
Exploring the Species Diversity of Edible Mushrooms in Yunnan, Southwestern China, by DNA Barcoding

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
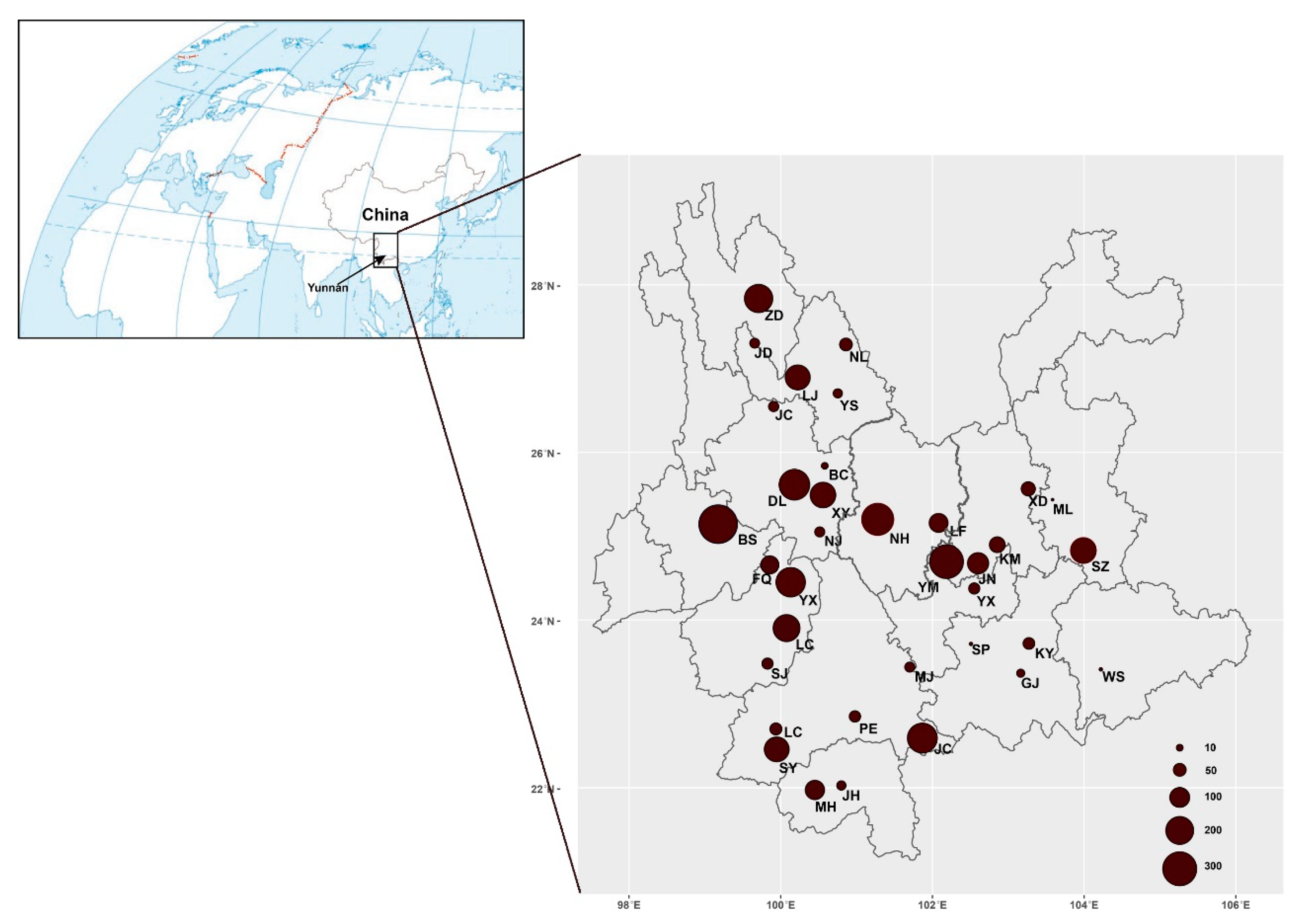
2.1. Sampling
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Data Analysis
2.3.1. Species Assignment
2.3.2. DNA Barcoding Assessment
2.3.3. Phylogenetic Species Identification in Selected Taxa
3. Results
3.1. Sequencing
3.2. Molecular Species Identification
3.2.1. Species Estimation
3.2.2. Potential New Species Based on Adjusted Criteria
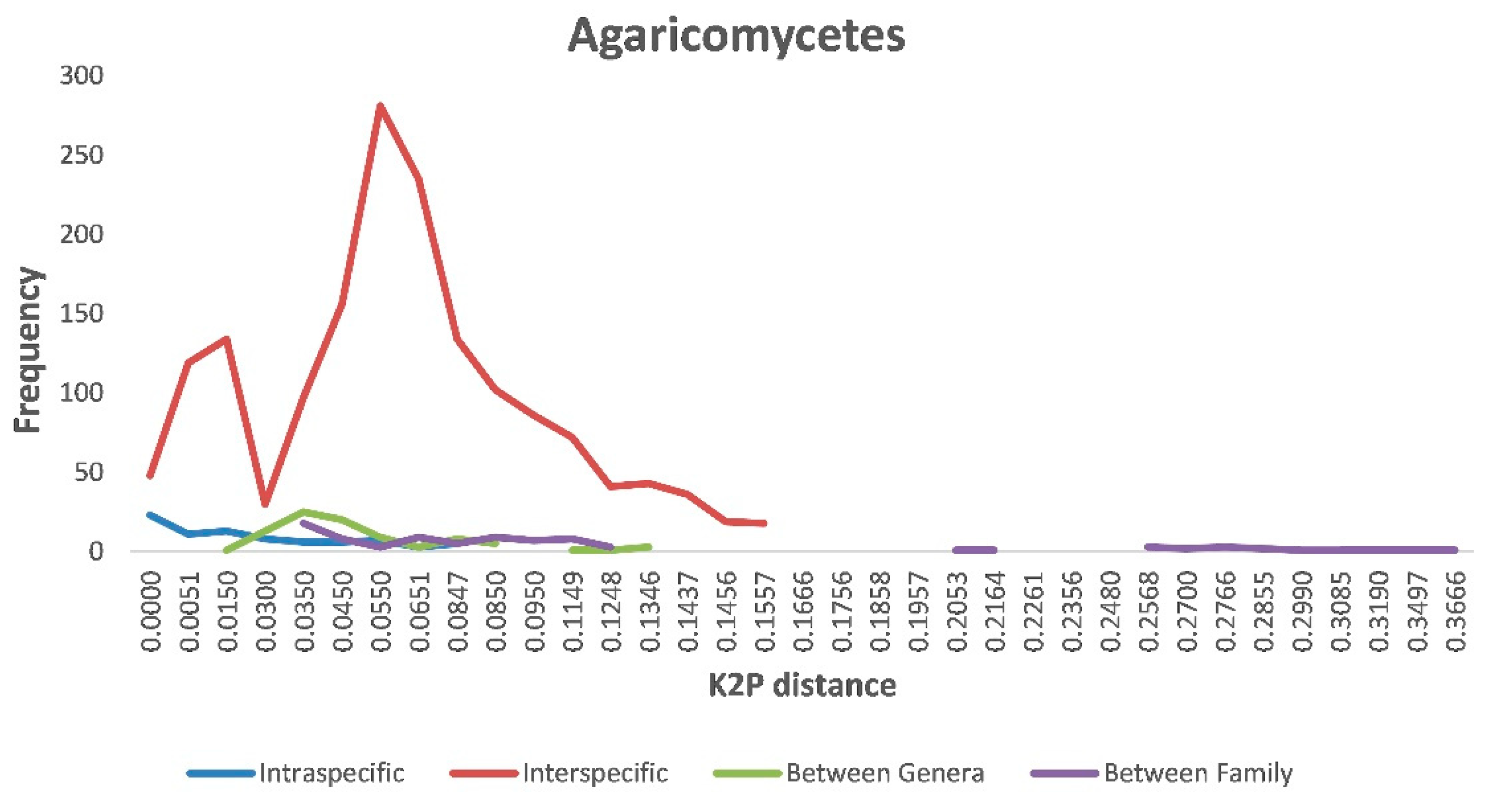
3.3. Intraspecific Variation, Interspecific Variation, and DNA Barcoding Gaps
4. Discussion
4.1. Species Number Estimation
4.2. Feasibility of ITS Sequence as Identification Marker of Wild Mushroom Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, N.G.; Ariyawansa, H.; Hyde, K.; Maharachchikumbura, S.; Zhao, R.L.; Phillips, A.; Jayawardena, R.; Thambugala, K.; Dissanayake, A.; Wijayawardene, N.; et al. Perspectives into the value of genera, families and orders in classification. Mycosphere 2016, 7, 1649–1668. [Google Scholar] [CrossRef]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lucking, R.; et al. A higher-level phylogenetic classification of the Fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- He, M.Q.; Zhao, R.L.; Hyde, K.; Begerow, D.; Kemler, M.; Yurkov, A.; McKenzie, E.; Raspé, O.; Kakishima, M.; Sanchez-Ramirez, S.; et al. Notes, outline and divergence times of Basidiomycota. Fungal Divers. 2019, 99, 105–367. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Sanchez-Ramirez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 2017, 5, FUNK-0052-2016. [Google Scholar] [CrossRef]
- Wu, B.; Hussain, M.; Zhang, W.; Stadler, M.; Liu, X.; Xiang, M. Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 2019, 10, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P.; Větrovský, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2021. [Google Scholar] [CrossRef]
- Hyde, K.D.; Jeewon, R.; Chen, Y.J.; Bhunjun, C.S.; Calabon, M.S.; Jiang, H.-B.; Lin, C.G.; Norphanphoun, C.; Sysouphanthong, P.; Pem, D.; et al. The numbers of fungi: Is the descriptive curve flattening? Fungal Divers. 2020, 103, 219–271. [Google Scholar] [CrossRef]
- Xu, J. Fungal species concepts in the genomics era. Genome 2020, 63, 459–468. [Google Scholar] [CrossRef]
- Wijayawardene, N.; Hyde, K.; Al-Ani, L.; Tedersoo, L.; Haelewaters, D.; Rajeshkumar, K.C.; Zhao, R.-L.; Aptroot, A.; Saxena, R.; Tokarev, Y.; et al. Outline of Fungi and fungus-like taxa. Mycosphere 2020, 11, 1160–1456. [Google Scholar] [CrossRef]
- Muggia, L.; Ametrano, C.G.; Sterflinger, K.; Tesei, D. An overview of genomics, phylogenomics and proteomics approaches in Ascomycota. Life 2020, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.; Abarenkov, K.; Koljalg, U.; Opik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of Fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Frøslev, T.G.; Kjøller, R.; Bruun, H.H.; Ejrnæs, R.; Hansen, A.J.; Læssøe, T.; Heilmann-Clausen, J. Man against machine: Do fungal fruitbodies and eDNA give similar biodiversity assessments across broad environmental gradients? Biol. Conserv. 2019, 233, 201–212. [Google Scholar] [CrossRef]
