
Spatial Genetic Structure and Pathogenic Race Composition at the Field Scale in the Sunflower Downy Mildew Pathogen, Plasmopara halstedii
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. P. halstedii Isolates Sampling
2.2. Soil Bioassay
2.3. Race Phenotyping and Nomenclature
2.4. DNA Extraction and Polymerase Chain Reaction (PCR)
2.5. Data Analyses
3. Results
3.1. Phenotypic Diversity
3.2. Genetic and Genotypic Diversity
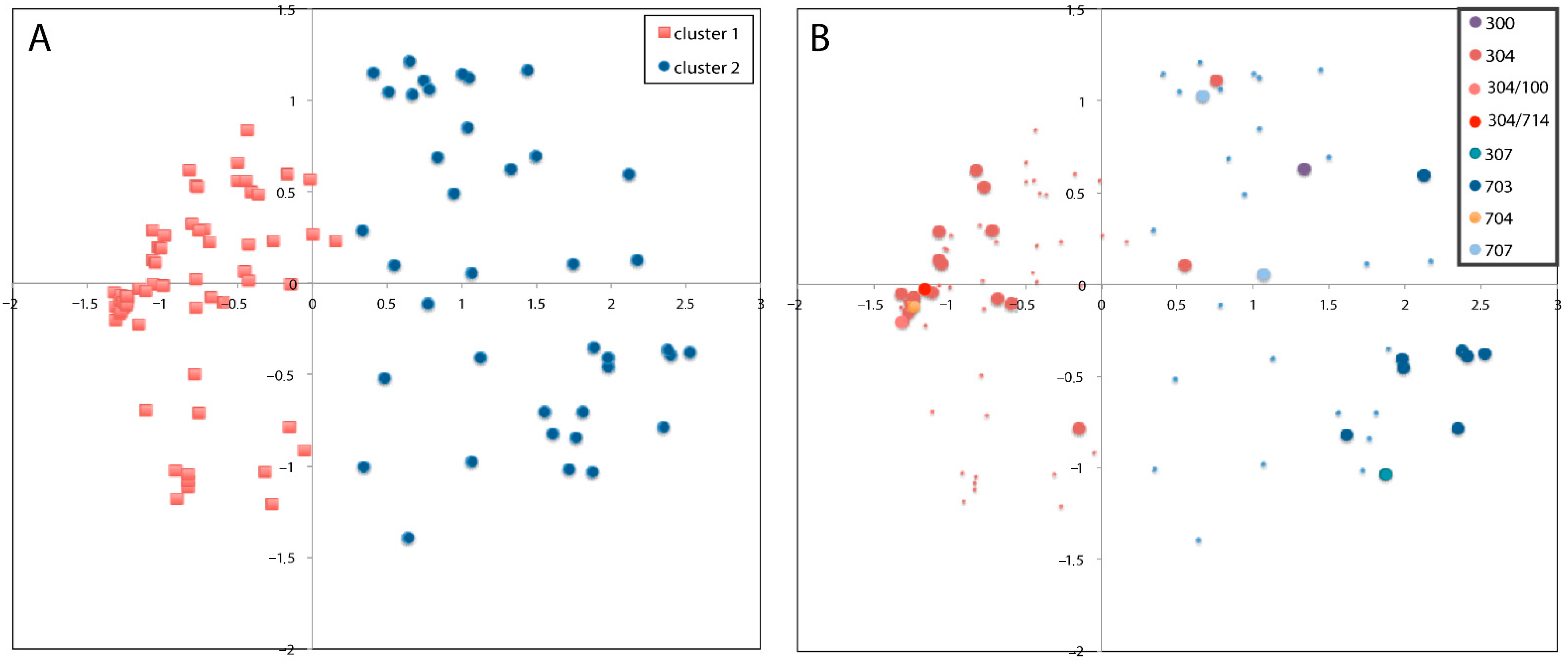
3.3. Principal Coordinate Analysis (PCO)
3.4. Individual Clustering Analysis
3.5. Clustering Differentiation
3.6. Spatial Genetic Structure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, P.A.; Morris, H.E. Plasmopara downy mildew of cultivated sunflowers. Am. J. Bot. 1927, 14, 551–552. [Google Scholar] [CrossRef]
- Jimenez, D. Notas sobre la presencia en Espana del mildiu del girasol. An. Inst. Nac. Investig. Agrarias. Ser. Prod. Veg. 1973, 3, 95–105. [Google Scholar]
- Spring, O.; Zipper, R. Isolation of oospores of sunflower downy mildew, Plasmopara halstedii, and microscopical studies on oospore germination. J. Phytopathol. 2000, 148, 227–231. [Google Scholar] [CrossRef]
- Spring, O.; Zipper, R. Evidence for asexual genetic recombination in sunflower downy mildew, Plasmopara halstedii. Mycol. Res. 2006, 110, 657–663. [Google Scholar] [CrossRef]
- Ban, R.; Kovacs, A.; Nisha, N.; Palinkas, Z.; Zalai, M.; Yousif, A.I.A.; Korosi, K. New and High Virulent Pathotypes of Sunflower Downy Mildew (Plasmopara halstedii) in Seven Countries in Europe. J. Fungi 2021, 7, 549. [Google Scholar] [CrossRef]
- Sharma, R.; Xia, X.J.; Cano, L.M.; Evangelisti, E.; Kemen, E.; Judelson, H.; Oome, S.; Sambles, C.; van den Hoogen, D.J.; Kitner, M.; et al. Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in downy mildews and Phytophthora. BMC Genom. 2015, 16, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Göre, M. Epidemic outbreaks of downy mildew caused by Plasmopara halstedii on sunflower in Thrace, part of the Marmara region of Turkey. Plant Pathol. 2009, 58, 396. [Google Scholar] [CrossRef]
- Gascuel, Q.; Bordat, A.; Sallet, E.; Pouilly, N.; Carrere, S.; Roux, F.; Vincourt, P.; Godiard, L. Effector Polymorphisms of the Sunflower Downy Mildew Pathogen Plasmopara halstedii and Their Use to Identify Pathotypes from Field Isolates. PLoS ONE 2016, 11, e0148513. [Google Scholar] [CrossRef]
- Gulya, T.; Harveson, R.; Mathew, F.; Block, C.; Thompson, S.; Kandel, H.; Berglund, D.; Sandbakken, J.; Kleingartner, L.; Markell, S. Comprehensive disease survey of US sunflower: Disease trends, research priorities and unanticipated impacts. Plant Dis. 2019, 103, 601–618. [Google Scholar] [CrossRef] [Green Version]
- Gascuel, Q.; Martinez, Y.; Boniface, M.C.; Vear, F.; Pichon, M.; Godiard, L. The sunflower downy mildew pathogen Plasmopara halstedii. Mol. Plant Pathol. 2015, 16, 109–122. [Google Scholar] [CrossRef]
