
Deciphering the Genome-Wide Transcriptomic Changes during Interactions of Resistant and Susceptible Genotypes of American Elm with Ophiostoma novo-ulmi

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials, Fungal Isolate, and Inoculation Conditions
2.2. RNA Extraction and Sequencing
2.3. Ulmus Americana Transcriptome Analysis
2.4. Comparative Analysis between U. americana and U. minor Transcriptomes
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways Enrichment Analysis
2.6. Validation of Candidate Genes Expression by Quantitative Real-Time PCR
3. Results
3.1. Development of Dutch Elm Disease (DED) Symptoms
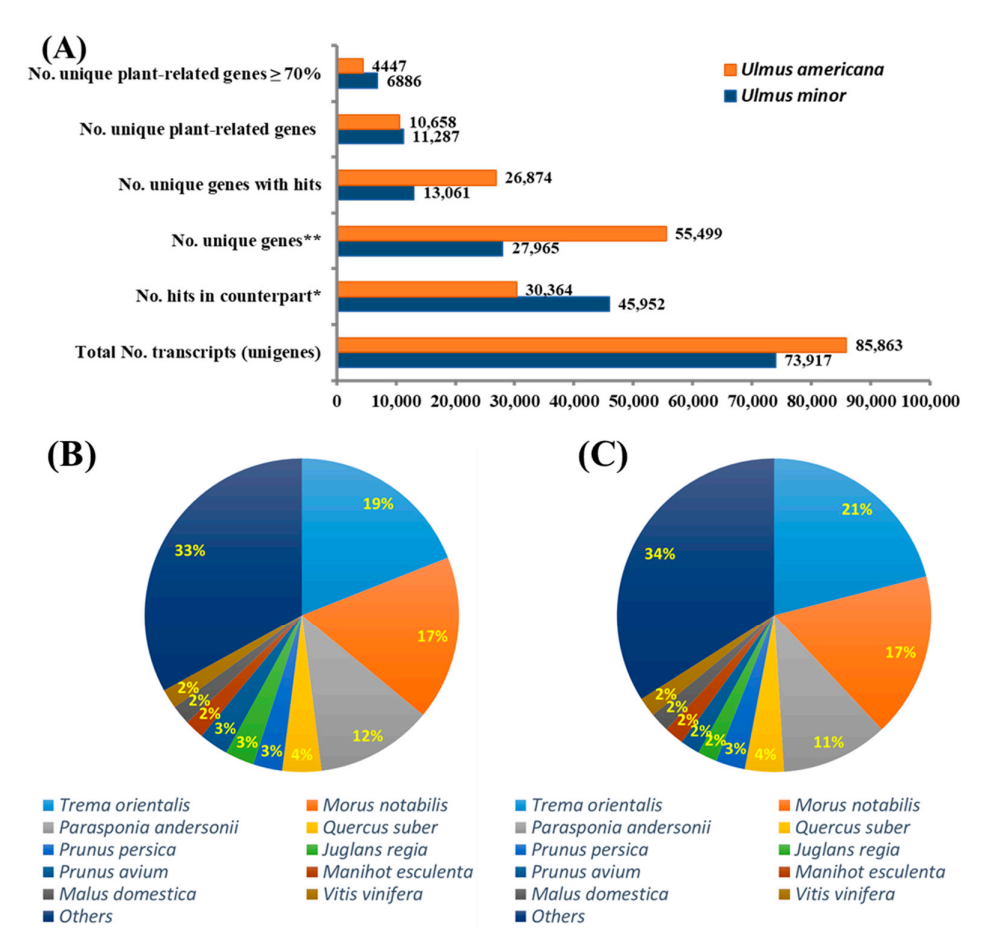
3.2. Analysis of the American Elm Transcriptomes
3.3. Comparative Transcriptome Analysis
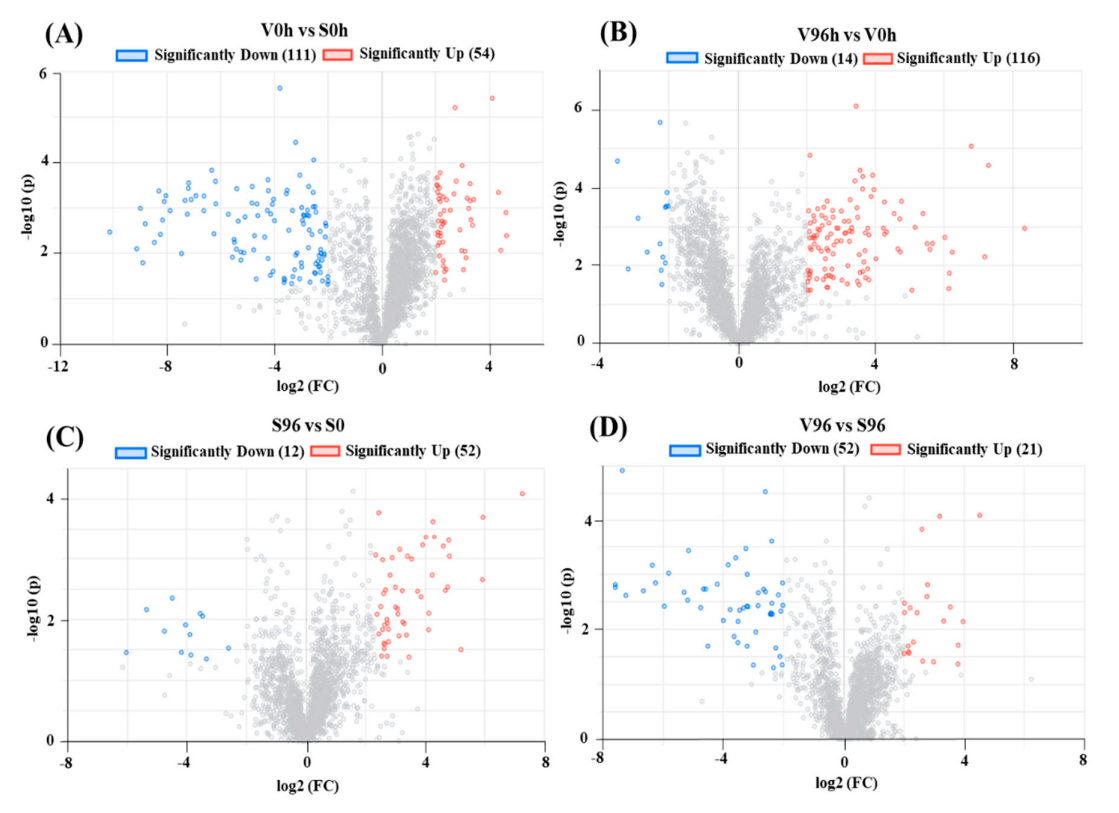
3.4. Differential Gene Expression Analysis
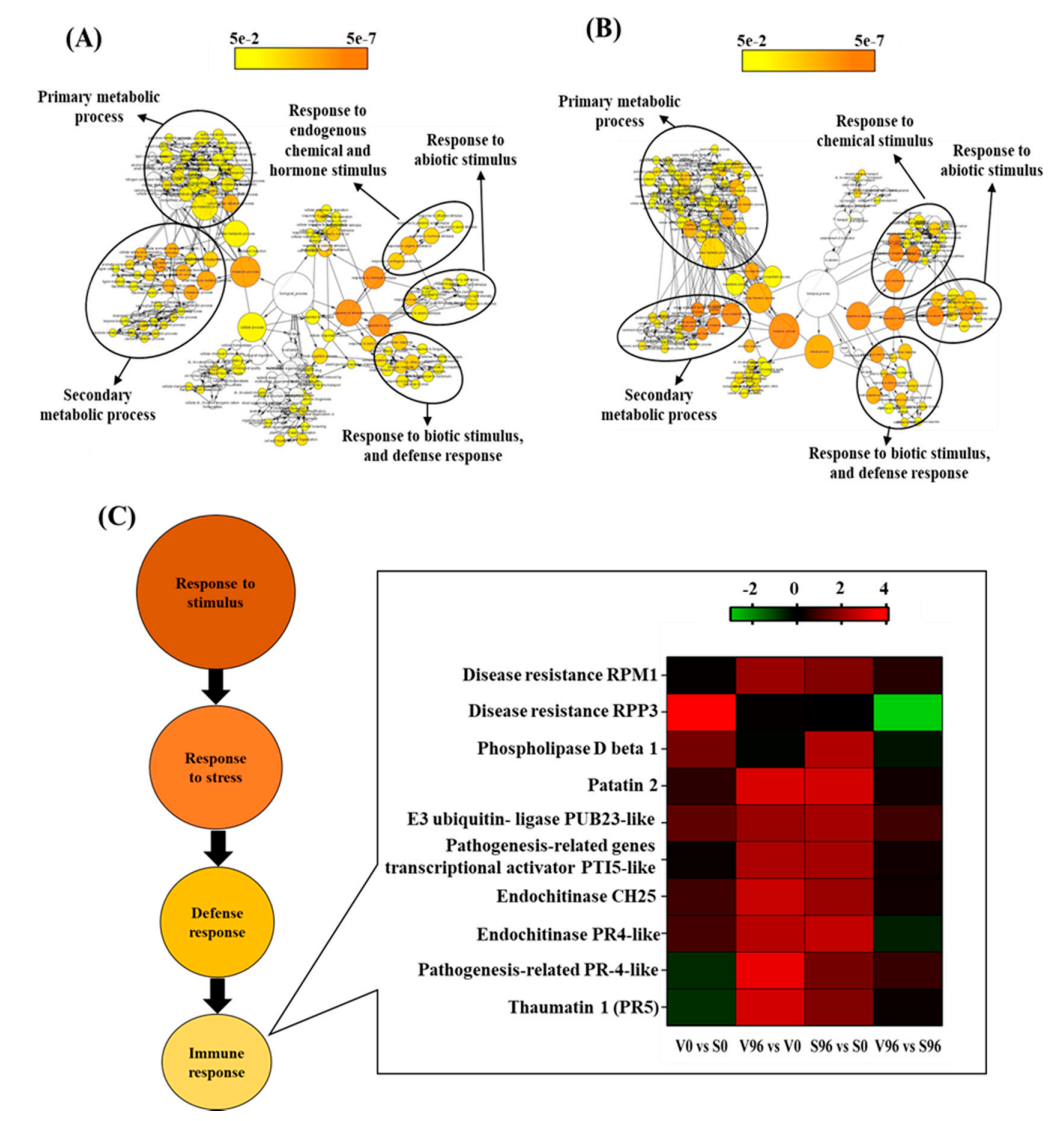
3.5. GO Enrichment Analysis
3.6. Pathway Enrichment Analysis
3.7. GO Network Analysis
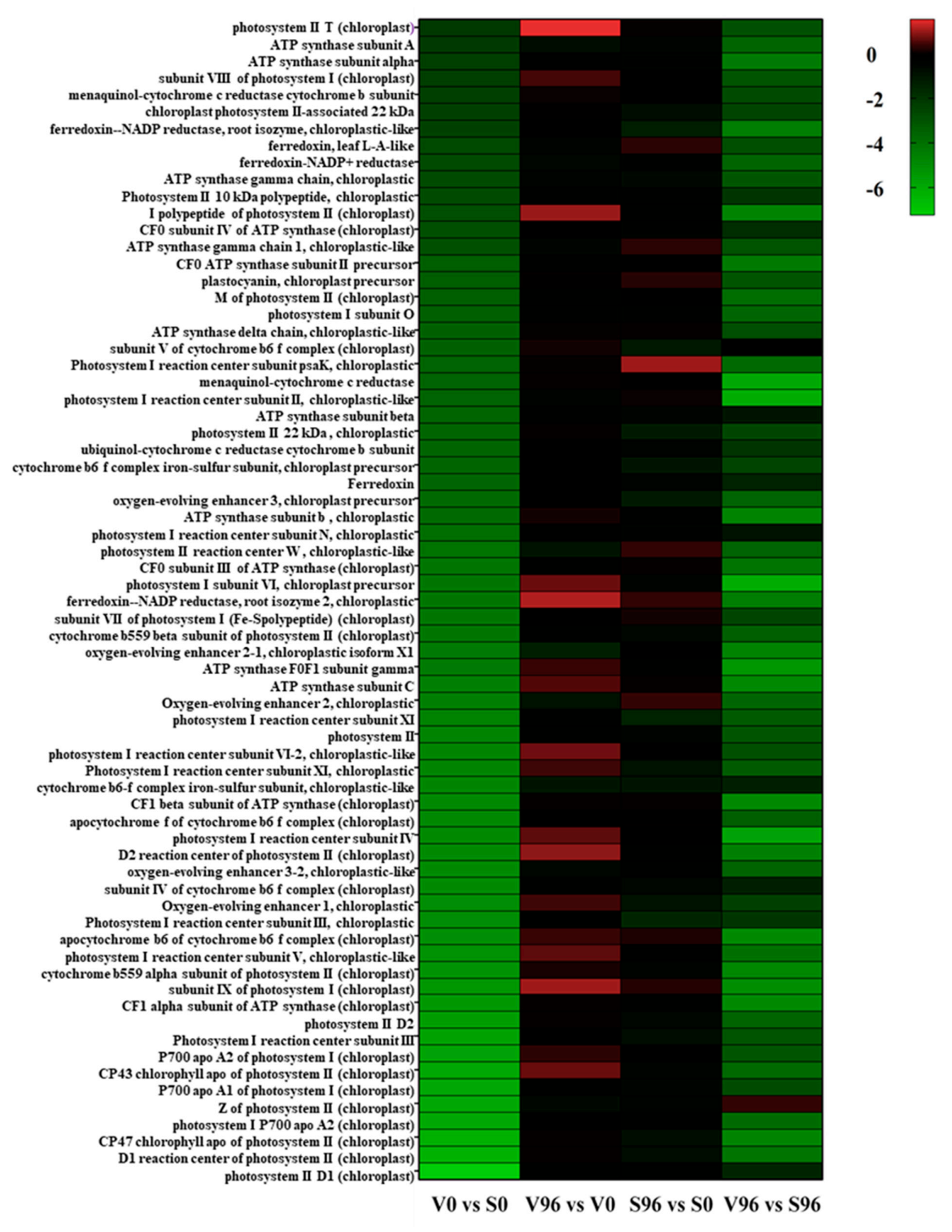
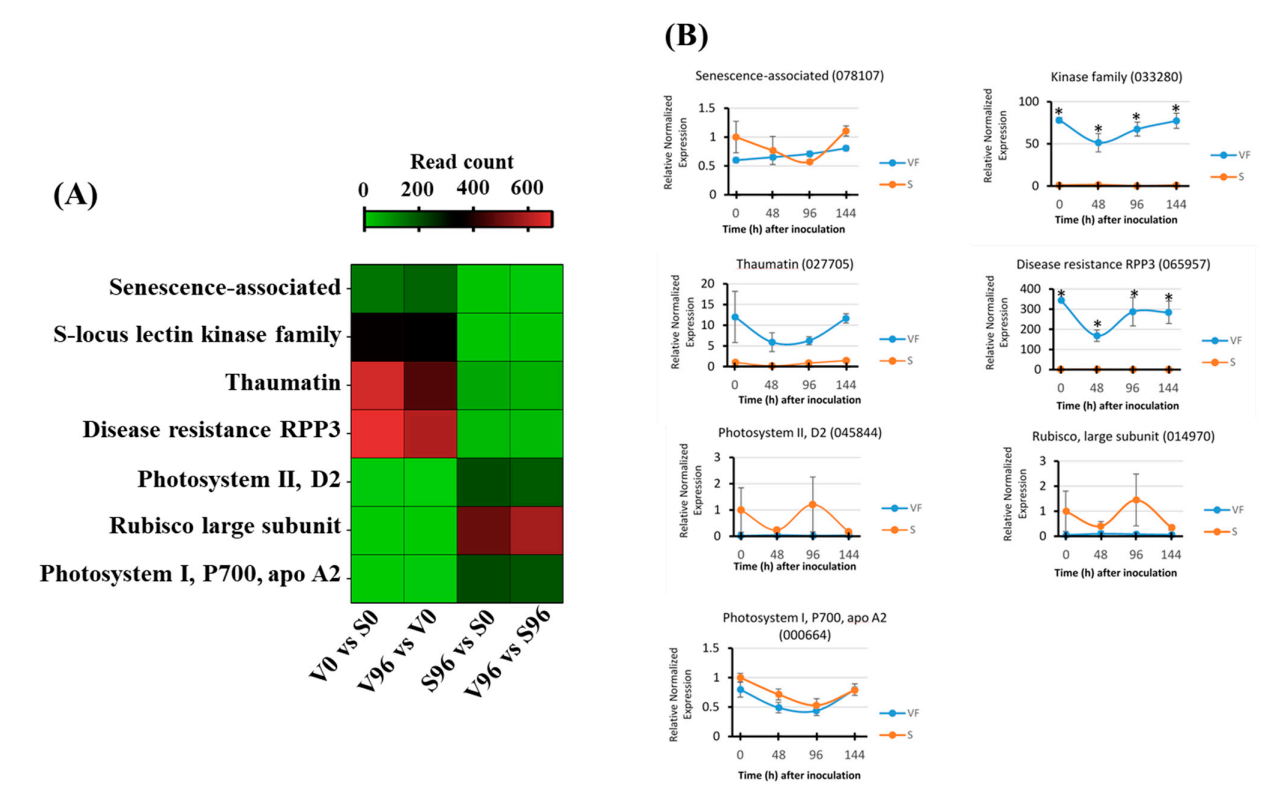
3.8. Expression Patterns of the DEGs Involved in Photosynthesis
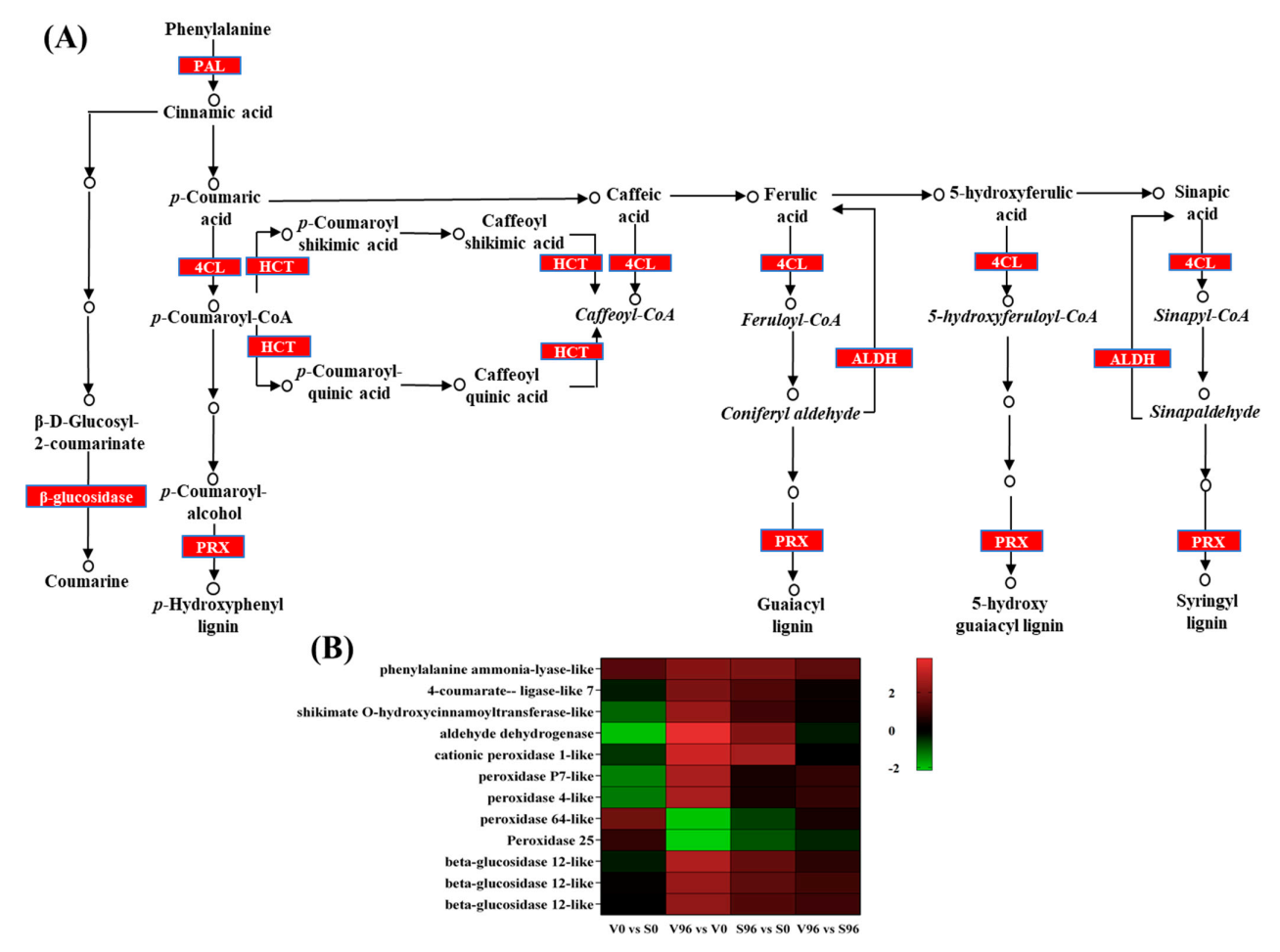
3.9. Expression Profile of the DEGs Involved in the Biosynthesis of Phenylpropanoids
3.10. Validation of RNA-Seq Results by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paoletti, M.; Buck, K.W.; Brasier, C.M. Selective acquisition of novel mating type and vegetative incompatibility genes via interspecies gene transfer in the globally invading eukaryote Ophiostoma novo-ulmi. Mol. Ecol. 2006, 15, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Ganley, R.J.; Bulman, L.S. Dutch elm disease in New Zealand: Impacts from eradication and management programmes. Plant Pathol. 2016, 65, 1047–1055. [Google Scholar] [CrossRef]