- Hoang, M.T.V.; Irinyi, L.; Chen, S.C.A.; Sorrell, T.C.; Meyer, W.; Arabatzis, M.; Arthur, I.; Cano-Lira, J.F.; Cardinali, G.; Castañón, L.R.; et al. Dual DNA barcoding for the molecular identification of the agents of invasive fungal infections. Front. Microb. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tian, Y.; Menolli, N., Jr.; Ye, L.; Karunarathna, S.; Perez, J.; Rahman, M.; Rashid, H.; Phengsintham, P.; Rizal, L.; et al. Reviewing the world’s edible mushroom species: A new evidence—Based classification system. Compr. Rev. Food Sci. F 2021, 20, 1982–2014. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Moreno, J.; Martínez-Reyes, M. Edible ectomycorrhizal mushrooms: Biofactories for sustainable development. In Biosystems Engineering: Biofactories for Food Production in the Century XXI; Guevara-Gonzalez, R., Torres-Pacheco, I., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 151–233. [Google Scholar] [CrossRef]
- Yang, X.; He, J.; Li, C.; Ma, J.; Yang, Y.; Xu, J. Matsutake trade in Yunnan province, China: An overview. Econ. Bot. 2008, 62, 269–277. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Yang, Z.L. Trends in fungal systematics: More and more taxa. Mycosystema 2020, 39, 1611–1616. [Google Scholar] [CrossRef]
- Feng, B.; Yang, Z. Studies on diversity of higher fungi in Yunnan, southwestern China: A review. Plant Divers. 2018, 40, 165–171. [Google Scholar] [CrossRef]
- Yang, Z. On wild mushroom resources and their utilization in Yunnan Province, Southwest China. J. Nat. Resour. 2002, 17, 463–469. [Google Scholar] [CrossRef]
- Xu, J. DNA barcoding, fungal diversity, and authentication of wild gourmet mushrooms. Acta Agric. Univ. Jiangxiensis 2010, 32, 1010–1017. [Google Scholar] [CrossRef]
- Xu, J.; Sha, T.; Li, Y.-C.; Zhao, Z.W.; Yang, Z.L. Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol. Ecol. 2008, 17, 1238–1247. [Google Scholar] [CrossRef]
- Li, M.; Liang, J.; Li, Y.; Feng, B.; Yang, Z.-L.; James, T.Y.; Xu, J. Genetic diversity of Dahongjun, the commercially important “Big Red Mushroom” from southern China. PLoS ONE 2010, 5, e10684. [Google Scholar] [CrossRef] [Green Version]
- Lam, K.Y.C.; Chan, G.K.L.; Xin, G.-Z.; Xu, H.; Ku, C.-F.; Chen, J.-P.; Yao, P.; Lin, H.-Q.; Dong, T.T.X.; Tsim, K.W.K. Authentication of Cordyceps sinensis by DNA Analyses: Comparison of ITS Sequence Analysis and RAPD-Derived Molecular Markers. Molecules 2015, 20, 22454–22462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhou, L.-W.; Yang, Z.-L.; Bau, T.; Li, T.-H.; Dai, Y.-C. Resource diversity of Chinese macrofungi: Edible, medicinal and poisonous species. Fungal Divers. 2019, 98, 1–76. [Google Scholar] [CrossRef]
- Li, H.; Zhang, H.; Zhou, J.; Yin, Y.; He, Q.; Jiang, S.; Ma, P.; Zhang, Y.; Wen, K.; Yuan, Y.; et al. Mushroom poisoning outbreaks—China, 2020. China CDC Wkly. 2021, 3, 41–45. [Google Scholar] [CrossRef]
- Stone, R. Will a midsummer’s nightmare return? Science 2010, 329, 132–134. [Google Scholar] [CrossRef]
- Yang, Z.; Li, Y.C.; Tang, L.P.; Shi, G.Q.; Zeng, G. Trogia venenata (Agaricales), a novel poisonous species which has caused hundreds of deaths in southwestern China. Mycol. Prog. 2012, 11, 937–945. [Google Scholar] [CrossRef]
- Balasundaram, S.V.; Engh, I.B.; Skrede, I.; Kauserud, H. How many DNA markers are needed to reveal cryptic fungal species? Fungal Biol. 2015, 119, 940–945. [Google Scholar] [CrossRef]