- Picard, C.; Afonso, T.; Benko-Beloglavec, A.; Karadjova, O.; Matthews-Berry, S.; Paunovic, S.; Pietsch, M.; Reed, P.; Van Der Gaag, D.; Ward, M. Recommended regulated non-quarantine pests (RNQP s), associated thresholds and risk management measures in the European and Mediterranean region. EPPO Bull. 2018, 48, 552–568. [Google Scholar] [CrossRef]
- Gulya, T.J. Distribution of Plasmopara halstedii races from sunflower around the world. Adv. Downy Mildew Res. 2007, 3, 121–134. [Google Scholar]
- Viranyi, F.; Gulya, T.J.; Tourvieille, D.L. Recent changes in the pathogenic variability of Plasmopara halstedii (sunflower downy mildew) populations from different continents. Helia 2015, 38, 149–162. [Google Scholar] [CrossRef]
- Spring, O. Spreading and global pathogenic diversity of sunflower downy mildew—Review. Plant Prot. Sci. 2019, 55, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Gilley, M.A.; Gulya, T.J.; Seiler, G.J.; Underwood, W.; Hulke, B.S.; Misar, C.G.; Markell, S.G. Determination of virulence phenotypes of Plasmopara halstedii in the United States. Plant Dis. 2020, 104, 2823–2831. [Google Scholar] [CrossRef]
- Miranda-Fuentes, P.; García-Carneros, A.B.; Molinero-Ruiz, L. Updated characterization of races of Plasmopara halstedii and entomopathogenic fungi as endophytes of sunflower plants in axenic culture. Agronomy 2021, 11, 268. [Google Scholar] [CrossRef]
- Virányi, F. CABI invasive species compendium. In Datasheet Plasmopara halstedii (Downy Mildew of Sunflower); CABI: Wallingford, UK, 2018. [Google Scholar]
- Molinero-Ruiz, L. Sustainable and efficient control of sunflower downy mildew by means of genetic resistance: A review. Theor. Appl. Genet. 2022, 1–15. [Google Scholar] [CrossRef]
- Delanoe, D. Contribution à la Connaissance du Mildiou du Tournesol: Systématique, Biologie et Épidémiologie de Plasmopara helianthi novot. Ph.D. Thesis, 1971. (In French). [Google Scholar]
- Korosi, K.; Kovacs, A.; Nisha, N.; Bota, I.; Perczel, M.; Yousif, A.I.A.; Kiss, J.; Ban, R. New data on pathotype distribution and mefenoxam tolerance of Plasmopara halstedii in Hungary. Plant Prot. Sci. 2021, 57, 31–37. [Google Scholar] [CrossRef]
- Martín-Sanz, A.; Rueda, S.; García-Carneros, A.B.; Molinero-Ruiz, L. First report of a new highly virulent pathotype of sunflower downy mildew (Plasmopara halstedii) overcoming the Pl8 resistance gene in Europe. Plant Dis. 2020, 104, 597. [Google Scholar] [CrossRef]
- Nisha, N.; Körösi, K.; Perczel, M.; Yousif, A.I.A.; Bán, R. First report on the occurrence of an aggressive pathotype, 734, of Plasmopara halstedii causing sunflower downy mildew in Hungary. Plant Dis. 2021, 105, 711. [Google Scholar] [CrossRef]