- Brasier, C.M.; Kirk, S.A. Designation of the EAN and NAN races of Ophiostoma novo-ulmi as subspecies. Mycol. Res. 2001, 105, 547–554. [Google Scholar] [CrossRef]
- Webber, J.F. Relative effectiveness of Scolytus scolytus, S. multistriatus and S. kirschi as vectors of Dutch elm disease. Eur. J. For. Pathol. 1990, 20, 184–192. [Google Scholar] [CrossRef]
- Webber, J.F. Experimental studies on factors influencing the transmission of Dutch elm disease. For. Syst. 2004, 13, 197–205. [Google Scholar]
- Lee, J.C.; Seybold, S.J. Host acceptance and larval competition in the banded and European elm bark beetles, Scolytus schevyrewi and S. multistriatus (Coleoptera: Scolytidae): Potential mechanisms for competitive displacement between invasive species. J. Insect Behav. 2010, 23, 19–34. [Google Scholar] [CrossRef]
- Townsend, A.M.; Bentz, S.E.; Johnson, G.R. Variation in response of selected American elm clones to Ophiostoma ulmi. J. Environ. Hortic. 1995, 13, 126. [Google Scholar] [CrossRef]
- Townsend, A.M.; Douglass, L.W. Variation among American elm clones in long-term dieback, growth, and survival following Ophiostoma inoculation. J. Environ. Hortic. 2001, 19, 100–103. [Google Scholar] [CrossRef]
- Sherif, S.M.; Shukla, M.R.; Murch, S.J.; Bernier, L.; Saxena, P.K. Simultaneous induction of jasmonic acid and disease-responsive genes signifies tolerance of American elm to Dutch elm disease. Sci. Rep. 2016, 6, 21934. [Google Scholar] [CrossRef] [Green Version]