- Peintner, U.; Kuhnert-Finkernagel, R.; Wille, V.; Biasioli, F.; Shiryaev, A.; Perini, C. How to resolve cryptic species of Polypores: An example in Fomes. IMA Fungus 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Zamora, J.C.; Svensson, M.; Kirschner, R.; Olariaga, I.; Ryman, S.; Parra, L.A.; Geml, J.; Rosling, A.; Adamčík, S.; Ahti, T.; et al. Considerations and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 2018, 9, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Badotti, F.; de Oliveira, F.S.; Garcia, C.F.; Vaz, A.B.M.; Fonseca, P.L.C.; Nahum, L.A.; Oliveira, G.; Góes-Neto, A. Effectiveness of ITS and sub-regions as DNA barcode markers for the identification of Basidiomycota (Fungi). BMC Microb. 2017, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Guo, H.; Li, J.; Li, W.; Wang, Q.; Yu, X. Evaluation of five regions as DNA barcodes for identification of Lepista species (Tricholomataceae, Basidiomycota) from China. PeerJ 2019, 7, e7307. [Google Scholar] [CrossRef] [Green Version]
- Fryssouli, V.; Zervakis, G.I.; Polemis, E.; Typas, M.A. A global meta-analysis of ITS rDNA sequences from material belonging to the genus Ganoderma (Basidiomycota, Polyporales) including new data from selected taxa. MycoKeys 2020, 75, 71–143. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Kõljalg, U.; Abarenkov, K. Identifying the ‘unidentified’ fungi: A global-scale long-read third-generation sequencing approach. Fungal Divers. 2020, 103, 273–293. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.H. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, 193–201. [Google Scholar] [CrossRef]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.A., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Deigo, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Dentinger, B.T.; Didukh, M.Y.; Moncalvo, J.-M. Comparing COI and ITS as DNA barcode markers for mushrooms and allies (Agaricomycotina). PLoS ONE 2011, 6, e25081. [Google Scholar] [CrossRef]
- El Karkouri, K.; Murat, C.; Zampieri, E.; Bonfante, P. Identification of internal transcribed spacer sequence motifs in truffles: A first step toward their DNA barcoding. Appl. Environ. Microb. 2007, 73, 5320–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.-H.; Zhao, Q.; Yang, Z.L.; Hansen, K.; Taşkin, H.; Büyükalaca, S.; Dewsbury, D.; Moncalvo, J.-M.; Douhan, G.W.; Robert, V.A.R.G.; et al. How well do ITS rDNA sequences differentiate species of true morels (Morchella)? Mycologia 2012, 104, 1351–1368. [Google Scholar] [CrossRef] [Green Version]
- Jargeat, P.; Martos, F.; Carriconde, F.; Gryta, H.; Moreau, P.A.; Gardes, M. Phylogenetic species delimitation in ectomycorrhizal fungi and implications for barcoding: The case of the Tricholoma scalpturatum complex (Basidiomycota). Mol. Ecol. 2010, 19, 5216–5230. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cadorin, M.; Liang, Y.; Yang, Z. DNA-based geographic typing of the gourmet mushroom Tricholoma matsutake traded in China. Mycoscience 2010, 51, 248–251. [Google Scholar] [CrossRef]
- Avin, F.A.; Bhassu, S.; Tan, Y.S.; Shahbazi, P.; Vikineswary, S. Molecular divergence and species delimitation of the cultivated oyster mushrooms: Integration of IGS1 and ITS. Sci. World J. 2014, 2014, 793414. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Xu, J.; Wu, G.; Zeng, N.-K.; Li, Y.-C.; Tolgor, B.; Kost, G.W.; Yang, Z.L. DNA sequence analyses reveal abundant diversity, endemism and evidence for Asian origin of the Porcini mushrooms. PLoS ONE 2012, 7, e37567. [Google Scholar] [CrossRef]
- Cai, Q.; Tang, L.-P.; Yang, Z.-L. DNA barcoding of economically important mushrooms: A case study on lethal Amanitas from China. Plant Divers. Resour. 2012, 34, 614. [Google Scholar] [CrossRef]
- Hughes, K.W.; Tulloss, R.H.; Petersen, R.H. Intragenomic nuclear RNA variation in a cryptic Amanita taxon. Mycologia 2018, 110, 93–103. [Google Scholar] [CrossRef]