- Iwebor, M.; Antonova, T.; Araslanova, N.; Saukova, S.; Pitinova, Y.; Eliseeva, K.; Belorutskiy, A. The first report of Plasmopara halstedii race 337 in the Russian Federation. Plant Prot. Sci. 2021, 58, 76–80. [Google Scholar] [CrossRef]
- Oros, G.; Virányi, F. Resistance of Plasmopara halstedii to metalaxyl in the greenhouse. Temp. Downy Mildew Newsl. 1984, 3, 22–23. [Google Scholar]
- Lafon, S.; Penaud, A.; Walser, P.; De Guenin, M.-C.; Molinero, V.; Mestres, R.; Tourvieille, D. Le mildiou du tournesol toujours sous surveillance. Phytoma-La Défense Des Végétaux 1996, 484, 35–36. [Google Scholar]
- Gulya, T.J.; Draper, M.; Harbour, J.; Holen, C.; Knodel, J.; Lamey, A.; Mason, P. Metalaxyl resistance in sunflower downy mildew in North America. In Proceedings of the 21st NSA Sunflower Research Workshop, Fargo, ND, USA, 14–15 January 1999; National Sunflower Association, Ed.; pp. 118–123. [Google Scholar]
- Molinero-Ruiz, M.L.; Domínguez, J.; Melero-Vara, J.M. Races of isolates of Plasmopara halstedii from Spain and studies on their virulence. Plant Dis. 2002, 86, 736–740. [Google Scholar] [CrossRef]
- Spring, O.; Bachofer, M.; Thines, M.; Riethmüller, A.; Göker, M.; Oberwinkler, F. Intraspecific relationship of Plasmopara halstedii isolates differing in pathogenicity and geographic origin based on ITS sequence data. Eur. J. Plant Pathol. 2006, 114, 309–315. [Google Scholar] [CrossRef]
- Vincourt, P.; As-sadi, F.; Bordat, A.; Langlade, N.B.; Gouzy, J.; Pouilly, N.; Lippi, Y.; Serre, F.; Godiard, L.; de Labrouhe, D.T.; et al. Consensus mapping of major resistance genes and independent QTL for quantitative resistance to sunflower downy mildew. Theor. Appl. Genet. 2012, 125, 909–920. [Google Scholar] [CrossRef]
- Gascuel, Q.; Buendia, L.; Pecrix, Y.; Blanchet, N.; Munos, S.; Vear, F.; Godiard, L. RXLR and CRN Effectors from the Sunflower Downy Mildew Pathogen Plasmopara halstedii Induce Hypersensitive-Like Responses in Resistant Sunflower Lines. Front. Plant Sci. 2016, 7, 1887. [Google Scholar] [CrossRef] [Green Version]
- Mestre, P.; Carrere, S.; Gouzy, J.; Piron, M.C.; de Labrouhe, D.T.; Vincourt, P.; Delmotte, F.; Godiard, L. Comparative analysis of expressed CRN and RXLR effectors from two Plasmopara species causing grapevine and sunflower downy mildew. Plant Pathol. 2016, 65, 767–781. [Google Scholar] [CrossRef]
- Pecrix, Y.; Buendia, L.; Penouilh-Suzette, C.; Marechaux, M.; Legrand, L.; Bouchez, O.; Rengel, D.; Gouzy, J.; Cottret, L.; Vear, F.; et al. Sunflower resistance to multiple downy mildew pathotypes revealed by recognition of conserved effectors of the oomycete Plasmopara halstedii. Plant J. 2019, 97, 730–748. [Google Scholar] [CrossRef] [Green Version]
