Caenorhabditis elegans as a Model Host to Monitor the Candida Infection Processes

Worcester Polytechnic Institute, Worcester, MA 01609, USA
*
Author to whom correspondence should be addressed.
†
These authors contribute equally to this paper.
J. Fungi 2018, 4(4), 123; https://0-doi-org.brum.beds.ac.uk/10.3390/jof4040123
Submission received: 6 October 2018
/
Revised: 31 October 2018
/
Accepted: 1 November 2018
/
Published: 7 November 2018
(This article belongs to the Special Issue Alternative Models for the Study of Fungal Host-Pathogen Interactions)
{kind=link}
{kind=link}
{kind=link}
Abstract
:C. elegans has several advantages as an experimental host for the study of infectious diseases. Worms are easily maintained and propagated on bacterial lawns. The worms can be frozen for long term storage and still maintain viability years later. Their short generation time and large brood size of thousands of worms grown on a single petri dish, makes it relatively easy to maintain at a low cost. The typical wild type adult worm grows to approximately 1.5 mm in length and are transparent, allowing for the identification of several internal organs using an affordable dissecting microscope. A large collection of loss of function mutant strains are readily available from the C. elegans genetic stock center, making targeted genetic studies in the nematode possible. Here we describe ways in which this facile model host has been used to study Candida albicans, an opportunistic fungal pathogen that poses a serious public health threat.
1. Introduction
Caenorhabditis elegans is a nematode that is naturally found in soil and compost [1] and has been used as a powerful model organism for more than 50 years. In the 1960s, South African biologist Sydney Brenner pioneered the use of C. elegans to study neuronal development and gene expression [2,3] and paved the way for a long lineage of “C. elegans scientists” that studied various aspects of cell and animal biology in nematodes. This lineage includes Nobel Prize Laureates Craig Mello and Andrew Fire for their RNAi work [4], Robert Horvitz and John Sulston for their work on organ development and apoptosis [5,6,7], and Martin Chalfie for his work on green fluorescent protein [8]. More recently, C. elegans have been used to study muscle development and mechanical function [9,10,11], and tumorigenesis and regulation [12]. Although traditionally used to study molecular and developmental biology, researchers in the past 20 years have also begun to use C. elegans to investigate the biology of various human bacterial pathogens including Pseudomonas aeruginosa, Staphylococcus aureus, Salmonella enterica, and Serratia marcescens [13,14,15,16,17,18,19], as well as fungal pathogens such as Cryptococcus neoformans and Candida albicans [20,21,22,23,24,25,26,27,28]. Together, this body of work revealed that many of the mechanisms involved in the human-pathogen interaction are conserved in nematodes, while other immunity mechanisms are unique to the nematode [29,30].
Innate immunity in nematodes is multi-tiered with physical barriers, biochemical, and genetic mechanisms to protect against pathogens. For example, the outer cuticle of the C. elegans protects its body from most pathogens. This soil dwelling animal is most susceptible to pathogens through ingestion since they are constantly feeding. The pharyngeal grinder of adult nematodes effectively grinds most bacteria such that no intact bacterial cells are found in their intestinal lumen [31]. Despite this, some microbes are able to colonize the C. elegans gut with varying success [32]. More recently, the native microbiome of C. elegans was studied and various bacterial strains were identified, with some strains exhibiting antifungal defense potential [33].
C. elegans are either self-fertilizing hermaphrodites or males, have a generation time of 2–4 days, a life span of 2–3 weeks and can be stored frozen almost indefinitely in liquid nitrogen. The nematode is transparent, making it possible to immobilize animals for live cell imaging (Figure 1A). This facilitated the developmental pattern of its 959 somatic cells to be traced through its transparent cuticle. Furthermore, its genome has been sequenced at high coverage and numerous experimental tools are readily available. All of the above qualities, in addition to the availability of functional mutants, fluorescently labeled transgenic strains and RNAi knockdown libraries, make C. elegans an extraordinarily powerful model organism.
In its natural habitat, C. elegans encounters a variety of threats from ingested pathogens present in the soil and this has provided a strong selective pressure to evolve and maintain a sophisticated innate immune system in its intestinal lumen. Many of the genes and mechanisms involved in the protection of intestinal lumen are orchestrated by highly-conserved elements that also exist in higher mammals [30,34,35]. C. elegans therefore represents a great model to study gastrointestinal pathogens such as Salmonella enterica [36], Shigella boydii [37], Salmonella typhimurium [38] or Vibrio cholera [39] and fungal species such as Candida albicans [26].
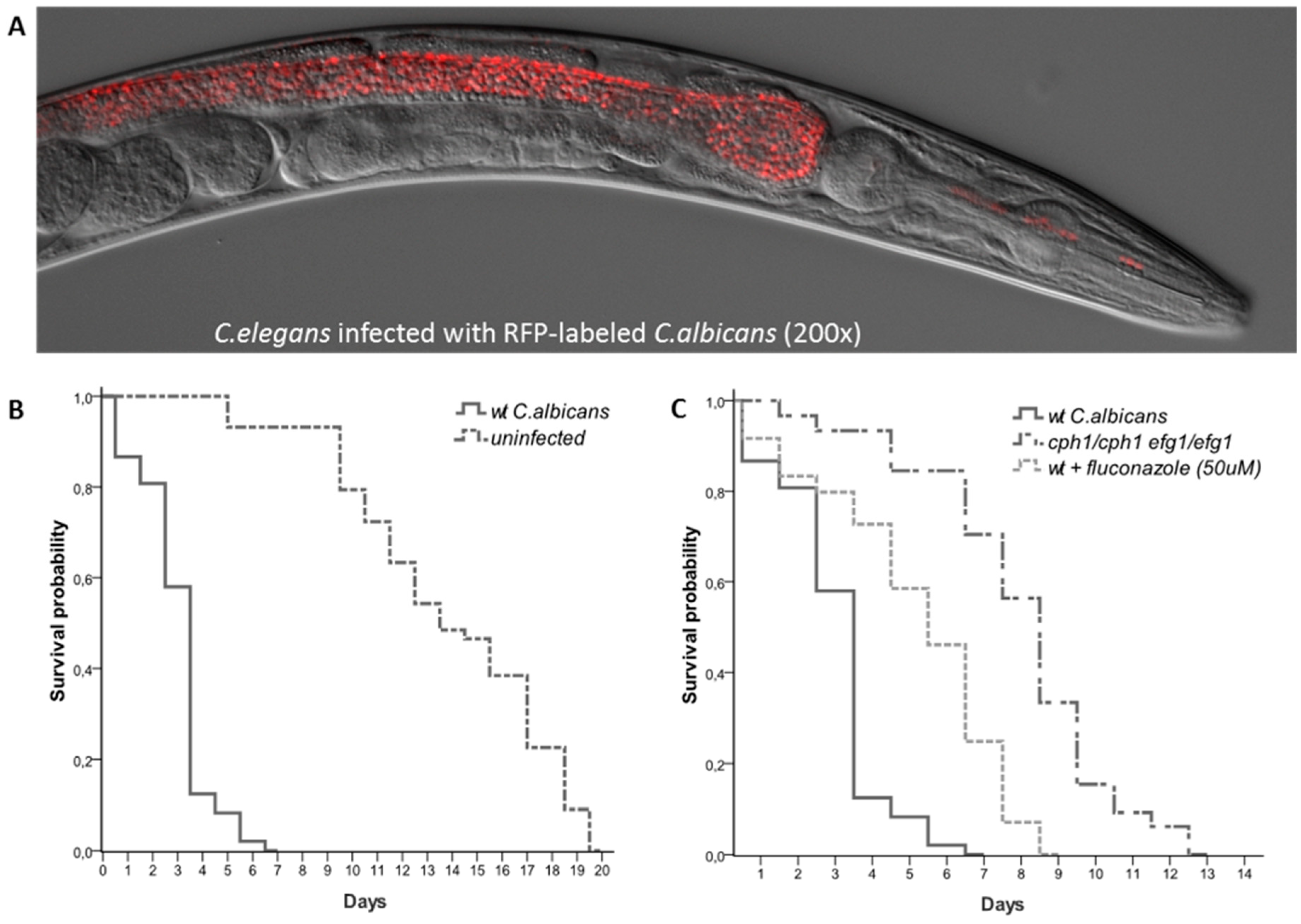
Recently our lab and others have shown that C. elegans is susceptible to C. albicans infections. When C. albicans is added to E.coli lawns it is ingested by the worms and colonizes the intestine. C. albicans infections cause a distinct swelling of the anal region (deformed anal region, Dar) a significantly shorter survival (Figure 1B) and, in certain conditions, C. albicans is capable of killing worms by piercing their cuticle [23,24,25,40].