- Jensen-Vargas, E.; Marizzi, C. DNA barcoding for identification of consumer-relevant fungi sold in New York: A powerful tool for citizen scientists? Foods 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Dentinger, B.T.M.; Suz, L.M. What’s for dinner? Undescribed species in commercial porcini from China. PeerJ 2014, 2, e423v421. [Google Scholar] [CrossRef] [Green Version]
- Raja, H.A.; Baker, T.R.; Little, J.G.; Oberlies, N.H. DNA barcoding for identification of consumer-relevant mushrooms: A partial solution for product certification? Food Chem. 2017, 214, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Li, Y.-C.; Zhu, X.-T.; Zhao, K.; Han, L.-H.; Cui, Y.-Y.; Li, F.; Xu, J.-P.; Yang, Z.L. One hundred noteworthy boletes from China. Fungal Divers. 2016, 81, 25–188. [Google Scholar] [CrossRef]
- Han, L.H.; Wu, G.; Horak, E.; Halling, R.; Xu, J.; Ndolo Ebika, S.; Sato, H.; Fechner, N.; Sharma, Y.; Yang, Z.L. Phylogeny and species delimitation of Strobilomyces (Boletaceae), with an emphasis on the Asian species. Persoonia 2019, 44, 113–139. [Google Scholar] [CrossRef]
- Li, J.; Han, L.-H.; Liu, X.-B.; Zhao, Z.-W. The saprotrophic Pleurotus ostreatus species complex: Late Eocene origin in East Asia, multiple dispersal, and complex speciation. IMA Fungus 2020, 11, 1–21. [Google Scholar] [CrossRef]
- Wang, P.M.; Liu, X.B.; Dai, Y.C.; Horak, E.; Steffen, K.; Yang, Z.L. Phylogeny and species delimitation of Flammulina: Taxonomic status of winter mushroom in East Asia and a new European species identified using an integrated approach. Mycol. Prog. 2018, 17, 1013–1030. [Google Scholar] [CrossRef]
- Feng, B.; Wang, X.H.; Ratkowsky, D.; Gates, G.; Lee, S.S.; Grebenc, T.; Yang, Z.L. Multilocus phylogenetic analyses reveal unexpected abundant diversity and significant disjunct distribution pattern of the Hedgehog Mushrooms (Hydnum L.). Sci. Rep. 2016, 6, 25586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Wang, H.C.; Xue, W.Q.; Zhao, J.; Yang, Z.L. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS ONE 2016, 11, e0154794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.Y.; Cai, Q.; Tang, L.-P.; Liu, J.-W.; Yang, Z.L. The family Amanitaceae: Molecular phylogeny, higher-rank taxonomy and the species in China. Fungal Divers. 2018, 91, 5–230. [Google Scholar] [CrossRef]
- Xu, J.; Yoell, H.J.; Anderson, J.B. An efficient protocol for isolating DNA from higher fungi. Trends Genet. 1994, 10, 226–227. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95. [Google Scholar]
- Hofstetter, V.; Buyck, B.; Eyssartier, G.; Schnee, S.; Gindro, K. The unbearable lightness of sequenced-based identification. Fungal Divers. 2019, 96, 243–284. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Paulay, G. DNA barcodes perform best with well-characterized taxa. PLoS Biol. 2005, 3, e435. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L.; Fazekas, A.J.; et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [Green Version]
- Buyck, B.; Hofstetter, V. The contribution of tef-1 sequences to species delimitation in the Cantharellus cibarius complex in the southeastern USA. Fungal Divers. 2011, 49, 35–46. [Google Scholar] [CrossRef]
- Olariaga, I.; Moreno, G.; Manjon, J.L.; Salcedo, I.; Hofstetter, V.; Rodriguez, D.; Buyck, B. Cantharellus (Cantharellales, Basidiomycota) revisited in Europe through a multigene phylogeny. Fungal Divers. 2017, 83, 263–292. [Google Scholar] [CrossRef]