- Pecrix, Y.; Penouilh-Suzette, C.; Munos, S.; Vear, F.; Godiard, L. Ten Broad Spectrum Resistances to Downy Mildew Physically Mapped on the Sunflower Genome. Front. Plant Sci. 2018, 9, 1780. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Song, Q.; Li, X.; Qi, L. High-Density Mapping and Candidate Gene Analysis of Pl 18 and Pl 20 in Sunflower by Whole-Genome Resequencing. Int. J. Mol. Sci. 2020, 21, 9571. [Google Scholar] [CrossRef]
- Qi, L.; Cai, X. Characterization and mapping of a downy mildew resistance gene, Pl36, in sunflower (Helianthus annuus L.). Mol. Breed. 2022, 42, 8. [Google Scholar] [CrossRef]
- Qi, L.; Ma, G. Marker-assisted gene pyramiding and the reliability of using SNP markers located in the recombination suppressed regions of sunflower (Helianthus annuus L.). Genes 2019, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Ma, G.; Seiler, G. Registration of HA-R14, HA-R15, HA-R16, and HA-R17 oilseed sunflower germplasm with broad resistance to rust and downy mildew. J. Plant Regist. 2022, 16, 137–146. [Google Scholar] [CrossRef]
- Gontcharov, S.; Goloschapova, N. Evaluation of horizontal resistance of sunflower (Helianthus annuus L.) to downy mildew (Plasmopara halstedii). OCL 2021, 28, 58. [Google Scholar] [CrossRef]
- Borovkova, G. Restriction fragment length polymorphisms and RAPD markers in DNA of Plasmopara halstedii, the downy mildew fungus of sunflower. In Proceedings of the 13th International Sunflower Conference Volume 2, Pisa, Italy, 7–11 September 1992; pp. 1420–1425. [Google Scholar]
- Roeckel-Drevet, P.; Coelho, V.; Nicolas, P.; Tourvieille, J.; Labrouhe, D.T.D. Lack of genetic variability in French identified races of Plasmopara halstedii, the cause of downy mildew in sunflower Helianthus annuus. Can. J. Microbiol. 1997, 43, 260–263. [Google Scholar] [CrossRef]
- Komjati, H.; Fekete, C.; Viranyi, F. Genetic and molecular characterisation of Plasmopara halstedii isolates from Hungary. In Advances in Downy Mildew Research—Volume 2; Springer: Berlin/Heidelberg, Germany, 2004; pp. 193–201. [Google Scholar]
- Intelmann, F.; Spring, O. Analysis of total DNA by minisatellite and simple-sequence repeat primers for the use of population studies in Plasmopara halstedii. Can. J. Microbiol. 2002, 48, 555–559. [Google Scholar] [CrossRef]
- Martínez, A.L.; Garayalde, A.; Quiroz, F.; Carrera, A. Genetic characterization of Plasmopara halstedii populations in Argentina using simple sequence repeats (SSR) and effector-based markers. In Phytopathology; The American Phytopathological Society: St Paul, MN, USA, 2021. [Google Scholar]
- Thines, M.; Komjáti, H.; Spring, O. Exceptional length of ITS in Plasmopara halstedii is due to multiple repetitions in the ITS-2 region. Eur. J. Plant Pathol. 2005, 112, 395–398. [Google Scholar] [CrossRef]
- Giresse, X.; Tourvieille de Labrouhe, D.; RICHARD-CERVERA, S.; Delmotte, F. Twelve polymorphic expressed sequence tags-derived markers for Plasmopara halstedii, the causal agent of sunflower downy mildew. Mol. Ecol. Notes 2007, 7, 1363–1365. [Google Scholar] [CrossRef]