Studies have also shown that many of the genes required for virulence in murine models of infection are also required for virulence in nematodes. For example RIM101, a C. albicans transcription factor required for alkaline-induced hyphal growth is required for both virulence in murine oropharyngeal candidiasis [41] and virulence in C. elegans [23]. Other genes such as NRG1, CAS5, ADA2, CPH1 and EFG1 have also shown to be required for virulence in both mice and nematodes [23,28,42] (Figure 1C). These studies clearly demonstrate that many of the conclusions drawn using this simpler organism remain valid in higher mammals, reiterating the utility of C. elegans as a model host for the study of infectious diseases. C. elegans can also be used for drug discovery (Figure 1C). A high-throughput semi-automated version of the C. elegans-C. albicans model system was recently used to screen a library of 3,228 compounds [43]. Seven of the 19 compounds capable of extending nematodes survival were known to have antimycotic activity, validating the approach. Taken together these studies show that C. elegans can be used as a powerful tool to study, not only the complex host-pathogen dynamics, but also for initial large-scale drug screening [44,45,46,47].
There are an estimated 750,000 hospitalizations and almost 9 million outpatient visits due to fungal infections every year, that amounts to costs of more than $7 billion. These infections can be caused by many Candida yeast species, the most common of which is C. albicans, and represent the fourth most common cause of healthcare-associated bloodstream infections in the United States [48,49,50,51,52,53]. This polymorphic fungus is part of the human microbiome and it usually lives as a harmless commensal on the skin and mucosa of healthy individuals. However, under certain circumstances, it can cause infections ranging from mild, superficial infections to life-threatening, systemic infections. Even though in the last two decades a large body of literature shed some light on C. albicans pathogenicity [54], our understanding of the basic mechanisms that result in this harmless commensal becoming pathogenic remain elusive. In addition, because drug resistance has dramatically increased in the last two decades [55], there is an urgent need to identify new targets to develop better diagnostics and therapeutics. C. elegans can be leveraged as the perfect go-between model host between in vitro studies and mammalian models.
2. Tools and Techniques
2.1. Maintenance of C. albicans and C. elegans
C. albicans strains are maintained in YPD (Yeast extract, Peptone, Dextrose) media with or without agar at 30 or 37 °C. Hernday et al. report more detailed methods in C. albicans maintenance [56].
Wild type C. elegans, N2 (Caenorhabditis Genetics Center, U. of Minnesota; https://cgc.umn.edu/strain/N2%20(ancestral)) are typically maintained in nematode growth media (NGM) with or without agar at 20 °C. More detailed methods are described in the WormBook [57]. NGM plates are seeded with E. coli OP50, the standard laboratory food for N2 wild type C. elegans [2,57]. To test the effects of gene knockdowns, the E. coli strain HT115 is used that maintains the RNA interference (RNAi) plasmid [4,10,40,58].
Typically a spot of the food is placed in the middle of the plate and worm eggs or larvae are placed on the pates. Animals freely roam around the plate and feed when they encounter the spot (“small lawn”). C. albicans may be mixed in with worm food (OP50). If worms are being tested for their attraction or aversion to a particular (microbial) food they can be exposed to each food by itself. Certain experiments might call for the entire plate to be covered (“big lawn”). This is particularly advantageous to ensure constant exposure to a treatment to identify the mechanism of pathogen resistance—whether it is due to avoidance behavior, or due to innate cellular immunity [59,60].
2.2. Measurement of Host Lifespan Post Infection
Overnight cultures of E. coli and C. albicans are grown overnight in appropriate media and conditions. The cultures are then centrifuged, washed and resuspended to achieve a particular cell density desired for seeding plates. Antibiotics may be used in the feeding mixture depending on experimental objectives and conditions. For example, the food can be a mixture of E. coli and C. albicans or they can be seeded independently, where the bacteria is used as control and the C. albicans as the variable tested. The limitation of the latter is in the age of the synchronized worms used. Since C. albicans are too large to be consumed by the larval stage nematode, only L4 and young adult nematodes can be transferred to plates containing C. albicans alone, without resulting in starved worms (aka dauer stage, where nematodes under harsh environmental conditions undergo arrest at the second molt [61]). The food is plated in the center of small petri dishes (3.5 cm in diameter) containing NGM agar and incubated overnight at room temperature. The following day, 20 young synchronized adult worms are transferred to the spotted plate (day 0).
Synchronous worm populations are obtained 2–3 days after the addition of C. elegans eggs to NGM plates seeded with E. coli OP50. In this time frame, the eggs hatch and the larvae reach young adulthood. The worms are scored daily by gentle prodding with a platinum wire and live animals are transferred to new seeded plates grown overnight at room temperature. Daily scoring includes live worms, dead worms and censored worms. Censored animals are those whose death cannot be attributed to the treatment test, like animals accidentally killed while transferring, found dead on the edges of the plates due to drying out, died due to secondary effects of infection, or simply “missing.” C. elegans survivals are examined using Kaplan-Meir method and statistical significance evaluated using the log-rank test or Gehan-Breslow-Wilcoxon test, depending on survival trends and fulfilling test assumptions. Additional resources for lifespan assay methods can be found in works by Wilkinson et al. and Amrit et al. [62,63].
2.3. Microscopy
The transparent nematodes can be easily visualized and enables live imaging of host-pathogen interactions [64,65]. Typically worms are anesthetisized using sodium azide and placed on an agarose pad containing a drop of M9 buffer. Immobilized worms may be observed under differential interference contrast (Nomarski) and epifluorescence optics. Immunofluorescence can also be used to visualize specific organs or regions of the body [66] for in vivo live visualization. Fluorescently tagged C. albicans can be used to infect C. elegans to visualize disease progression [25]. As a note of caution, C. elegans produce lipofuscin under stress or as they age. This compound fluoresces in blue-green range [67] and may interfere if the tagged proteins are also emitting in the similar range. Therefore, the red/far red fluorophore is effective for visualizing short-term infection [25].
Short-term microscopy of living worms can be conducted using analgesics or anesthetics to immobilize worms and capture images. This method is limited to short-term imaging since worms cannot feed, or will dry out if left for longer periods of time, and then often worms are not recoverable [65]. Alternate methods are necessary for image feeding, locomotion behavioral or physiological responses to stimuli. Different automated approaches have been developed to track the movement and process images of mobile live worms, as reviewed by Husson et al. [68]. Microfluidics is a new approach to handling and capturing images of worms over prolonged periods, and can be used for high throughput assays of RNAi libraries, for instance. Longitudinal observations of individual worms can also be carried out using chambers or droplets. These microfluidics devices are reviewed by San-Miguel and Lu [69]. Recent protocols have also been developed to use multi-channel time-lapse confocal imaging to monitor cell invasion through C. elegans basement membranes [70]. Luke et al. also developed some non-microfluidic protocols that enable phase and fluorescent imaging of motile worms [65,71].
3. C. elegans as a Model Host to Study Virulence of C. albicans
3.1. Using C. elegans to Study Host Innate Immunity against Fungal Pathogens
The C. elegans intestine faithfully mimics mammalian intestines. Like humans, C. elegans encounters a variety of threats from ingested pathogens, posing a constant challenge to the innate immune system in its intestinal lumen. Like mammalian intestines, microvilli in C. elegans intestine assimilate and digest food while the intestinal luminal cells synthesize and store macromolecules. In addition, the C. elegans intestinal microbiota has emerged as a powerful experimental system to study microbial interactions [72,73]. For these reasons, C. elegans represents a relevant model to study gastrointestinal pathogens.
