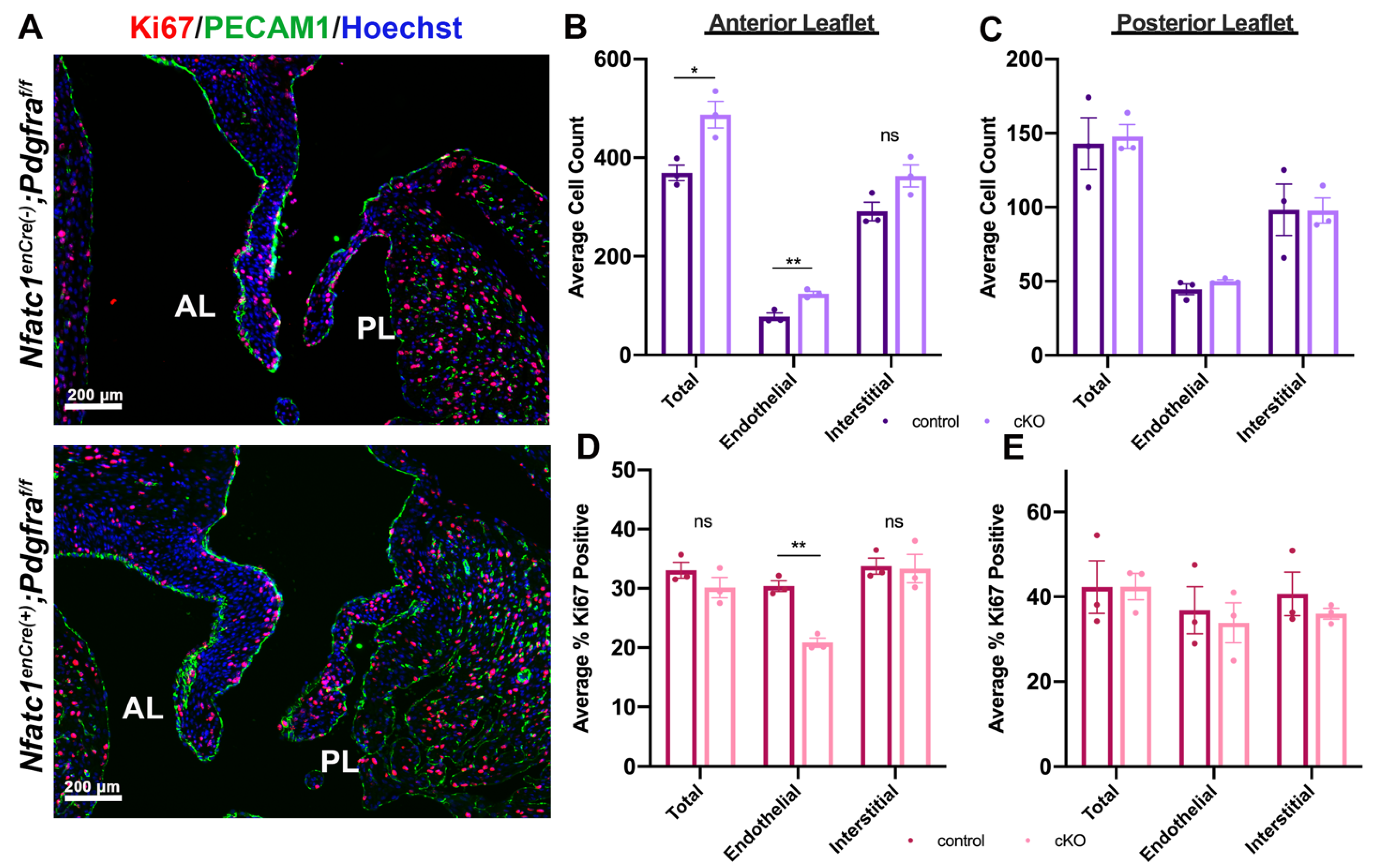
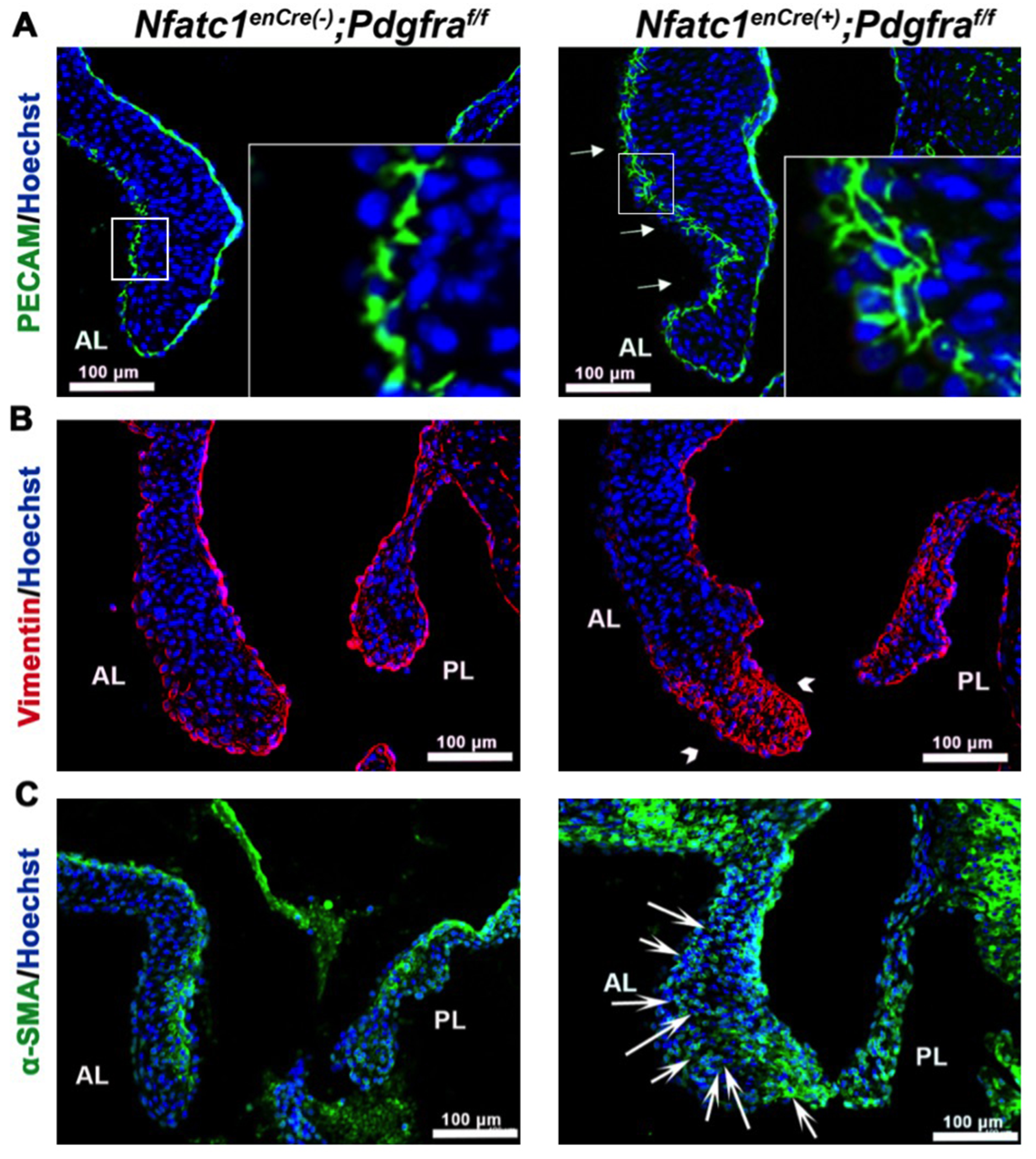
3.2. Genetic Ablation of Pdgfra in the Valve Endocardium Leads to Abnormal Valve Morphogenesis
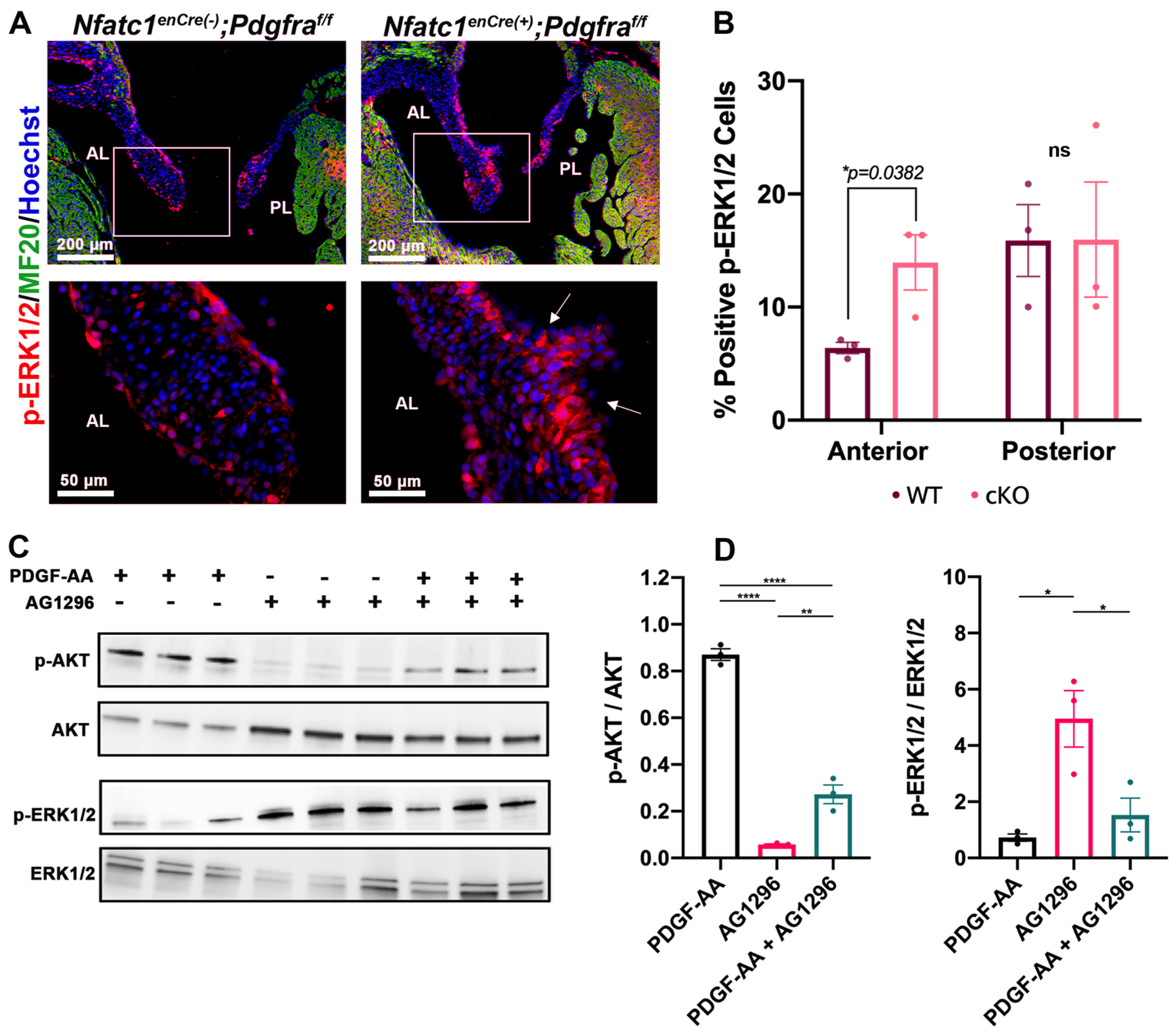
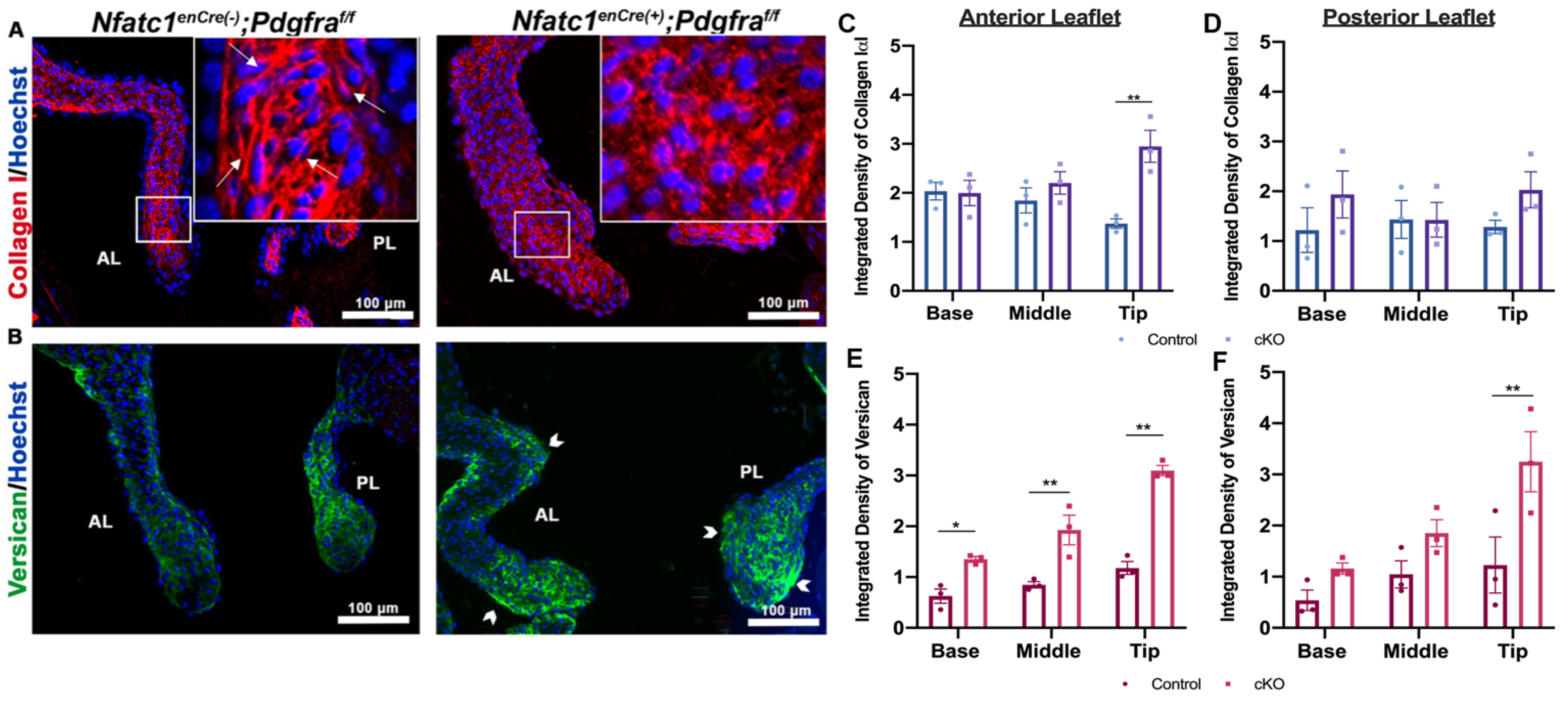
3.3. Perturbation of Pdgfra Results in Hyperplastic Valves, Altered Downstream Signaling and Changes in ECM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Arcy, J.L.; Prendergast, B.D.; Chambers, J.B.; Ray, S.G.; Bridgewater, B. Valvular heart disease: The next cardiac epidemic. Heart 2010, 97, 91–93. [Google Scholar] [CrossRef]
- Gammie, J.S.; Chikwe, J.; Badhwar, V.; Thibault, D.P.; Vemulapalli, S.; Thourani, V.H.; Gillinov, M.; Adams, D.H.; Rankin, J.S.; Ghoreishi, M.; et al. Isolated Mitral Valve Surgery: The Society of Thoracic Surgeons Adult Cardiac Surgery Database Analysis. Ann. Thorac. Surg. 2018, 106, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Pagnozzi, L.A.; Butcher, J.T. Mechanotransduction Mechanisms in Mitral Valve Physiology and Disease Pathogenesis. Front. Cardiovasc. Med. 2017, 4, 83. [Google Scholar] [CrossRef] [Green Version]
- Toomer, K.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Drayton, K.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 2019, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Durst, R.; Sauls, K.; Peal, D.S.; DeVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nat. Cell Biol. 2015, 525, 109–113. [Google Scholar] [CrossRef]