- Martín, J.A.; Domínguez, J.; Solla, A.; Brasier, C.M.; Webber, J.F.; Santini, A.; Martínez-Arias, C.; Bernier, L.; Gil, L. Complexities underlying the breeding and deployment of Dutch elm disease resistant elms. New For. 2021, 1–36. [Google Scholar] [CrossRef]
- Martín, J.A.; Sobrino-Plata, J.; Rodríguez-Calcerrada, J.; Collada, C.; Gil, L. Breeding and scientific advances in the fight against Dutch elm disease: Will they allow the use of elms in forest restoration? New For. 2019, 50, 183–215. [Google Scholar] [CrossRef] [Green Version]
- Smalley, E.B.; Guries, R.P. Asian elms: Sources ofdiseaseand insect resistance. In The Elms; Dunn, C.P., Ed.; Springer: Berlin, Germany, 2000; pp. 215–230. [Google Scholar]
- Santini, A.; Pecori, F.; Pepori, A.; Brookes, A. ‘Morfeo’ Elm: A new variety resistant to Dutch elm disease. For. Pathol. 2011, 42, 171–176. [Google Scholar] [CrossRef]
- Venturas, M.; López, R.; Martín, J.; Gascó, A.; Gil, L. Heritability of Ulmus minor resistance to Dutch elm disease and its relationship to vessel size, but not to xylem vulnerability to drought. Plant Pathol. 2014, 63, 500–509. [Google Scholar] [CrossRef]
- Perdiguero, P.; Venturas, M.; Cervera, M.T.; Gil, L.; Collada, C. Massive sequencing of Ulmus minor’s transcriptome provides new molecular tools for a genus under the constant threat of Dutch elm disease. Front. Plant Sci. 2015, 6, 541. [Google Scholar] [CrossRef] [Green Version]