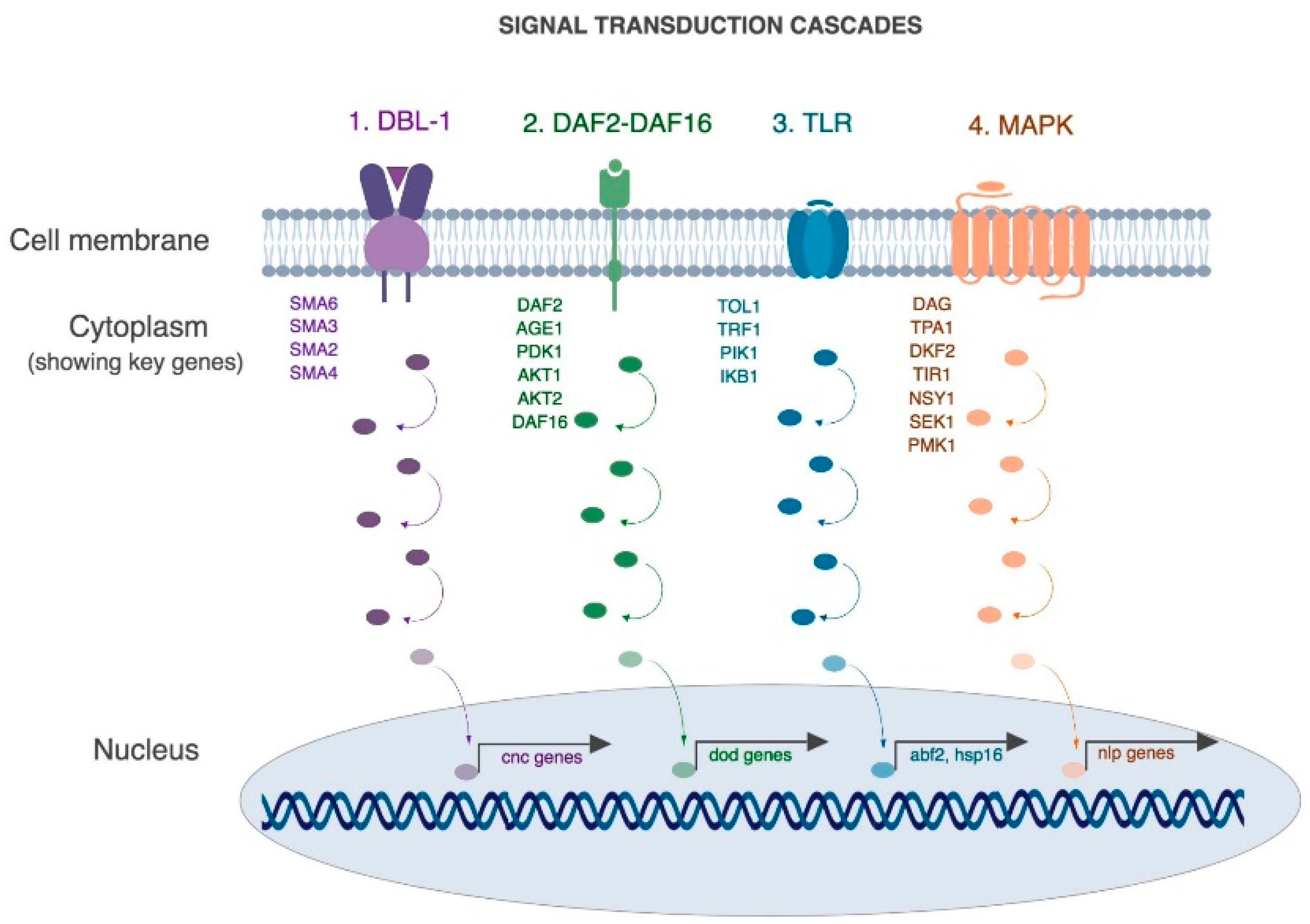
A rich body of literature has revealed that many of the mechanisms involved in the human-pathogen interaction are conserved in nematodes [29,30]. For example, four conserved pathways have been shown to be involved in the protection of intestinal lumen in C. elegans as well as higher mammals (Figure 2, [30,36,74,75]). A brief description of these pathways as they relate to infection and immunity is described here (Figure 2).
First is the Transforming Growth Factor-β pathway (TGF aka DBL-1 pathway), that regulates the expression of key antimicrobial effectors such as lysozymes and lectins [80]. DBL-1 functions as a dose-dependent ligand that binds heterodimeric receptor DAF-4/SMA-6 triggering a phosphorylation cascade involving SMAD proteins (SMA-2,3,4) and activates SMA-9. SMA-9 controls expression of a key innate immune effector lysozyme (lys-1 and lys-8) [81].
Second, the insulin signaling pathway (aka DAF-2/DAF-16 pathway), is a general stress response pathway that is linked to the expression of multiple antimicrobial proteins such as saponins, thaumatin-like proteins and lysozymes [81,82,83,84]. Briefly, in the absence of microbial threats, the insulin-like peptide DAF-28 binds DAF-2 and triggers a phosphorylation cascade that leads to the cytoplasmic retention of the transcription factor DAF-16. However, in the presence of a microbial threat, the pathway is repressed and DAF-16 phosphorylation triggers expression of antimicrobial peptides.
Third, the Toll pathway (aka TLR pathway) is required for the expression of heat-shock proteins and other defense-like molecules [77] and is implicated in inflammatory microbial interactions [78]. During infection with certain pathogens such as Serratia mercenses and Salmonella enterica it is required to mount an appropriate avoidance-like immune response [77]. However, in the presence of other pathogens such as Pseudomonas aeruginosa, Microbacterium nematophilum and Drechmeria coniospora this pathway is not required for resistance [79].
Fourth is the mitogen-activated protein kinase (MAPK), which is a well characterized signal transduction cascade in nematodes. It has various biological functions including regulating resistance to bacterial pathogens such as P. aeruginosa and D. consispora [30,36,74,76]. Briefly, upon activation, DKF-2 activates TIR-1, NSY-1, SEK-1 and PMK-1 in a linear phosphotransfer cascade that leads to the activation of antimicrobial peptides, nlp-29 and nlp-31. The MAPK pathway is a highly conserved pathway and NSY-1, SEK-1 and PMK-1 are the C. elegans homologs of human ASK-1, MKK3 and p38 which play major roles in mammalian cellular immune response by controlling the expression of pro-inflammatory cytokines like IL-1 and TNFα.
3.2. Other Host Pathways
It is becoming increasingly clear that the resident microbiota plays a significant role in human health and disease [86,87,88,89]. C. elegans with its microbial diet is an effective system to study interspecies interactions as well as host-microbe-drug interactions. For example, a systems biology approach was employed to delineate the effects of micronutrients derived from the microbiome on physiology and metabolism of the host. The study connected the flux of micronutrients propionic acid and Vitamin B12 into methionine and folic acid. Interestingly, known orthologs of these genes are present in humans [90,91], suggesting that C. elegans is a relevant model for microbiome studies.