- Shao, S.C.; Liu, P.G.; Wei, T.Z.; Herrera, M. New insights into the taxonomy of the genus Cantharellus in China: Epityfication of C. yunnanensis W.F. Chiu and the first record of C. cibarius Fr. Cryptogam. Mycol. 2021, 42, 25–37. [Google Scholar] [CrossRef]
- Wang, K.; Su, J.H.; Yang, L.; Deng, H.; Wang, Y.H.; Wu, H.J.; Li, Y.; Wu, H.M.; Wei, X.D.; Wei, T.Z.; et al. The use of Checklist of Fungi in China database in the red list assessment of macrofungi in China. Biodivers. Sci. 2020, 28, 74–98. [Google Scholar] [CrossRef]
- Dai, Y.; Yang, Z.; Cui, B.; Zhou, L. Species diversity and utilization of medicinal mushrooms and Fungi in China. Int. J. Med. Mushrooms 2009, 11, 287–302. [Google Scholar] [CrossRef]
- Zhuang, W.Y. Higher Fungi of Tropical China; Mycotaxon LTD: Ithaca, NY, USA, 2001. [Google Scholar]
- Cao, B.; He, M.Q.; Ling, Z.L.; Zhang, M.Z.; Zhao, R.L. A revision of Agaricus section Arvenses with nine new species from China. Mycologia 2020, 113, 191–211. [Google Scholar] [CrossRef]
- Wu, G.; Feng, B.; Xu, J.; Zhu, X.T.; Li, Y.C.; Zeng, N.K.; Hosen, M.I.; Yang, Z.L. Molecular phylogenetic analyses redefine seven major clades and reveal 22 new generic clades in the fungal family Boletaceae. Fungal Divers. 2014, 69, 93–115. [Google Scholar] [CrossRef]
- Feng, B.; Zhao, Q.; Xu, J.; Qin, J.; Yang, Z.L. Drainage isolation and climate change-driven population expansion shape the genetic structures of Tuber indicum complex in the Hengduan Mountains region. Sci. Rep. 2016, 6, 21811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, B.K.; Li, H.J.; Ji, X.; Zhou, J.L.; Song, J.; Si, J.; Yang, Z.L.; Dai, Y.C. Species diversity, taxonomy and phylogeny of Polyporaceae (Basidiomycota) in China. Fungal Divers. 2019, 97, 137–392. [Google Scholar] [CrossRef]
- Wang, X.H.; Buyck, B.; Verbeken, A.; Hansen, K. Revisiting the morphology and phylogeny of Lactifluus with three new lineages from southern China. Mycologia 2015, 107, 941–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Zhang, Y.; Yu, Z.; Mi, F.; Liu, C.; Tang, X.; Long, Y.; He, X.; Wang, P.; Xu, J. Structure, gene flow, and recombination among geographic populations of a Russula virescens ally from southwestern China. PLoS ONE 2013, 8, e73174. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Oberwinkler, F. Fruit body development of Amanita muscaria (Basidiomycetes). Nova Hedwig. 1999, 68, 441–468. [Google Scholar] [CrossRef]
- Yao, Y.J.; Zhuang, W.Y.; Wei, T.Z.; Li, L.; Wei, X.L.; Deng, H.; Liu, D.M.; Cai, L.; Li, J.S.; Wang, K.; et al. Threatened species list of China’s macrofungi. Biodivers. Sci. 2020, 28, 20–25. [Google Scholar] [CrossRef]
- Alhawatema, M.; Alqudah, A.; Al Tawaha, A.R. Application of using DNA barcoding genes in identification of fungi Species, A review. Biosci. Res. 2019, 16, 1763–1775. [Google Scholar]



{kind=link}
{kind=link}
| Genus | Sample Size | ITS Amplicons | Amplification Success (%) | Heterozygotic Sequences |
|---|---|---|---|---|
| Amanita | 26 | 26 | 100 | 0 |
| Auricularia | 37 | 33 | 88.6 | 0 |
| Boletus | 94 | 94 | 100 | 0 |
| Butyriboletus | 27 | 27 | 100 | 0 |
| Cantharellus | 380 | 277 | 72.8 | 177 |
| Catathelasma | 22 | 22 | 100 | 0 |
| Cortinarius | 20 | 20 | 100 | 0 |
| Hygrophorus | 39 | 39 | 100 | 0 |
| Lactarius | 102 | 89 | 87.2 | 0 |
| Leucopaxillus | 26 | 23 | 86.2 | 0 |
| Lyophyllum | 176 | 125 | 71 | 0 |
| Ramaria | 252 | 118 | 47.2 | 82 |
| Russula | 294 | 281 | 95.7 | 0 |
| Termitomyces | 608 | 396 | 65.1 | 118 |
| Thelephora | 494 | 494 | 100 | 0 |
| Tricholoma | 35 | 35 | 100 | 0 |