- Aoun, M.; Jacobi, V.; Boyle, B.; Bernier, L. Identification and monitoring of Ulmus americana transcripts during in vitro interactions with the Dutch elm disease pathogen Ophiostoma novo-ulmi. Physiol. Mol. Plant Pathol. 2010, 74, 254–266. [Google Scholar] [CrossRef]
- Martínez-Arias, C.; Sobrino-Plata, J.; Gil, L.; Rodríguez-Calcerrada, J.; Martín, J.A. Priming of Plant Defenses against Ophiostoma novo-ulmi by Elm (Ulmus minor Mill.) Fungal Endophytes. J. Fungi 2021, 7, 687. [Google Scholar] [CrossRef]
- Ding, L.; Xu, H.; Yi, H.; Yang, L.; Kong, Z.; Zhang, L.; Xue, S.; Jia, H.; Ma, Z. Resistance to hemi-biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS ONE 2011, 6, e19008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tian, L.; Yan, D.H.; He, W. Genome-Wide Transcriptome Analysis Reveals the Comprehensive Response of Two Susceptible Poplar Sections to Marssonina brunnea Infection. Genes 2018, 9, 154. [Google Scholar] [CrossRef] [Green Version]
- Pedro, H.; Maheswari, U.; Urban, M.; Irvine, A.G.; Cuzick, A.; McDowall, M.D.; Staines, D.M.; Kulesha, E.; Hammond-Kosack, K.E.; Kersey, P.J. PhytoPath: An integrative resource for plant pathogen genomics. Nucleic Acids Res. 2016, 44, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Townsend, A.M.; Bentz, S.E.; Douglass, L.W. Evaluation of 19 American elm clones for tolerance to Dutch elm disease. J. Environ. Hortic. 2005, 23, 21–24. [Google Scholar] [CrossRef]