- A Toomer, K.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, D.; Toomer, K.; Guo, L.; Moore, K.; Glover, J.; Moore, R.; Stairley, R.; Lobo, G.; Zuo, X.; Dang, Y.; et al. Defects in the Exocyst-Cilia Machinery Cause Bicuspid Aortic Valve Disease and Aortic Stenosis. Circation 2019, 140, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, D.; Toomer, K.A.; Glover, J.; Guo, L.; Moore, K.; Moore, R.; Stairley, R.; Gensemer, C.; Abrol, S.; Rumph, M.K.; et al. Desert hedgehog-primary cilia cross talk shapes mitral valve tissue by organizing smooth muscle actin. Dev. Biol. 2020, 463, 26–38. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Hoch, R.V.; Soriano, P. Roles of PDGF in animal development. Development 2003, 130, 4769–4784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Wu, X.; Boström, H.; Kim, I.; Wong, N.; Tsoi, B.; O’Rourke, M.; Koh, G.Y.; Soriano, P.; Betsholtz, C.; et al. A specific requirement for PDGF-C in palate formation and PDGFR-α signaling. Nat. Genet. 2004, 36, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Fantauzzo, K.A.; Soriano, P. PI3K-mediated PDGFR signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 2014, 28, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Bahm, I.; Barriga, E.H.; Frolov, A.; Theveneau, E.; Frankel, P.; Mayor, R. PDGF controls contact inhibition of locomotion by regulating N-cadherin during neural crest migration. Development 2017, 144, 2456–2468. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, S.; Ishii, Y.; Hamashima, T.; Yamamoto, S.; Mori, H.; Fujimori, T.; Shen, J.; Inoue, R.; Nishizono, H.; Itoh, H.; et al. PDGFRalpha plays a crucial role in connective tissue remodeling. Sci. Rep. 2015, 5, 17948. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, C.; Lier, J.M.; Kuschel, S.; Rüther, U. The Ciliary Protein Ftm Is Required for Ventricular Wall and Septal Development. PLoS ONE 2013, 8, e57545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, F.M.; Schou, K.B.; Vilhelm, M.J.; Holm, M.S.; Breslin, L.; Farinelli, P.; Larsen, L.A.; Andersen, J.S.; Pedersen, L.B.; Christensen, S.T. IFT20 modulates ciliary PDGFRalpha signaling by regulating the stability of Cbl E3 ubiquitin ligases. J. Cell Biol. 2018, 217, 151–161. [Google Scholar] [CrossRef]
- Qian, C.; Wong, C.W.Y.; Wu, Z.; He, Q.; Xia, H.; Tam, P.K.H.; Wong, K.K.Y.; Lui, V.C.H. Stage specific requirement of platelet-derived growth factor receptor-α in embryonic development. PLoS ONE 2017, 12, e0184473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallquist, M.D.; Soriano, P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 2003, 130, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomekatz, J.; Singh, R.; Prall, O.W.; Dunn, A.C.; Vaughan, M.; Loo, C.-S.; Harvey, R.P.; Yelon, D. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. eLife 2017, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Wang, Y.; Lui, W.; Langworthy, M.; Tompkins, K.L.; Hatzopoulos, A.K.; Baldwin, H.S.; Zhou, B. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circ. Res. 2011, 109, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visel, A.; Thaller, C.; Eichele, G. GenePaint.org: An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004, 32, D552–D556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zovein, A.C.; Luque, A.; Turlo, K.A.; Hofmann, J.J.; Yee, K.M.; Becker, M.S.; Fassler, R.; Mellman, I.; Lane, T.F.; Iruela-Arispe, M.L. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Gu, Y.; Li, P.; Johnson, B.L.; Sucov, H.M.; Thomas, P.S. PDGF-A as an epicardial mitogen during heart development. Dev. Dyn. 2008, 237, 692–701. [Google Scholar] [CrossRef]
- Chong, J.J.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult Cardiac-Resident MSC-like Stem Cells with a Proepicardial Origin. Cell Stem Cell 2011, 9, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Avi Orr-Urtreger, M.T.B.; Do, M.S.; Eisenbach, L.; Lonai, P. Developmental expression of the alpha receptor for platelet-derived growth factor, which is deleted in the embryonic lethal Patch mutation. Development 1992, 115, 289–303. [Google Scholar]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-α is essential for cardiac fibroblast survival. Am. J. Physiol. Circ. Physiol. 2019, 317, H330–H344. [Google Scholar] [CrossRef]
- Aghajanian, H.; Cho, Y.K.; Rizer, N.W.; Wang, Q.; Li, L.; Degenhardt, K.; Jain, R. Pdgfralpha functions in endothelial-derived cells to regulate neural crest cells and the development of the great arteries. Dis. Model Mech. 2017, 10, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Goddard, L.M.; Duchemin, A.-L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289.e5. [Google Scholar] [CrossRef]
- Bergeron, F.; Bagu, E.T.; Tremblay, J.J. Transcription of platelet-derived growth factor receptor in Leydig cells involves specificity protein 1 and 3. J. Mol. Endocrinol. 2011, 46, 125–138. [Google Scholar] [CrossRef]