3.3. Using C. elegans as in vivo Model to Study C. albicans Virulence Mechanisms
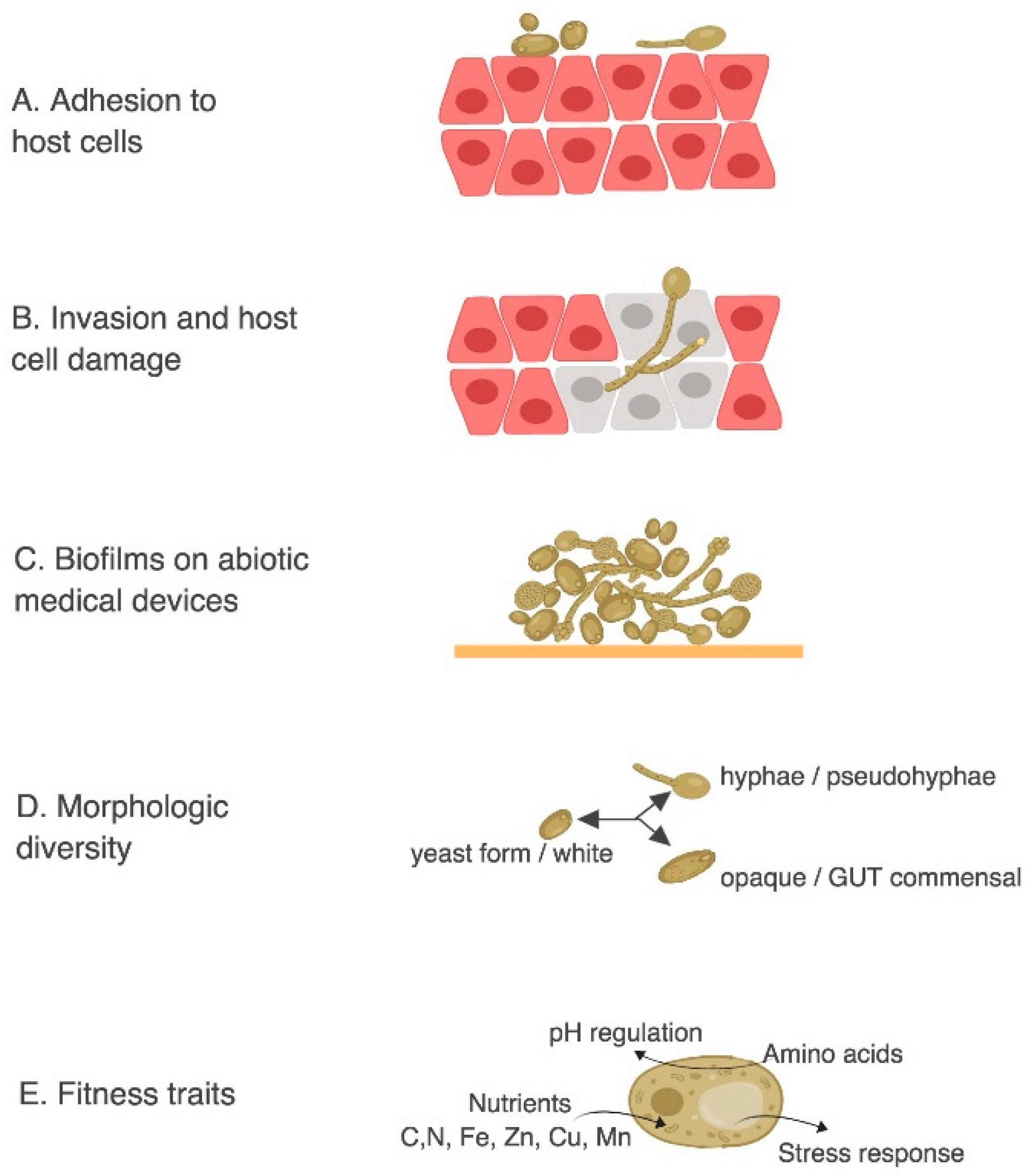
Powerful reverse genetic approaches have been employed to study C. albicans mutants that alter the virulence phenotype in C. elegans [23,24]. The most important factor that controls C. albicans infection is the physiological status of the host since C. albicans infections are commonly found in immunocompromised patients. However, C. albicans is also found in approximately 50% of the population as part of the individual’s microflora and it has been shown that slight alterations in the host can turn this normally harmless commensal into a life-threatening pathogen [92]. The transition from harmless commensal to pathogen is a fine line in C. albicans and it is attributable to its extensive arsenal of virulence factors. These factors are selectively expressed under suitable predisposing conditions such as the ability to: adhere and penetrate host tissues; form biofilm; switch from yeast-to-hypha; switch from white to opaque state; switch from commensal gut state to virulent state; resist stress, uptake amino acids, adapt to pH fluctuations and utilize extracellular carbon, nitrogen and essential trace metals, just to name a few (Figure 3).
C. albicans infections begin with the adhesion of yeast cells to host tissues (Figure 3A). This process is mediated by adhesins proteins such as Als (Agglutinin-Like Sequence) [93]. Als are a family of eight glycosylphosphatidylinositol (GPI)-anchored cell surface proteins that protrude from the C. albicans cell wall and that are capable of binding host surface proteins and extracellular matrix [94,95].
Once C. albicans attaches to host cells, thigmotropism (contact sensing) [96] triggers C. albicans filamentation and allows the pathogen to penetrate deeper host tissues (Figure 3B) via the secretion of hydrolytic enzymes such as Saps (Secreted Aspartyl Proteases) and phospholipases [97,98]. The ability to switch from yeast to hyphae has been shown to be critical for virulence. C. albicans can grow in a planktonic yeast form (ovoid shape) that is thought to promote dispersion while hyphal growth of C. albicans has an elongated ellipsoid form and can form long filaments that can penetrate deeper tissues (Figure 3B–D). The hyphal form is considered more invasive than the yeast form [99] but the ability to switch from one form to the other is required for virulence. In particular, strains locked in one form or the other have a significantly reduced virulence compared to strains that can freely switch from one form to the other [100]. The hyphal forms are important for tissue invasion in epithelial infections, while yeast form, which are smaller in size and can travel more effectively in the blood stream, are required for systemic infections [101].
C. albicans also has a remarkable ability to form biofilms on abiotic surfaces such as catheters, dentures and other implants (Figure 3C). Upon adhesion C. albicans proliferates, forms hyphal cells in the upper part of the biofilms, and finally produces a complex extracellular matrix that renders the biofilm more resistant to antimycotic agents [102]. Recent gene expression profiling and ChIP-chip experiments suggest that the cellular circuit controlling biofilm formation is regulated by six transcriptional regulators (TEC1, EFG1, BCR1, NDT80, ROB1 and BRG1) that tightly control the expression of ~1000 target genes [103].
Also, C. albicans is polymorphic and capable of switching from the an ovoid yeast morphology (white) to distinct elongated cell types that are either mating-competent (opaquea/α) or commensal Gastrointestinally-Induced Transition (GUT) forms. These polymorphic forms not only exhibit differences in vitro and in animal models of commensalism and disease but also influence its ability to colonize the host [104]. In particular, white and opaque cells are differentially adapted to specific host niches, with opaque cells being more virulent in the hypoxic environment of the mammalian intestine [105].
In addition to these direct virulence factors, C. albicans has various “fitness traits” (Figure 3D) that allow it to grow and resist host immunity. These mechanisms include a robust stress response mediated by heat shock proteins (Hsps) as well as an arsenal of detoxifying enzymes and efflux pumps. Moreover, C. albicans has a remarkable ability to adapt to the different nutritional environments of the host. Modulating glycolysis, gluconeogenesis, amino acid uptake and starvation responses are all believed to be important for virulence. For example, in the gastrointestinal tract the concentration of nutrients is relatively high but C. albicans competes for nutrients with other members of the microbiome. If the gastrointestinal flora is imbalanced, for example in patients undergoing systemic antibiotics therapies, C. albicans has access to additional nutrients and can outgrow the other organisms [106]. On the other hand, when C. albicans is in the blood stream, glucose is highly abundant (6–8 mM), but as soon as it is phagocytized, then the nutritional environment changes completely and C. albicans faces nutrient starved conditions [107]. Inside the macrophage, C. albicans promptly switches from glycolysis to gluconeogenesis and downregulated energetically demanding processes such as ribosome biogenesis [108]. Since the environments that C. albicans faces during infection are various, a prompt and efficient metabolic plasticity is required for adapting to such different conditions.
3.4. C. elegans as a Live Animal Model for Antifungal Drug Discovery
The increased use of immunosuppressive therapies, potent chemotherapeutic agents and the surge of AIDS patients caused a dramatic increase in the number of immunocompromised patients, which led to a dramatic rise in the number of patients suffering from C. albicans infections [109]. In addition to the increased number of C. albicans infected patients, the clinical outcome of these infections is complicated by the ability of the strain to develop drug resistance and by the limited number of antimycotics currently available [110,111,112,113]. C. albicans developed various clever mechanisms for drug resistance, including genetic alteration of the targeted proteins and overexpression of efflux pumps [114]. Patients infected with drug resistant strains do not respond to the pharmacological therapy and, because the number of antimycotic alternatives is limited, the prognosis can be poor. The large majority of antimycotics currently prescribed have been FDA approved in the 1960s and 1990s and there are not very many drugs in company pipelines that have promising safety and efficacy profiles [115], with the exception of echinocandins such as caspofungin [116,117].
The strength of the nematode model system is that it can be pharmacologically modulated (Figure 1). Fluconazole, amphotericin B and caspofungin [44,118,119,120] alter the course of C. elegans infection as measured by extended worm life spans and decreased Dar. Fluconazole and amphotericin B are the most commonly prescribed drugs against C. albicans infections, but they have very different mechanisms of action. Fluconazole reduces cell growth by inhibiting the synthesis of ergosterol, a sterol that maintains membrane fluidity. It is considered fungistatic because it can inhibit growth but not cause cell death. On the other hand, amphotericin B is fungicidal because it ultimately kills yeast cells. Amphotericin B directly binds ergosterol, depolarizing the cell membrane, causing leakage of intracellular cations and leading to cell death. A third family of antimycotics was recently discovered: echinocandins, like caspofungin, inhibit 1-3-β-D-glucan synthase [117]—an enzyme that produces 1-3-β-D-glucan, an essential protein in the cell wall of many yeast species including Candida [116,121]. Caspofungin has been found to have fungicidal [122,123] or fungistatic effects [124,125] on various fungal species. Fluconazole is more commonly prescribed (can be delivered orally) but poses the threat of generating drug resistance; amphotericin B and caspofungin are more potent but can only be administrated intravenously, where amphotericin B is associated with high nephrotoxicity and caspofungin was associated with relatively low nephrotoxicity and hepatotoxicity [126,127].