- Comeau, A.M.; Dufour, J.; Bouvet, G.F.; Nigg, M.; Jacobi, V.; Henrissat, B.; Laroche, J.; Levesque, R.C.; Bernier, L. Functional annotation of the Ophiostoma novo-ulmi genome: Insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biol. Evol. 2015, 7, 410–430. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: http://www.Rproject.org/ (accessed on 1 March 2018).
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community 2017 update. Nucleic Acids Res. 2017, 45, 122–129. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Curr. Protoc. Mol. Biol. 2013, 103, 4–19. [Google Scholar]
- Li, S.; Tighe, S.W.; Nicolet, C.M.; Grove, D.; Levy, S.; Farmerie, W.; Viale, A.; Wright, C.; Schweitzer, P.A.; Gao, Y.; et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol. 2014, 32, 915. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, R.J.; Elgersma, D.M. A scanning electron microscope study of cell wall degradation in elm wood by aggressive and non-aggressive isolates of Ophiostoma ulmi. Eur. J. Plant Pathol. 1982, 12, 25–28. [Google Scholar] [CrossRef]
- Newbanks, D.; Bosch, A.; Zimmermann, M.H. Evidence for xylem dysfunction by embolization in Dutch elm disease. Phytopathology 1983, 73, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [PubMed]
- Perdiguero, P.; Sobrino-Plata, J.; Venturas, M.; Martín, J.A.; Gil, L.; Collada, C. Gene expression trade-offs between defence and growth in English elm induced by Ophiostoma novo-ulmi. Plant Cell Environ. 2018, 41, 198–214. [Google Scholar] [CrossRef]
- Karasov, T.L.; Chae, E.; Herman, J.J.; Bergelson, J. Mechanisms to Mitigate the Trade-Off between Growth and Defense. Plant Cell 2017, 29, 666–680. [Google Scholar] [CrossRef] [Green Version]