- Ahmed, S.; de Labrouhe, D.T.; Delmotte, F. Emerging virulence arising from hybridisation facilitated by multiple introductions of the sunflower downy mildew pathogen Plasmopara halstedii. Fungal Genet. Biol. 2012, 49, 847–855. [Google Scholar] [CrossRef]
- Sakr, N. Pathogenic, morphological and genetic diversity in Plasmopara halstedii, the causal agent of sunflower downy mildew. Acta Scientiarum. Agron. 2013, 35, 9–19. [Google Scholar] [CrossRef] [Green Version]
- As-Sadi, F.; Carrere, S.; Gascuel, Q.; Hourlier, T.; Rengel, D.; Le Paslier, M.-C.; Bordat, A.; Boniface, M.-C.; Brunel, D.; Gouzy, J. Transcriptomic analysis of the interaction between Helianthus annuus and its obligate parasite Plasmopara halstedii shows single nucleotide polymorphisms in CRN sequences. BMC Genom. 2011, 12, 498. [Google Scholar] [CrossRef] [Green Version]
- Kösoğlu, K.; Yumuk, S.; Aydin, Y.; Evci, G.; Uncuoğlu, A.A. Use of SNP markers by KASP assay for MAS studies in sunflower against Plasmopara halstedii. Turk. J. Agric. For. 2017, 41, 480–489. [Google Scholar] [CrossRef]
- Delmotte, F.; Giresse, X.; Richard-Cervera, S.; M’Baya, J.; Vear, F.; Tourvieille, J.; Walser, P.; de Labrouhe, D.T. Single nucleotide polymorphisms reveal multiple introductions into France of Plasmopara halstedii, the plant pathogen causing sunflower downy mildew. Infect. Genet. Evol. 2008, 8, 534–540. [Google Scholar] [CrossRef]
- Suffert, F.; Delmotte, F.; Delos, M.; Grimault, V.; de Labrouhe, D.T.; Vincourt, P.; Wilson, V.; Reignault, P.; Augustin, S.; Bréda, N. Analyse de Risque Phytosanitaire Plasmopara halstedii agent Responsable de la Maladie du Mildiou du Tournesol; Agence Nationale de Sécurité Sanitaire de l’Alimentation de l’Environnement: Paris, France, 2014. [Google Scholar]
- Tourvieille de Labrouhe, D.T.; Walser, P.; Jolivot, D.; Roche, S.; Serre, F.; Leguillon, M.; Delmotte, F.; Bordat, A.; Godiard, L.; Vincourt, P. Proposal for improvement of sunflower downy mildew race nomenclature. In Proceedings of the 18th International Sunflower Conference, Mar del Plata, Argentina, 27 February–1 March 2012; pp. 322–327. [Google Scholar]
- Torto-Alalibo, T.; Tian, M.; Gajendran, K.; Waugh, M.E.; Van West, P.; Kamoun, S. Expressed sequence tags from the oomycete fish pathogen Saprolegnia parasitica reveal putative virulence factors. BMC Microbiol. 2005, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Gulya, T.J.; de Labrouhe, D.T.; Maširević, S.; Penaud, A.; Rashid, K.; Viranyi, F. Proposal for standardized nomenclature and identification of races of Plasmopara halstedii (sunflower downy mildew). In Proceedings of the ISA Symposium III: Sunflower Downy Mildew, Fargo, ND, USA, 13–14 January 1998; pp. 13–14. [Google Scholar]