| Class | Order | Family | Genus | Species | ≥97% | 95–97% | 90–95% | ≤90% | Sample Size | Edibility Status | Intraspecific Distance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sordariomycetes | Hypocreales | Cordycipitaceae | Cordyceps | C. militaris | 4 | 4 | E | 0 | |||
| Agaricomycetes | Agaricales | Agaricaceae | Agaricus | A. rubescens | 2 | 1 | 3 | E | 0.029 | ||
| A. virgineoides | 2 | 2 | P | n/a | |||||||
| Amanitaceae | Amanita | A. caojizong | 1 | 2 | 2 | 5 | E | 0.099 | |||
| A. citrinoannulata | 1 | 1 | U | n/a | |||||||
| A. cupreobrunneus | 3 | 1 | 4 | E | 0.005 | ||||||
| A. depauperatus | 1 | 1 | U, N | n/a | |||||||
| A. imazekii | 6 | 6 | E | 0.057 | |||||||
| A. masasiensis | 5 | 5 | U, N | 0.049 | |||||||
| A. pantherina | 2 | 2 | N | n/a | |||||||
| A. pseudoporphyria | 1 | 1 | 2 | E | n/a | ||||||
| Biannulariaceae | Catathelasma | C. ventricosum | 6 | 14 | 20 | E | 0.012 | ||||
| Cortinariaceae | Cortinarius | C. balteatoalbus | 8 | 2 | 10 | U | 0.077 | ||||
| C. caperatus | 3 | 3 | E | 0 | |||||||
| C. flavescentipes | 1 | 5 | 6 | U, N | 0.003 | ||||||
| C. vernus | 1 | 1 | U | n/a | |||||||
| Hydnangiaceae | Laccaria | L. laccata | 1 | 1 | E | n/a | |||||
| Laccaria | L. vinaceoavellanea | 8 | 4 | 1 | 13 | E | 0.029 | ||||
| Hygrophoraceae | Hygrophorus | H. agathosmus | 3 | 3 | E | 0.002 | |||||
| H. hypothejus | 1 | 1 | E | n/a | |||||||
| H. purpurascens | 13 | 1 | 14 | E | 0.002 | ||||||
| H. russula | 1 | 1 | E | n/a | |||||||
| Hymenogastraceae | Naucoria | N. fellea | 1 | 1 | U, N | n/a | |||||
| Psilocybe | P. semilanceata | 14 | 14 | P | 0.061 | ||||||
| Lyophyllaceae | Lyophyllum | L. decastes | 1 | 1 | 2 | E | n/a | ||||
| L. fumosum | 5 | 5 | 6 | 16 | E | 0.084 | |||||
| L. shimeji | 77 | 4 | 3 | 84 | E | 0.048 | |||||
| Termitomyces | T. bulborhizus | 10 | 10 | E | 0.003 | ||||||
| T. clypeatus | 13 | 176 | 189 | E | 0.011 | ||||||
| T. eurrhizus | 66 | 66 | E | 0.003 | |||||||
| T. heimii | 5 | 4 | 9 | E | 0.012 | ||||||
| T. medius | 5 | 8 | 13 | E | 0.008 | ||||||
| T. microcarpus | 1 | 44 | 89 | 134 | E | 0.055 | |||||
| T. radicatus | 4 | 4 | E | 0.045 | |||||||
| Tephrocybe | T. ancida | 3 | 3 | U, N | 0.04 | ||||||
| Omphalotaceae | Lentinula | L. lateritia | 1 | 2 | 3 | U, N | 0.065 | ||||
| Physalacriaceae | Hymenopellis | H. radicata | 1 | 1 | E | n/a | |||||
| Pleurotaceae | Pleurotus | P. giganteus | 3 | 3 | U | 0.001 | |||||
| Pseudoclitocybaceae | Pseudoclitocybe | P. cyathiformis | 1 | 1 | E | n/a | |||||
| Tricholomataceae | Tricholoma | T. albobrunneum | 3 | 1 | 4 | M | 0.009 | ||||
| T. dulciolens | 2 | 2 | U, N | n/a | |||||||
| T. equestre | 1 | 1 | 2 | M | n/a | ||||||
| T. joachimii | 3 | 1 | 4 | U | 0.014 | ||||||
| T. matsutake | 18 | 18 | E | 0.002 | |||||||
| T. stans | 2 | 1 | 3 | E | 0.001 | ||||||
| T. terreum | 4 | 4 | E | 0.006 | |||||||
| Agaricales genera incertae sedis | Clitocybe | C. nebularis | 24 | 1 | 1 | 26 | E | 0.101 | |||
| Lepista | L. sordida | 2 | 2 | E | n/a | ||||||
| Leucocybe | L. connata | 2 | 1 | 3 | M | 0.013 | |||||
| Auriculariale | Auriculariaceae | Auricularia | A. cornea | 22 | 8 | 1 | 31 | E | 0.028 | ||
| Boletales | Boletaceae | Aureoboletus | A. moravicus | 1 | 1 | U | n/a | ||||
| Austroboletus | A. gracilis | 1 | 1 | U, N | n/a | ||||||
| Baorangia | B. bicolor | 3 | 2 | 5 | U | 0.02 | |||||
| Boletus | B. aereus | 1 | 1 | E | n/a | ||||||
| B. eastwoodiae | 2 | 2 | U | n/a | |||||||
| B. edulis s.l. | 2 | 2 | E | n/a | |||||||
| B. pseudosulphureus | 4 | 4 | E | 0.022 | |||||||
| B. reticulatus | 1 | 15 | 45 | 6 | 67 | E | 0.075 | ||||