- Huot, B.; Yao, J.; Montgomery, B.L.; He, S.Y. Growth-defense tradeoffs in plants: A balancing act to optimize fitness. Mol. Plant. 2014, 7, 1267–1287. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef]
- Van Loon, L.C.; Rep, M.; Pieterse, C.M. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef] [Green Version]
- Baggs, E.; Dagdas, G.; Krasileva, K.V. NLR diversity, helpers and integrated domains: Making sense of the NLR ID entity. Curr. Opin. Plant Biol. 2017, 38, 59–67. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–222. [Google Scholar] [CrossRef]
- Islam, M.T.; Mamun, M.A.; Lee, B.R.; La, H.V.; Kim, T.H. Role of salicylic acid signaling in the biotrophy-necrotrophy transition of Xanthomonas campestris pv. campestris infection in Brassica napus. Physiol. Mol. Plant Pathol. 2021, 113, 101578. [Google Scholar] [CrossRef]
- Delaure, S.L.; Hemelrijck, W.V.; Bolle, M.F.C.D.; Cammue, B.P.A.; De Coninck, B.M.A. Building up plant defenses by breaking down proteins. Plant Sci. 2008, 174, 375–385. [Google Scholar] [CrossRef]
- Norman-Setterblad, C.; Vidal, S.; Palva, E.T. Interacting signal pathways control defense gene expression in Arabidopsis in response to cell wall-degrading enzymes from Erwinia carotovora. Mol. Plant Microbe Interact. 2000, 13, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, P.J.; Lee, A.K.; Xiang, F.; Park, C.M. Molecular and functional profiling of Arabidopsis Pathogenesis-related genes: Insights into their roles in salt response of seed germination. Plant Cell Physiol. 