The C. albicans—C. elegans virulence model has been used effectively to screen small molecule libraries combined with a chemical structure analysis to identify novel antifungal agents [43,128,129,130,131,132,133]. Typically, C. elegans is infected with C. albicans and treated with a library of small molecules. This model host can also be used to test for drug efficacy and toxicity. For example, Filastatin was discovered using C. elegans as a live host and has been shown to alleviate disease and improve nematode survival to levels comparable to fluconazole [129] using the C. elegans infection model. Furthermore, Filastatin-coated surfaces have been shown to decrease C. albicans adhesion [134].
3.5. Studying the in vivo Evolution of C. albicans
The nematode virulence model can be used to study the evolution of drug resistance in clinical isolates of C. albicans. C. elegans has been used as a facile animal model to study the in vivo fitness of several C. albicans clinical isolates. Isolates are obtained before and after fluconazole treatment and patients may be sampled multiple times. This serves as a powerful time series to study the progressive evolution of C. albicans drug resistance in humans [42].
This study [42] identified single-nucleotide polymorphisms (SNPs), copy number variations (CNV) and loss of heterozygosity (LOH) events that alter C. albicans drug resistance and other phenotypes associated with virulence. The ability of each of these strains to form biofilms was determined by testing adhesion to polystyrene, to transition from yeast-to-hyphae and to infect and kill nematodes. These three phenotypes are highly linked to virulence as the ability to form biofilm on medical devices, to transition from yeast-to-hyphae and to kill nematodes, which have been all connected to mammalian virulence [25,135,136].
This study [42] also revealed that LOH events were recurrent and tracked with increased in drug resistance while aneuploidies were transient and did not correlate with fluconazole resistance. The study also identified 240 genes that accumulate persistent SNPs that recur between patients, suggesting that these genes might play a primary role in the complex process of host adaptation and fluconazole exposure. In general, there were substantial variations in the phenotypes observed between isolates. However, C. albicans in vitro fitness was shown to be anti-correlated with phenotypic markers of virulence. Furthermore, a progressive increase in drug resistance in vitro was mirrored by a progressive increase in virulence supporting the notion that C. albicans adaption during antimycotic therapy relies on its genotypic and phenotypic plasticity [42].
A contributor to this plasticity was recently discovered through the presence of viable haploid and polyploid Candida cells in culture. Candida was long thought to be an “obligate diploid” but was recently shown to also exist in a viable state as haploid where it can form true hyphae, pseudohayphae and chlamydospores, as well as switch between white-opaque forms [137]. Though haploids grow at slower rates than heterozygous diploids, the haploid cells enabled mating and the production of more adaptable, viable heterozygous diploids. Candida in its parasexual phase can develop multi-ploidy which potentiates genetic variation and drives population diversity, increasing probability of adaptation and selection [138] and leading to its remarkable plasticity.
More recently, the C. elegans model has been leveraged to study host fitness associated with fungal infection. This study [139] demonstrates that exposure to C. albicans as well as other non-albicans Candida species affects fecundity, specifically, delayed reproduction, early mortality, reduced brood sizes and delayed reproduction relative to infected healthy hosts.
4. Conclusions
C. elegans is a good host model organism to study innate immune responses to Candida pathogenesis. Its response to infection can often be easily visualized under a dissecting microscope or through immunofluorescence tagging. It is also used for genetic studies and can be used to better understand mechanistic pathways of host-pathogen interaction. Another advantage to using C. elegans is the large array of mutants easily attainable from CGC, where high throughput assays can be used for drug screens and to identify pathways of interest. Naturally, there are some limitations to using C. elegans as a model host organism since it does not have adaptive immunity and its innate immunity may be less complex than higher organisms. Also, visualization techniques are continuously developing to enhance capturing internal processes in live animals, tracking of motile animals, and automation of high throughput processes.
Author Contributions
Funding
A.B.E. was supported on a grant from NIH NCCIH 1R15AT009926-01 to R.P.R.
Acknowledgments
This work is supported by the National Institutes of Health grant 1R15AT009926-01 to R.P.R.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schulenburg, H.; Felix, M.A. The Natural Biotic Environment of Caenorhabditis elegans. Genetics 2017, 206, 55–86. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Riddle, D.L.; Blumenthal, T.; Meyer, B.J.; Priess, J.R. Introduction to C. elegans. In C. elegans II, 2nd ed.; Riddle, D.L., Blumenthal, T., Meyer, B.J., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.M.; Horvitz, H.R. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef]
- Hengartner, M.O.; Ellis, R.E.; Horvitz, H.R. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 1992, 356, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Ruijtenberg, S.; Van Den Heuvel, S. G1/S Inhibitors and the SWI/SNF Complex Control Cell-Cycle Exit during Muscle Differentiation. Cell 2015, 162, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bass, J.J.; Szewczyk, N.J. Knockdown of the C. elegans Kinome identifies Kinases required for normal protein Homeostasis, Mitochondrial network structure, and Sarcomere structure in muscle. Cell Commun. Signal. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Ono, S. Regulation of structure and function of sarcomeric actin filaments in striated muscle of the nematode Caenorhabditis elegans. Anat. Rec. 2014, 297, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chin-Sang, I.D.C. C. elegans as a model to study PTEN’s regulation and function. Methods 2015, 77, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.W.; Mahajan-Miklos, S.; Ausubel, F.M. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Irazoqui, J.E.; Troemel, E.R.; Feinbaum, R.L.; Luhachack, L.G.; Cezairliyan, B.O.; Ausubel, F.M. Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog. 2010, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Yehye, W.A.; Abd Rahman, N.; Tan, M.W.; Nathan, S. Discovery of potential anti-infectives against Staphylococcus aureus using a Caenorhabditis elegans infection model. BMC Complement. Altern. Med. 2014, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.K.; May, R.C. Caenorhabditis elegans, a model organism for investigating immunity. Appl. Environ. Microbiol. 2012, 78, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Sem, X.; Rhen, M. Pathogenicity of Salmonella enterica in Caenorhabditis elegans relies on disseminated oxidative stress in the infected host. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Sifri, C.D.; Begun, J.; Ausubel, F.M.; Calderwood, S.B. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect. Immun. 2003, 71, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Ausubel, F.M.; Perfect, J.R.; Heitman, J.; Calderwood, S.B. Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 15675–15680. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Idnurm, A.; Moreno, R.; El Khoury, J.; Rottman, J.B.; Ausubel, F.M.; Heitman, J.; Calderwood, S.B. Cryptococcus neoformans Kin1 protein kinase homologue, identified through a Caenorhabditis elegans screen, promotes virulence in mammals. Mol. Microbiol. 2004, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.C.; Woerlee, J.Z.; Ma, H.; May, R.C. Sex-dependent resistance to the pathogenic fungus Cryptococcus neoformans. Genetics 2006, 173, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Peleg, A.Y.; Tampakakis, E.; Mylonakis, E. Candida albicans hyphal formation and virulence assessed using a Caenorhabditis elegans infection model. Eukaryot. Cell 2009, 8, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Ausubel, F.M.; Mylonakis, E. Candida albicans infection of caenorhabditis elegans induces antifungal immune defenses. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Pastor, K.; Gonzalez, A.Y.; Lorenz, M.C.; Rao, R.P. The role of Candida albicans AP-1 protein against host derived ROS in in vivo models of infection. Virulence 2013, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Issi, L.; Rioux, M.; Rao, R. The Nematode Caenorhabditis Elegans—A Versatile In Vivo Model to Study Host-microbe Interactions. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Desalermos, A.; Tan, X.; Rajamuthiah, R.; Arvanitis, M.; Wang, Y.; Li, D.; Kourkoumpetis, T.K.; Fuchs, B.B.; Mylonakis, E. A multi-host approach for the systematic analysis of virulence factors in Cryptococcus neoformans. J. Infect. Dis. 2015, 211, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Desalermos, A.; Fuchs, B.B.; Mylonakis, E. Selecting an invertebrate model host for the study of fungal pathogenesis. PLoS Pathog. 2012, 8, 8–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, T.; Banger, A.K.; Wallace, A.; Glass, E.M.; Aslund, F.; Schneewind, O.; Missiakas, D.M. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 2004, 101, 12312–12317. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Feinbaum, R.; Alloing, G.; Emerson, F.E.; Garsin, D.A.; Inoue, H.; Tanaka-Hino, M.; Hisamoto, N.; Matsumoto, K.; Tan, M.W.; Ausubel, F.M. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 2002, 297, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Aballay, A. Worms and flies as genetically tractable animal models to study host-pathogen interactions. Infect. Immun. 2005, 73, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Portal-Celhay, C.; Blaser, M.J. Competition and resilience between founder and introduced bacteria in the Caenorhabditis elegans gut. Infect. Immun. 2012, 80, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P.; Felix, M.A.; Schulenburg, H. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Couillault, C.; Pujol, N.; Reboul, J.; Sabatier, L.; Guichou, J.F.; Kohara, Y.; Ewbank, J.J. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat. Immunol. 2004, 5, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Aballay, A.; Ausubel, F.M. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr. Opin. Microbiol. 2002, 5, 97–101. [Google Scholar] [CrossRef]
- Aballay, A.; Drenkard, E.; Hilbun, L.R.; Ausubel, F.M. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 2003, 13, 47–52. [Google Scholar] [CrossRef]
- Kesika, P.; Balamurugan, K. Studies on Shigella boydii infection in Caenorhabditis elegans and bioinformatics analysis of immune regulatory protein interactions. Biochim. Biophys. Acta 2012, 1824, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Aballay, A.; Yorgey, P.; Ausubel, F.M. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr. Biol. 2000, 10, 1539–1542. [Google Scholar] [CrossRef]
- Cinar, H.N.; Kothary, M.; Datta, A.R.; Tall, B.D.; Sprando, R.; Bilecen, K.; Yildiz, F.; McCardell, B. Vibrio cholerae hemolysin is required for lethality, developmental delay, and intestinal vacuolation in Caenorhabditis elegans. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Yun, M.; Politz, S.M.; Rao, R.P. A Pathogenesis Assay Using Saccharomyces cerevisiae and Caenorhabditis elegans Reveals Novel Roles for Yeast AP-1, Yap1, and Host Dual Oxidase BLI-3 in Fungal Pathogenesis. Eukaryot. Cell 2009, 8, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Solis, N.; Myers, C.L.; Fay, A.J.; Deneault, J.S.; Nantel, A.; Mitchell, A.P.; Filler, S.G. Candida albicans transcription factor Rim101 mediates pathogenic interactions through cell wall functions. Cell. Immunol. 2008, 1, 2180–2196. [Google Scholar]
- Ford, C.B.; Funt, J.M.; Abbey, D.; Issi, L.; Guiducci, C.; Martinez, D.A.; Delorey, T.; Li, B.Y.; White, T.C.; Cuomo, C.; Rao, R.P.; Berman, J.; Thompson, D.A.; Regev, A. The evolution of drug resistance in clinical isolates of Candida albicans. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Okoli, I.; Coleman, J.J.; Tempakakis, E.; An, W.F.; Holson, E.; Wagner, F.; Conery, A.L.; Larkins-Ford, J.; Wu, G.; Stern, A.; Ausubel, F.M.; Mylonakis, E. Identification of antifungal compounds active against Candida albicans using an improved high-throughput Caenorhabditis elegans assay. PLoS ONE 2009, 4, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breger, J.; Fuchs, B.B.; Aperis, G.; Moy, T.I.; Ausubel, F.M.; Mylonakis, E. Antifungal chemical compounds identified using a C. elegans pathogenicity assay. PLoS Pathog. 2007, 3, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammed, M.; Arvanitis, M.; Mylonakis, E. Whole animal HTS of small molecules for antifungal compounds. Expert Opin. Drug Discov. 2016, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Desalermos, A.; Muhammed, M.; Glavis-Bloom, J.; Mylonakis, E. Using C. elegans for antimicrobial drug discovery. Expert Opin. Drug Discov. 2011, 6, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Luca, V.; Olivi, M.; Di Grazia, A.; Palleschi, C.; Uccelletti, D.; Mangoni, M.L. Anti-Candida activity of 1-18 fragment of the frog skin peptide esculentin-1b: In vitro and in vivo studies in a Caenorhabditis elegans infection model. Cell. Mol. Life Sci. 2014, 71, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Rentz, A.M.; Halpern, M.T.; Bowden, R. The impact of candidemia on length of hospital stay, outcome, and overall cost of illness. Clin. Infect. Dis. 1998, 27, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.S.; Reyes, C.M.; Stolpman, M.; Speckman, J.; Allen, K.; Beney, J. The direct cost and incidence of systemic fungal infections. Value Health 2002, 5, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of direct healthcare costs of fungal diseases in the United States. Clin. Infect. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, L.T.; Premji, A.; Tybout, C.; Hunt, A.; Cutshaw, D.; Elsamadicy, A.A.; Yang, S.; Xie, J.; Giamberardino, C.; Pagadala, P.; et al. Prevalence, healthcare resource utilization and overall burden of fungal meningitis in the United States. J. Med. Microbiol. 2018, 67, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Dodds Ashley, E.; Drew, R.; Johnson, M.; Danna, R.; Dabrowski, D.; Walker, V.; Prasad, M.; Alexander, B.; Papadopoulos, G.; Perfect, J. Cost of invasive fungal infections in the era of new diagnostics and expanded treatment options. Pharmacotherapy 2012, 32, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowen, L.E. The evolution of fungal drug resistance: Modulating the trajectory from genotype to phenotype. Nat. Rev. Microbiol. 2008, 6, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Hernday, A.D.; Noble, S.M.; Mitrovich, Q.M.; Johnson, A.D. Genetics and Molecular Biology in Candida albicans. Methods Enzymol. 2010, 470, 737–758. [Google Scholar] [PubMed]
- Stiernagle, T. Maintenance of C. elegans. WormBook 2006. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Chun, L.; Ronan, E.A.; Friedman, D.I.; Liu, J.; Xu, X.Z. RNAi Interrogation of Dietary Modulation of Development, Metabolism, Behavior, and Aging in C. elegans. Cell. Rep. 2015, 11, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.C.; Andersen, E.C.; Kruglyak, L.; Kim, D.H. A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science 2009, 323, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Chiu, H.C.; Kuo, C.J.; Wu, C.M.; Syu, W.J.; Chiu, W.T.; Chen, C.S. Enterohaemorrhagic Escherichia coli O157:H7 Shiga-like toxin 1 is required for full pathogenicity and activation of the p38 mitogen-activated protein kinase pathway in Caenorhabditis elegans. Cell. Immunol. 2013, 15, 82–97. [Google Scholar]
- Hu, P.J. Dauer. In WormBook; Riddle, D.L., Ed.; WormBook: Pasadena, CA, USA, 2007. [Google Scholar]
- Wilkinson, D.S.; Taylor, R.C.; Dillin, A. Analysis of aging in Caenorhabditis elegans. Methods Cell Biol. 2012, 107, 353–381. [Google Scholar] [PubMed]
- Amrit, F.R.G.; Ratnappan, R.; Keith, S.A.; Ghazi, A. The C. elegans lifespan assay toolkit. Methods 2014, 68, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Podbilewicz, B.; Gruenbaum, Y. Live Imaging of Caenorhabditis elegans: Preparation of Samples. Cold Spring Harb. Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Luke, C.J.; Niehaus, J.Z.; O’Reilly, L.P.; Watkins, S.C. Non-microfluidic methods for imaging live C. elegans. Methods 2014, 68, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, C.J.; Bass, J.J.; Barratt, T.F.; Szewczyk, N.J. Methods to Assess Subcellular Compartments of Muscle in C. elegans. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Pincus, Z.; Mazer, T.C.; Slack, F.J. Autofluorescence as a measure of senescence in C. elegans: Look to red, not blue or green. Aging 2016, 8, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Husson, S.J.; Costa, W.S.; Schmitt, C.; Gottschalk, A. Keeping track of worm trackers. WormBook 2013. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, A.; Lu, H. Microfluidics as a tool for C. elegans research. WormBook 2013. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Wang, Z.; Hagedorn, E.J.; Wang, L.; Shen, W.; Lei, S.; Johnson, S.A.; Sherwood, D.R. Live-cell confocal microscopy and quantitative 4D image analysis of anchor-cell invasion through the basement membrane in Caenorhabditis elegans. Nat. Protoc. 2017, 12, 2081–2096. [Google Scholar] [CrossRef] [PubMed]