| B. rhodopurpureus | 1 | 1 | U | n/a | |||||||
| B. satanas | 5 | 5 | P | 0.016 | |||||||
| Butyriboletus | B. appendiculatus | 2 | 2 | U | n/a | ||||||
| B. pseudospeciosus | 12 | 3 | 15 | U | 0.075 | ||||||
| B. subappendiculatus | 6 | 4 | 10 | U, N | 0.049 | ||||||
| Caloboletus | C. radicans | 2 | 2 | U | n/a | ||||||
| Harrya | H. chromapes | 3 | 3 | U, N | 0.005 | ||||||
| Heimioporus | H. japonicus | 2 | 2 | P | n/a | ||||||
| Neoboletus | N. multipunctatus | 4 | 2 | 3 | 9 | U | 0.045 | ||||
| N. obscureumbrinus | 1 | 1 | U | n/a | |||||||
| Pulveroboletus | P. brunneopunctatus | 1 | 1 | U | n/a | ||||||
| Retiboletus | R. retipes | 2 | 2 | E | n/a | ||||||
| Rugiboletus | R. extremiorientalis | 4 | 1 | 5 | U | 0.01 | |||||
| Sutorius | S. uridiformis | 1 | 8 | 9 | U | 0.078 | |||||
| Tylopilus | T. microsporus | 4 | 4 | P | 0.014 | ||||||
| T. neofelleus | 1 | 1 | p | n/a | |||||||
| T. obscurus | 1 | 1 | U, N | n/a | |||||||
| Boletinellaceae | Phlebopus | P. portentosus | 1 | 1 | E | n/a | |||||
| Gomphidiaceae | Chroogomphus | C. rutilus | 1 | 1 | 2 | 4 | E | 0.039 | |||
| Gyroporaceae | Gyroporus | G. ballouii | 2 | 2 | U | n/a | |||||
| Sclerodermataceae | Scleroderma | S. yunnanense | 14 | 14 | E | 0.002 | |||||
| Suillaceae | Suillus | S. bovinus | 2 | 2 | M | n/a | |||||
| Cantharellales | Hydnaceae | Cantharellus | C. amethysteus | 2 | 3 | 1 | 1 | 7 | E | 0 | |
| C. cibarius | 62 | 7 | 19 | 2 | 90 | E | 0 | ||||
| C. cinereus | 4 | 4 | E | 0 | |||||||
| C. cinnabarinus | 18 | 1 | 3 | 5 | 27 | E | 0.034 | ||||
| C. enelensis | 4 | 1 | 4 | 9 | U, N | 0.011 | |||||
| C. formosus | 1 | 0 | 1 | 0 | 2 | E | n/a | ||||
| C. friesii | 2 | 2 | 4 | U | 0.025 | ||||||
| C. lateritius | 2 | 0 | 0 | 1 | 3 | E | 0.009 | ||||
| C. lewisii | 4 | 0 | 7 | 2 | 13 | U, N | 0.051 | ||||
| C. pallens | 2 | 2 | E | 0.078 | |||||||
| C. roseocanus | 2 | 1 | 3 | U | 0.006 | ||||||
| C. subalbidus | 13 | 8 | 0 | 2 | 23 | E | 0.034 | ||||
| C. tenuithrix | 1 | 1 | U | n/a | |||||||
| Craterellus | C. cornucopioides | 0 | 4 | 0 | 4 | E | 0.011 | ||||
| C. luteus | 2 | 0 | 0 | 2 | 4 | E | 0.006 | ||||
| Gomphales | Gomphaceae | Gomphus | G. clavatus | 5 | 5 | E | 0.024 | ||||
| Ramaria | R. apiculata | 5 | 5 | E | 0.0026 | ||||||
| R. araiospora | 5 | 5 | E | 0.061 | |||||||
| R. aurantiisiccescens | 2 | 2 | U | n/a | |||||||
| R. aurea | 1 | 1 | M | n/a | |||||||
| R. botrytis | 8 | 8 | 16 | E | 0.064 | ||||||
| R. conjunctipes | 1 | 1 | E | n/a | |||||||
| R. cystidiophora | 3 | 3 | U | 0.095 | |||||||
| R. flavobrunnescens | 1 | 1 | E | n/a | |||||||
| R. formosa | 7 | 7 | M | 0.041 | |||||||
| R. fumigata | 5 | 5 | P | 0.051 | |||||||
| R. obtusissima | 13 | 13 | E | 0.044 | |||||||
| R. pinicola | 1 | 1 | U, N | n/a | |||||||
| R. rubrievanescens | 1 | 18 | 5 | 2 | 26 | U, N | 0.012 | ||||
| Hymenochaetales genera incertae sedis | Trichaptum | T. abietinum | 2 | 2 | U | n/a | |||||
| Polyporales | Cerrenaceae | Cerrena | C. unicolor | 2 | 2 | E | n/a | ||||
| Grifolaceae | Grifola | G. frondosa | 1 | 1 | 2 | E | n/a | ||||
| Polyporaceae | Amauroderma | A. rugosum | 16 | 3 | 19 | U | 0.055 | ||||
| Russulales | Albatrellaceae | Albatrellus | A. confluens | 4 | 4 | E | 0.028 | ||||
| Russulaceae | Lactarius | L. deliciosus | 7 | 7 | E | 0 | |||||
| L. deterrimus | 5 | 1 | 2 | 8 | E | 0.02 | |||||
| L. fulvissimus | 1 | 1 | E | n/a | |||||||
| L. hatsudake | 10 | 10 | E | 0.015 | |||||||
| L. piperatus | 1 | 1 | M | n/a | |||||||
| L. quieticolor | 3 | 3 | E | 0.026 | |||||||