- De Labrouhe, D.T.; Gulya, T.J.; Masirevic, S.; Penaud, A.; Rashid, K.Y.; Viranyi, F. New nomenclature of races of Plasmopara halstedii (sunflower downy mildew). In Proceedings of the 15th International Sunflower Conference, Toulouse, France, 12–16 June 2000; pp. 61–66. [Google Scholar]
- Cohen, Y.; Sackston, W. Seed infection and latent infection of sunflowers by Plasmopara halstedii. Can. J. Bot. 1974, 52, 231–238. [Google Scholar] [CrossRef]
- Mouzeyar, S.; Labrouhe, D.T.; Vear, F. Histopathological studies of resistance of sunflower (Helianthus annuus L.) to downy mildew (Plasmopara halstedii). J. Phytopathol. 1993, 139, 289–297. [Google Scholar] [CrossRef]
- Delmotte, F.; Chen, W.; Richard-Cervera, S.; Greif, C.; Papura, D.; Giresse, X.; Mondor-Genson, G.; Corio-Costet, M. Microsatellite DNA markers for Plasmopara viticola, the causal agent of downy mildew of grapes. Mol. Ecol. Notes 2006, 6, 379–381. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Arnud-Haond, S.; Belkhir, K. Genclone: A computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol. Ecol. Notes 2007, 7, 15–17. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Weir, S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Haubold, B.; Hudson, R.R. LIAN 3.0: Detecting linkage disequilibrium in multilocus data. Bioinformatics 2000, 16, 847–849. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Schneider, S.; Roessli, D.; Excoffier, L. Arlequin: A software for population genetics data analysis. User Man. Ver 2000, 2, 2496–2497. [Google Scholar]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Roeckel-Drevet, P.; Tourvieille, J.; Gulya, T.; Charmet, G.; Nicolas, P.; de Labrouhe, D.T. Molecular variability of sunflower downy mildew, Plasmopara halstedii, from different continents. Can. J. Microbiol. 2003, 49, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Sedlářová, M.; PoSPíchalová, R.; Trojanová, Z.D.; Bartůšek, T.; Slobodianová, L.; Lebeda, A. First report of Plasmopara halstedii new races 705 and 715 on sunflower from the Czech Republic—Short communication. Plant Prot. Sci. 2016, 52, 182–187. [Google Scholar]
- Drábková Trojanová, Z.; Sedlářová, M.; Pospíchalová, R.; Lebeda, A. Pathogenic variability of Plasmopara halstedii infecting sunflower in the Czech Republic. Plant Pathol. 2018, 67, 136–144. [Google Scholar] [CrossRef]
- Bazzalo, E.; Huguet, N.; Romano, C.; Bock, F.; Piubello, S.; Quiroz, F.; Erreguerena, I.; Zuil, S.; Bertero, A. History and present state of downy mildew in Argentina. In Proceedings of the 19th International Sunflower Conference, Edirne, Turkey, 29 May–3 June 2016; pp. 828–832. [Google Scholar]
- Goodwin, S.B.; Sujkowski, L.S.; Fry, W.E. Rapid evolution of pathogenicity within clonal lineages of the potato late blight disease fungus. Phytopathology 1995, 85, 669–676. [Google Scholar] [CrossRef]