- Lockhart, M.M.; Phelps, A.L.; Hoff, M.J.B.V.D.; Wessels, A. The Epicardium and the Development of the Atrioventricular Junction in the Murine Heart. J. Dev. Biol. 2014, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Runyan, R.B.; Markwald, R.R. Invasion of Mesenchyme into Three-Dimensional Collagen Gels: A Regional and Temporal Analysis of Interaction in Embryonic Heart Tissue. Dev. Biol. 1983, 95, 108–114. [Google Scholar] [CrossRef]
- Person, A.D.; Klewer, S.E.; Runyan, R.B. Cell Biology of Cardiac Cushion Development. Adv. Appl. Microbiol. 2005, 243, 287–335. [Google Scholar]
- Akiyama, H.; Chaboissier, M.-C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; De Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, F.; Gentile, A.; Gauvrit, S.; Stainier, D.Y.; Bensimon-Brito, A. Nfatc1 Promotes Interstitial Cell Formation During Cardiac Valve Development in Zebrafish. Circ. Res. 2020, 126, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Yutzey, K.E. VEGF and RANKL Regulation of NFATc1 in Heart Valve Development. Circ. Res. 2009, 105, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranger, A.M.; Grusby, M.J.; Hodge, M.R.; Gravallese, E.M.; De La Brousse, F.C.; Hoey, T.; Mickanin, C.; Baldwin, H.S.; Glimcher, L.H. The transcription factor NF-ATc is essential for cardiac valve formation. Nat. Cell Biol. 1998, 392, 186–190. [Google Scholar] [CrossRef]