- Luke, C.J.; O’Reilly, L.P. Microscopic Investigation of Protein Function in C. elegans Using Fluorescent Imaging. Curr. Protoc. Cytom. 2015, 74. [Google Scholar] [CrossRef]
- Garsin, D.A.; Sifri, C.D.; Mylonakis, E.; Qin, X.; Singh, K.V.; Murray, B.E.; Calderwood, S.B.; Ausubel, F.M. A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 2001, 98, 10892–10897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, M.R.; Graham, C.E.; Gagliano, B.C.; Lorenz, M.C.; Garsin, D.A. Enterococcus faecalis inhibits hyphal morphogenesis and virulence of Candida albicans. Infect. Immun. 2013, 81, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Pujol, N.; Cypowyj, S.; Ziegler, K.; Millet, A.; Astrain, A.; Goncharov, A.; Jin, Y.; Chisholm, A.D.; Ewbank, J.J. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr. Biol. 2008, 18, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Kurz, C.L.; Cypowyj, S.; Couillault, C.; Pophillat, M.; Pujol, N.; Ewbank, J.J. Antifungal innate immunity in C. elegans: PKCdelta links G protein signaling and a conserved p38 MAPK cascade. Cell. Host Microbe 2009, 5, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenor, J.L.; Aballay, A. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 2008, 9, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Michelsen, K.S.; Eri, R.; Thomas, L.S.; Hu, B.; Lukasek, K.; Nast, C.C.; Lechago, J.; Xu, R.; Naiki, Y.; et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1055-65. [Google Scholar] [CrossRef] [PubMed]
- Pujol, N.; Link, E.M.; Liu, L.X.; Kurz, C.L.; Alloing, G.; Tan, M.W.; Ray, K.P.; Solari, R.; Johnson, C.D.; Ewbank, J.J. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr. Biol. 2001, 11, 809–821. [Google Scholar] [CrossRef]
- Mallo, G.V.; Kurz, C.L.; Couillault, C.; Pujol, N.; Granjeaud, S.; Kohara, Y.; Ewbank, J.J. Inducible antibacterial defense system in C. elegans. Curr. Biol. 2002, 12, 1209–1214. [Google Scholar] [CrossRef]
- Alper, S.; McBride, S.J.; Lackford, B.; Freedman, J.H.; Schwartz, D.A. Specificity and complexity of the Caenorhabditis elegans innate immune response. Mol. Cell. Biol. 2007, 27, 5544–5553. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; Hu, P.J. Insulin/insulin-like growth factor signaling in C. elegans. WormBook 2013. [Google Scholar] [CrossRef] [PubMed]
- Lamitina, S.T.; Strange, K. Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am. J. Physiol. Cell Physiol. 2005, 288, C467–C474. [Google Scholar] [CrossRef] [PubMed]
- Ewbank, J.J.; Pujol, N. Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr. Opin. Immunol. 2016, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santelmann, H.; Howard, J.M. Yeast metabolic products, yeast antigens and yeasts as possible triggers for irritable bowel syndrome. Eur. J. Gastroenterol. Hepatol. 2005, 17, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, M.N.; Elliott, D.E. Immunologic and molecular mechanisms in inflammatory bowel disease. Surg. Clin. North Am. 2007, 87, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, L.S.; Walhout, A.J. Worms, bacteria, and micronutrients: An elegant model of our diet. Trends Genet. 2014, 30, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, L.S.; Walhout, A.J. A Caenorhabditis elegans Genome-Scale Metabolic Network Model. Cell Syst. 2016, 2, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.R.; Challacombe, S.J.; Hube, B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 400–428. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L.; Fundyga, R.; Hecht, J.E.; Kapteyn, J.C.; Klis, F.M.; Arnold, J. Characterization of agglutinin-like sequence genes from non-albicans Candida and phylogenetic analysis of the ALS family. Genetics 2001, 157, 1555–1567. [Google Scholar] [PubMed]
- Desai, J.V.; Cheng, S.; Ying, T.; Nguyen, M.H.; Clancy, C.J.; Lanni, F.; Mitchell, A.P. Coordination of Candida albicans Invasion and Infection Functions by Phosphoglycerol Phosphatase Rhr2. Pathogens 2015, 4, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Wachtler, B.; Wilson, D.; Haedicke, K.; Dalle, F.; Hube, B. From attachment to damage: Defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.C.; Brand, A.C. Thigmo Responses: The Fungal Sense of Touch. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Aoki, W.; Kitahara, N.; Miura, N.; Morisaka, H.; Yamamoto, Y.; Kuroda, K.; Ueda, M. Comprehensive characterization of secreted aspartic proteases encoded by a virulence gene family in Candida albicans. J. Biochem. 2011, 150, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, I.D.; Wilson, D.; Wachtler, B.; Brunke, S.; Naglik, J.R.; Hube, B. Candida albicans dimorphism as a therapeutic target. Expert Rev. Anti-Infect. Ther. 2012, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.J.; Kohler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous C. albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef]
- Saville, S.P.; Lazzell, A.L.; Monteagudo, C.; Lopez-Ribot, J.L. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot. Cell 2003, 2, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Finkel, J.S.; Mitchell, A.P. Genetic control of Candida albicans biofilm development. Nat. Rev. Microbiol. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Sasse, C.; Hasenberg, M.; Weyler, M.; Gunzer, M.; Morschhauser, J. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot. Cell 2013, 12, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Zavala, B.; Reuss, O.; Park, Y.N.; Ohlsen, K.; Morschhauser, J. Environmental induction of white-opaque switching in Candida albicans. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Brock, M. Fungal metabolism in host niches. Curr. Opin. Microbiol. 2009, 12, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Tucey, T.M.; Verma, J.; Harrison, P.F.; Snelgrove, S.L.; Lo, T.L.; Scherer, A.K.; Barugahare, A.A.; Powell, D.R.; Wheeler, R.T.; Hickey, M.J.; et al. Glucose Homeostasis Is Important for Immune Cell Viability during Candida Challenge and Host Survival of Systemic Fungal Infection. Cell Metab. 2018, 27, 988–1006. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.C.; Bender, J.A.; Fink, G.R. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell 2004, 3, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.L. Bacterial peptidoglycan-derived molecules activate Candida albicans hyphal growth. Commun. Integr. Biol. 2008, 1, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Silva, S.; Faria-Oliveira, F.; Pinho, E.; Henriques, M.; Lucas, C. Candida albicans virulence and drug-resistance requires the O-acyltransferase Gup1p. BMC Microbiol. 2010, 10, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnowski, R.; Sanchez, H.; Covelli, A.S.; Dominguez, E.; Jaromin, A.; Berhardt, J.; Heiss, C.; Azadi, P.; Mitchell, A.; Andes, D.R. Candida albicans biofilm-induced vesicles confer drug resistance through matrix biogenesis. PLoS Biol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Ksiezopolska, E.; Gabaldon, T. Evolutionary Emergence of Drug Resistance in Candida Opportunistic Pathogens. Genes 2018, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Bonus, M.; Tomiyama, S.; Miyoshi, K.; Nagi, M.; Niimi, K.; Chindamporn, A.; Gohlke, H.; Schmitt, L.; Cannon, R.D.; et al. FK506 resistance of Saccharomyces cerevisiae Pdr5 and Candida albicans Cdr1 involves mutations in the transmembrane domains and extracellular loops. Antimicrob. Agents Chemother. 2018. [Google Scholar] [CrossRef] [PubMed]