| L. sanguifluus | 3 | 4 | 1 | 8 | E | 0.029 | |||||
| L. semisanguifluus | 1 | 1 | U | n/a | |||||||
| L. bertillonii | 1 | 1 | U | n/a | |||||||
| L. dwaliensis | 1 | 1 | U, N | n/a | |||||||
| L. glaucescens | 1 | 3 | 4 | U, N | 0.034 | ||||||
| L. leae | 1 | 1 | U | n/a | |||||||
| L. piperatus | 2 | 3 | 2 | 1 | 8 | M | 0.018 | ||||
| L. rugatus | 3 | 1 | 4 | E | 0.005 | ||||||
| L. subvolemus | 1 | 1 | U, N | n/a | |||||||
| L. tenuicystidiatus | 3 | 3 | E | 0.002 | |||||||
| L. volemus | 10 | 4 | 12 | 8 | 34 | E | 0.016 | ||||
| Russula | R. aeruginea | 2 | 2 | E | n/a | ||||||
| R. albonigra | 2 | 2 | E | n/a | |||||||
| R. amoenolens | 1 | 1 | U | n/a | |||||||
| R. aquosa | 1 | 1 | U | n/a | |||||||
| R. aurea | 2 | 1 | 3 | M | 0.017 | ||||||
| R. aurora | 4 | 4 | E | 0.006 | |||||||
| R. cyanoxantha | 1 | 15 | 22 | 5 | 43 | E | 0.017 | ||||
| R. densifolia | 1 | 1 | E | n/a | |||||||
| R. graminea | 3 | 3 | U | 0.07 | |||||||
| R. minutula | 1 | 1 | U | n/a | |||||||
| R. nobilis | 2 | 2 | P | n/a | |||||||
| R. paludosa | 1 | 1 | E | n/a | |||||||
| R. pubescens | 1 | 2 | 3 | P | 0.104 | ||||||
| R. renidens | 1 | 1 | U | n/a | |||||||
| R. rosea | 1 | 3 | 1 | 5 | E | 0.059 | |||||
| R. stenocystidiata | 5 | 1 | 1 | 7 | E | 0.059 | |||||
| R. turci | 1 | 1 | E | n/a | |||||||
| R. versicolor | 2 | 2 | U | n/a | |||||||
| R. vinosa | 2 | 2 | E | n/a | |||||||
| R. vinosobrunnea | 1 | 1 | U | n/a | |||||||
| R. virescens | 190 | 8 | 3 | 4 | 205 | E | 0 | ||||
| Scutiger | S. pes-caprae | 9 | 9 | E | 0.029 | ||||||
| Thelephorales | Bankeraceae | Sarcodon | S. leucopus | 8 | 8 | E | 0.002 | ||||
| Bankeraceae | S. squamosus | 2 | 2 | U, N | n/a | ||||||
| Thelephoraceae | Pseudotomentella | P. mucidula | 1 | 1 | U | n/a | |||||
| Thelephora | T. aurantiotincta | 1 | 8 | 9 | E | 0.068 | |||||
| T. ganbajun | 410 | 3 | 413 | E | 0.032 | ||||||
| T. vialis | 14 | 99 | 1 | 114 | E | 0 | |||||
| Total | 11 | 31 | 56 | 159 | 1074 | 293 | 492 | 339 | 2198 |
| No. Sequences | No. Genotypes | No. Species by ITS Blast | Cutoff Values for the Phylogenetic Species Identification | No. Provisional Species | No. New Phylogenetic Species | |
|---|---|---|---|---|---|---|
| Boletus and allies | 162 | N/A | 24 | 0.05 | 33 | 2 |
| Cantharellus cibarius species complex | 95 | 15 | 6 | 0.028 | 6 | 1 |
| Lactarius | 88 | N/A | 17 | 0.022 | 32 | 5 |
| Lyophyllum | 96 | N/A | 3 | 0.014 | 20 | 2 |
| Russula virescens and allies | 226 | 19 | 1 | 0.01 | 5 | 4 |
| Termitomyces clypeatus species complex | 380 | 110 | 7 | 0.035 | 10 | 6 |
| Thelephora ganbajun and allies | 489 | 94 | 1 | 0.025 | 4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Mo, M.; Yang, L.; Mi, F.; Cao, Y.; Liu, C.; Tang, X.; Wang, P.; Xu, J. Exploring the Species Diversity of Edible Mushrooms in Yunnan, Southwestern China, by DNA Barcoding. J. Fungi 2021, 7, 310. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040310
Zhang Y, Mo M, Yang L, Mi F, Cao Y, Liu C, Tang X, Wang P, Xu J. Exploring the Species Diversity of Edible Mushrooms in Yunnan, Southwestern China, by DNA Barcoding. Journal of Fungi. 2021; 7(4):310. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040310
Chicago/Turabian StyleZhang, Ying, Meizi Mo, Liu Yang, Fei Mi, Yang Cao, Chunli Liu, Xiaozhao Tang, Pengfei Wang, and Jianping Xu. 2021. "Exploring the Species Diversity of Edible Mushrooms in Yunnan, Southwestern China, by DNA Barcoding" Journal of Fungi 7, no. 4: 310. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7040310