- Otmar, S.; Reinhard, Z. New highly aggressive pathotype 354 of Plasmopara halstedii in German sunflower fields. Plant Prot. Sci. 2018, 54, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Mboup, M.; Leconte, M.; Gautier, A.; Wan, A.; Chen, W.; de Vallavieille-Pope, C.; Enjalbert, J. Evidence of genetic recombination in wheat yellow rust populations of a Chinese oversummering area. Fungal Genet. Biol. 2009, 46, 299–307. [Google Scholar] [CrossRef]
- Milgroom, M.G. Recombination and the multilocus structure of fungal populations. Annu. Rev. Phytopathol. 1996, 34, 457–477. [Google Scholar] [CrossRef]
- Baumgartner, K.; Baker, B.R.; Korhonen, K.; Zhao, J.; Hughes, K.W.; Bruhn, J.; Bowman, T.S.; Bergemann, S.E. Evidence of natural hybridization among homothallic members of the Basidiomycete. Armillaria Mellea Sensu Stricto. Fungal Biol. 2012, 116, 677–691. [Google Scholar] [CrossRef]
- Milgroom, M.G.; Sotirovski, K.; Risteski, M.; Brewer, M.T. Heterokaryons and parasexual recombinants of Cryphonectria parasitica in two clonal populations in southeastern Europe. Fungal Genet. Biol. 2009, 46, 849–854. [Google Scholar] [CrossRef]
- Tourvieille de Labrouhe, D.; Bordat, A.; Tourvieille, J.; Mestries, E.; Walser, P.; Sakr, N.; Ducher, M.; Delmotte, F.; Vear, F. Impact of major gene resistance management for sunflower on fitness of Plasmopara halstedii (downy mildew) populations. OCL 2010, 17, 56–64. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | Virulence Profiles According to the Differential Sunflower Lines | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| D1 | D2 | D3 | D4 | D5 | D6 | D7 | D8 | D9 | |
| Score | 1 | 2 | 4 | 1 | 2 | 4 | 1 | 2 | 4 |
| races | |||||||||
| 100 | S 1 | R 2 | R | R | R | R | R | R | R |
| 703 | S | S | S | R | R | R | S | S | R |
| 300 | S | S | R | R | R | R | R | R | R |
| 304 | S | S | R | R | R | R | R | R | S |
| 307 | S | S | R | R | R | R | S | S | S |
| 704 | S | S | S | R | R | R | R | R | S |
| 714 | S | S | S | S | R | R | R | R | S |
| 707 | S | S | S | R | R | R | S | S | S |
| Site | Percentage of Emerged Seedlings (%) | Percentage of Infected Seedlings (%) |
|---|---|---|
| 1 | 95.0 | 33.0 |
| 2 | 92.5 | 41.6 |
| 3 | 93.3 | 22.1 |
| 4 | 93.3 | 12.0 |
| 5 | 90.8 | 31.8 |
| 6 | 92.5 | 43.1 |
| 7 | 91.7 | 35.5 |
| 8 | 90.8 | 45.9 |
| 9 | 85.8 | 49.1 |
| 10 | 92.5 | 54.1 |
| 11 | 90.8 | 18.7 |
| 12 | 92.5 | 38.3 |
| 13 | 87.5 | 39.1 |
| 14 | 89.2 | 30.7 |
| 15 | 89.2 | 16.7 |
| 16 | 95.0 | 4.3 |
| 17 | 91.7 | 26.5 |
| 18 | 96.7 | 48.0 |
| 19 | 90.0 | 22.1 |
| 20 | 89.2 | 15.5 |
| Site | Number of Isolates | Number of MLG | Race | Genetic Diversity |
|---|---|---|---|---|
| S1 | 6 | 4 | 304(6) | 0.1527 |
| S2 | 30 | 6 | 304(6) | 0.1908 |