- White, T.C.; Holleman, S.; Dy, F.; Mirels, L.F.; Stevens, D.A. Resistance mechanisms in clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 2002, 46, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Ostrosky-Zeichner, L.; Casadevall, A.; Galgiani, J.N.; Odds, F.C.; Rex, J.H. An insight into the antifungal pipeline: Selected new molecules and beyond. Nat. Rev. Drug Discov. 2010, 9, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Rybowicz, J.; Gurk-Turner, C. Caspofungin: The first agent available in the echinocandin class of antifungals. Proc. (Bayl. Univ. Med. Cent.) 2002, 15, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Balkovec, J.M.; Hughes, D.L.; Masurekar, P.S.; Sable, C.A.; Schwartz, R.E.; Singh, S.B. Discovery and development of first in class antifungal caspofungin (CANCIDAS(R.))—A case study. Nat. Prod. Rep. 2014, 31, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Troskie, A.M.; Rautenbach, M.; Delattin, N.; Vosloo, J.A.; Dathe, M.; Cammue, B.P.; Thevissen, K. Synergistic activity of the tyrocidines, antimicrobial cyclodecapeptides from Bacillus aneurinolyticus, with amphotericin B and caspofungin against Candida albicans biofilms. Antimicrob. Agents Chemother. 2014, 58, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Delattin, N.; De Brucker, K.; Vandamme, K.; Meert, E.; Marchand, A.; Chaltin, P.; Cammue, B.P.; Thevissen, K. Repurposing as a means to increase the activity of amphotericin B and caspofungin against Candida albicans biofilms. J. Antimicrob. Chemother. 2014, 69, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.R.; Fuchs, B.B.; Alves, V.S.; Jayamani, E.; Colombo, A.L.; Mylonakis, E. Pathogenesis of the Candida parapsilosis Complex in the Model Host Caenorhabditis elegans. Genes 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Eschenauer, G.; Depestel, D.D.; Carver, P.L. Comparison of echinocandin antifungals. Ther. Clin. Risk Manag. 2007, 3, 71–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchiesi, F.; Spreghini, E.; Tomassetti, S.; Arzeni, D.; Giannini, D.; Scalise, G. Comparison of the fungicidal activities of caspofungin and amphotericin B against Candida glabrata. Antimicrob. Agents Chemother. 2005, 49, 4989–4992. [Google Scholar] [CrossRef] [PubMed]
- Elefanti, A.; Mouton, J.W.; Krompa, K.; Al-Saigh, R.; Verweij, P.E.; Zerva, L.; Meletiadis, J. Inhibitory and fungicidal effects of antifungal drugs against Aspergillus species in the presence of serum. Antimicrob. Agents Chemother. 2013, 57, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.A.; Lee, K.K.; Munro, C.A.; Gow, N.A. Caspofungin Treatment of Aspergillus fumigatus Results in ChsG-Dependent Upregulation of Chitin Synthesis and the Formation of Chitin-Rich Microcolonies. Antimicrob. Agents Chemother. 2015, 59, 5932–5941. [Google Scholar] [CrossRef] [PubMed]
- Doman, M.; Kovacs, R.; Perlin, D.S.; Kardos, G.; Gesztelyi, R.; Juhasz, B.; Bozo, A.; Majoros, L. Dose escalation studies with caspofungin against Candida glabrata. J. Med. Microbiol. 2015, 64, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.J.; Teppler, H.; Donowitz, G.R.; Maertens, J.A.; Baden, L.R.; Dmoszynska, A.; Cornely, O.A.; Bourque, M.R.; Lupinacci, R.J.; Sable, C.A.; et al. Caspofungin versus liposomal amphotericin B for empirical antifungal therapy in patients with persistent fever and neutropenia. N. Engl. J. Med. 2004, 351, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Maertens, J.; Raad, I.; Petrikkos, G.; Boogaerts, M.; Selleslag, D.; Petersen, F.B.; Sable, C.A.; Kartsonis, N.A.; Ngai, A.; Taylor, A.; et al. Efficacy and safety of caspofungin for treatment of invasive aspergillosis in patients refractory to or intolerant of conventional antifungal therapy. Clin. Infect. Dis. 2004, 39, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Tampakakis, E.; Okoli, I.; Mylonakis, E.A.C. C. elegans-based, whole animal, in vivo screen for the identification of antifungal compounds. Nat. Protoc. 2008, 3, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Fazly, A.; Jain, C.; Dehner, A.C.; Issi, L.; Lilly, E.A.; Ali, A.; Cao, H.; Fidel, P.L., Jr.; Rao, R.P.; Kaufman, P.D. Chemical screening identifies filastatin, a small molecule inhibitor of Candida albicans adhesion, morphogenesis, and pathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 13594–13599. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Mylonakis, E. From the outside in and the inside out: Antifungal immune responses in Caenorhabditis elegans. Virulence 2010, 1, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Tucci, M.L.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem. 2012, 287, 4107–4120. [Google Scholar] [CrossRef] [PubMed]
- Plaza, D.F.; Schmieder, S.S.; Lipzen, A.; Lindquist, E.; Künzler, M. Identification of a Novel Nematotoxic Protein by Challenging the Model Mushroom Coprinopsis cinerea with a Fungivorous Nematode. G3 (Bethesda) 2015, 6, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Blanco, D.; Lynn, A.; Rosch, J.; Noreldin, R.; Salerni, A.; Lambert, C.; Rao, R.P. A pre-therapeutic coating for medical devices that prevents the attachment of Candida albicans. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Kuhn, D.M.; Mukherjee, P.K.; Hoyer, L.L.; McCormick, T.; Ghannoum, M.A. Biofilm formation by the fungal pathogen Candida albicans: Development, architecture, and drug resistance. J. Bacteriol. 2001, 183, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Su, C.; Wang, A.; Liu, H. Hyphal development in Candida albicans requires two temporally linked changes in promoter chromatin for initiation and maintenance. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef]
- Hickman, M.A.; Zeng, G.; Forche, A.; Hirakawa, M.P.; Abbey, D.; Harrison, B.D.; Wang, Y.-M.; Su, C.-H.; Bennett, R.J.; Wang, Y.; et al. The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature 2013, 494, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hickman, M.A.; Paulson, C.; Dudley, A.; Berman, J. Parasexual Ploidy Reduction Drives Population Heterogeneity Through Random and Transient Aneuploidy in Candida albicans. Genetics 2015, 200, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Feistel, D.J.; Elmostafa, R.; Nguyen, N.; Penley, M.; Morran, L.; Hickman, M.A. A Novel Virulence Phenotype Rapidly Assesses Candida Fungal Pathogenesis in Healthy and Immunocompromised Caenorhabditis elegans Hosts. Available online: https://www.biorxiv.org/content/early/2018/07/16/370403 (accessed on 16 July 2018).
Figure 1.
C. albicans—C. elegans infection model can be pharmacologically or genetically modulated. (A) Live cell imaging of C. elegans shows C. albicans accumulation in the nematode intestinal lumen day 3 post infection. Yeast cells are quickly ingested by the worms and accumulate in the intestinal lumen completely intact indicating that they are able to survive the mechanical crushing of the pharynx. (B) Survival curves of nematodes challenged with C. albicans versus uninfected controls. (C) Survival curves of nematodes challenged with either wild-type C. albicans, cph1/cph1 efg1/efg1 double mutant or wild-type C. albicans + 50 mM of fluconazole.
Figure 1.
C. albicans—C. elegans infection model can be pharmacologically or genetically modulated. (A) Live cell imaging of C. elegans shows C. albicans accumulation in the nematode intestinal lumen day 3 post infection. Yeast cells are quickly ingested by the worms and accumulate in the intestinal lumen completely intact indicating that they are able to survive the mechanical crushing of the pharynx. (B) Survival curves of nematodes challenged with C. albicans versus uninfected controls. (C) Survival curves of nematodes challenged with either wild-type C. albicans, cph1/cph1 efg1/efg1 double mutant or wild-type C. albicans + 50 mM of fluconazole.

Figure 2.
Molecular pathways that play central roles in mounting an immune response in C. elegans. (1) TGF-β (or DBL-1) pathway, (2) Insulin signaling pathway, (3) Toll pathway and (4) MAPK pathway. (Collated from [36,74,76,77,78,79,80,81,82,83,84,85]).

Figure 3.
C. albicans common virulence mechanisms. Adapted from Mayer et al. [54] (A) adhesion to host cells. (B) Invasion and host cell damage. (C) Biofilms on abiotic medical devices. (D) Morphologic diversity. (E) Fitness traits: amino acid and nutrient uptake, pH regulation, and stress response.
Figure 3.
C. albicans common virulence mechanisms. Adapted from Mayer et al. [54] (A) adhesion to host cells. (B) Invasion and host cell damage. (C) Biofilms on abiotic medical devices. (D) Morphologic diversity. (E) Fitness traits: amino acid and nutrient uptake, pH regulation, and stress response.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Elkabti, A.B.; Issi, L.; Rao, R.P. Caenorhabditis elegans as a Model Host to Monitor the Candida Infection Processes. J. Fungi 2018, 4, 123. https://0-doi-org.brum.beds.ac.uk/10.3390/jof4040123
AMA Style
Elkabti AB, Issi L, Rao RP. Caenorhabditis elegans as a Model Host to Monitor the Candida Infection Processes. Journal of Fungi. 2018; 4(4):123. https://0-doi-org.brum.beds.ac.uk/10.3390/jof4040123
Chicago/Turabian StyleElkabti, Asmaa B., Luca Issi, and Reeta P. Rao. 2018. "Caenorhabditis elegans as a Model Host to Monitor the Candida Infection Processes" Journal of Fungi 4, no. 4: 123. https://0-doi-org.brum.beds.ac.uk/10.3390/jof4040123
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.