- Chang, C.-P.; Neilson, J.R.; Bayle, J.; Gestwicki, J.E.; Kuo, A.; Stankunas, K.; Graef, I.A.; Crabtree, G.R. A Field of Myocardial-Endocardial NFAT Signaling Underlies Heart Valve Morphogenesis. Cell 2004, 118, 649–663. [Google Scholar] [CrossRef] [Green Version]
- Dal-Bianco, J.P.; Aikawa, E.; Bischoff, J.; Guerrero, J.L.; Handschumacher, M.D.; Sullivan, S.; Johnson, B.; Titus, J.S.; Iwamoto, Y.; Wylie-Sears, J.; et al. Active adaptation of the tethered mitral valve: Insights into a compensatory mechanism for functional mitral regurgitation. Circulation 2009, 120, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.A.; Network, F.T.L.M.T.; Hagége, A.A.; Judge, D.P.; Padala, M.; Dal-Bianco, J.P.; Aikawa, E.; Beaudoin, J.; Bischoff, J.; Bouatia-Naji, N.; et al. Mitral valve disease—Morphology and mechanisms. Nat. Rev. Cardiol. 2015, 12, 689–710. [Google Scholar] [CrossRef] [Green Version]
- Anstine, L.J.; Bobba, C.; Ghadiali, S.; Lincoln, J. Growth and maturation of heart valves leads to changes in endothelial cell distribution, impaired function, decreased metabolism and reduced cell proliferation. J. Mol. Cell. Cardiol. 2016, 100, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Hulin, A.; Anstine, L.J.; Kim, A.J.; Potter, S.J.; DeFalco, T.; Lincoln, J.; Yutzey, K.E. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arter. Thromb. Vasc. Biol. 2018, 38, 636–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, J.; Casanovas, G.; Wylie-Sears, J.; Kim, D.-H.; Bartko, P.E.; Guerrero, J.L.; Dal-Bianco, J.P.; Beaudoin, J.; Garcia, M.L.; Sullivan, S.M.; et al. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ. Res. 2016, 119, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Sauls, K.; Toomer, K.; Williams, K.; Johnson, A.J.; Markwald, R.R.; Hajdu, Z.; Norris, R.A. Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. J. Cardiovasc. Dev. Dis. 2015, 2, 200–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toomer, K.; Sauls, K.; Fulmer, D.; Guo, L.; Moore, K.; Glover, J.; Stairley, R.; Bischoff, J.; Levine, R.A.; Norris, R.A. Filamin-A as a Balance between Erk/Smad Activities During Cardiac Valve Development. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2019, 302, 117–124. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, K.; Fulmer, D.; Guo, L.; Koren, N.; Glover, J.; Moore, R.; Gensemer, C.; Beck, T.; Morningstar, J.; Stairley, R.; et al. PDGFRα: Expression and Function during Mitral Valve Morphogenesis. J. Cardiovasc. Dev. Dis. 2021, 8, 28. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8030028
Moore K, Fulmer D, Guo L, Koren N, Glover J, Moore R, Gensemer C, Beck T, Morningstar J, Stairley R, et al. PDGFRα: Expression and Function during Mitral Valve Morphogenesis. Journal of Cardiovascular Development and Disease. 2021; 8(3):28. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8030028
Chicago/Turabian StyleMoore, Kelsey, Diana Fulmer, Lilong Guo, Natalie Koren, Janiece Glover, Reece Moore, Cortney Gensemer, Tyler Beck, Jordan Morningstar, Rebecca Stairley, and et al. 2021. "PDGFRα: Expression and Function during Mitral Valve Morphogenesis" Journal of Cardiovascular Development and Disease 8, no. 3: 28. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8030028