2008, 49, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, M.R.; Godiard, L.; Straube, E.; Ashfield, T.; Lewald, J.; Sattler, A.; Innes, R.W.; Dangl, J.L. Structure of the Arabidopsis RPM1 gene enabling dual specificity disease resistance. Science 1995, 269, 843–846. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, Z.; Huang, J.; Xuan, H.; Gao, Z. Phospholipidase Dδ Negatively Regulates the Function of Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1). Front. Plant Sci. 2019, 9, 1991. [Google Scholar] [CrossRef]
- Bargmann, B.O.R.; Laxalt, A.M.; Ter Riet, B.; Schouten, E.; Van Leeuwen, W.; Dekker, H.L.; De Koster, C.G.; Haring, M.A.; Munnik, T. LePLDβ1 activation and relocalization in suspension-cultured tomato cells treated with xylanase. Plant J. 2006, 45, 358–368. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kuroda, M.; Yamakawa, H.; Ashizawa, T.; Hirayae, K.; Kurimoto, L.; Shinya, T.; Shibuya, N. Suppression of a phospholipase D gene, OsPLDβ1, activates defense responses and increases disease resistance in rice. Plant Physiol. 2009, 150, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Devaiah, S.P.; Wang, C.; Li, M.; Welti, R.; Wang, X. Arabidopsis phospholipase Dβ1 modulates defense responses to bacterial and fungal pathogens. New Phytol. 2013, 199, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Cvikrová, M.; Malá, J.; Hrubcová, M. Eder; J. Soluble and cell wall-bound phenolics and lignin in Ascocalyx abietina infected Norway spruces. Plant Sci. 2006, 170, 563–570. [Google Scholar] [CrossRef]