| S3 | 20 | 5 | 304(4), 703(2) | 0.1523 |
| S4 | 12 | 6 | 100(1), 304(2), 703(2) | 0.2635 |
| S5 | 6 | 4 | 304(6) | 0.0758 |
| S6 | 31 | 9 | 304(4), 703(2), 704(1) | 0.2045 |
| S7 | 6 | 4 | 304(1) | 0.3298 |
| S8 | 6 | 1 | 304(2) | 0.0314 |
| S9 | 25 | 14 | 300(1), 707(1) | 0.2726 |
| S10 | 6 | 4 | 304(2) | 0.2913 |
| S11 | 8 | 6 | 304(1) | 0.2217 |
| S12 | 6 | 3 | 304(2) | 0.1433 |
| S13 | 29 | 14 | 304(1), 703(3), 704(1), 707(1), 714(1) | 0.2316 |
| S14 | 6 | 4 | 703(1) | 0.2540 |
| S15 | 6 | 2 | 304(2) | 0.0555 |
| S16 | 4 | 2 | 304(1) | 0.0576 |
| S17 | 21 | 8 | 304(1), 707(1) | 0.1742 |
| S18 | 6 | 2 | 304(1) | 0.0629 |
| S19 | 6 | 5 | 304(2) | 0.1982 |
| S20 | 10 | 6 | 304(1), 307(1) | 0.2465 |
| Global | 250 | 109 | 100(1), 300(1), 304(45), 307(1), 703(10), 704(2), 707(3), 714(1) | 0.1776 |
| MLG Name | Number of Isolates | Race Characterization | |
|---|---|---|---|
| Cluster 1 | MLG91 | 26 | 304(6), 714(1) |
| MLG95 | 15 | 100(1), 304(3), 703(1) | |
| MLG96 | 48 | 304(7), 704(1) | |
| MLG99 | 33 | 304(8) | |
| Cluster 2 | MLG5 | 7 | 703(1) |
| Nuclear Cluster | Number of Isolates | Number of MLG | Number of Races | Races | Ar | Gd | FIS |
|---|---|---|---|---|---|---|---|
| Cluster 1 | 198 | 66 | 4 | 304, 714, 704, 100 | 1.54 | 0.093 | 0.815 * |
| Cluster 2 | 52 | 43 | 4 | 703, 307, 300, 707 | 2.05 | 0.189 | 0.682 * |
| Total | 250 | 109 | 8 | 2.16 | 0.177 | 0.764 * |
| Regions | d.f. | Sum of Squares | Variance Components | % of Variation | FST Value |
|---|---|---|---|---|---|
| A | |||||
| Among clusters | 1 | 254.331 | 3.25632 | 65.11 | 0.650 |
| Within clusters | 107 | 432.825 | 1.74526 | 34.89 | |
| Total | 687.156 | 5.00158 | |||
| B | |||||
| Among sites | 19 | 81.792 | 0.13199 | 4.66 | 0.077 |
| Within sites | 230 | 621.684 | 2.70297 | 95.34 | |
| Total | 703.476 | 2.83496 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elameen, A.; de Labrouhe, D.T.; Bret-Mestries, E.; Delmotte, F. Spatial Genetic Structure and Pathogenic Race Composition at the Field Scale in the Sunflower Downy Mildew Pathogen, Plasmopara halstedii. J. Fungi 2022, 8, 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8101084
Elameen A, de Labrouhe DT, Bret-Mestries E, Delmotte F. Spatial Genetic Structure and Pathogenic Race Composition at the Field Scale in the Sunflower Downy Mildew Pathogen, Plasmopara halstedii. Journal of Fungi. 2022; 8(10):1084. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8101084
Chicago/Turabian StyleElameen, Abdelhameed, Denis Tourvieille de Labrouhe, Emmanuelle Bret-Mestries, and Francois Delmotte. 2022. "Spatial Genetic Structure and Pathogenic Race Composition at the Field Scale in the Sunflower Downy Mildew Pathogen, Plasmopara halstedii" Journal of Fungi 8, no. 10: 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8101084