- Islam, M.T.; Lee, B.R.; Park, S.H.; La, V.H.; Jung, W.J.; Bae, D.W.; Kim, T.H. Hormonal regulations in soluble and cell-wall bound phenolics accumulation in two cultivars of Brassica napus contrasting susceptibility to Xanthomonas campestris pv. campestris. Plant Sci. 2019, 285, 132–140. [Google Scholar] [CrossRef]
- Dixon, R.A.; Barros, J. Lignin biosynthesis: Old roads revisited and new roads explored. Open Biol. 2019, 9, 190215. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.C.; Li, Q.; Shuford, C.M.; Liu, J.; Muddiman, D.C.; Sederoff, R.R.; Chiang, V.L. Membrane protein complexes catalyze both 4- and 3-hydroxylation of cinnamic acid derivatives in monolignol biosynthesis. Proc. Natl Acad. Sci. USA 2011, 108, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Duroux, L.; Welinder, K.G. The peroxidase gene family in plants: A phylogenetic overview. J. Mol. Evol. 2003, 57, 397–407. [Google Scholar] [CrossRef]
- Zhao, Q.; Nakashima, J.; Chen, F.; Yin, Y.; Fu, C.; Yun, J.; Shao, H.; Wang, X.; Wang, Z.; Dixon, R.A. LACCASE is necessary and nonredundant with PEROXIDASE for lignin polymerization during vascular development in Arabidopsis thaliana. Plant Cell 2013, 25, 3976–3987. [Google Scholar] [CrossRef] [Green Version]
- Douaiher, M.N.; Nowak, E.; Durand, R.; Halama, P.; Reignault, P. Correlative analysis of Mycosphaerella graminicola pathogenicity and cell wall-degrading enzymes produced in vitro: The importance of xylanase and polygalacturonase. Plant Pathol. 2007, 56, 79–86. [Google Scholar] [CrossRef]
- Kikot, G.E.; Hours, R.A.; Alconada, T.M. Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: A review. J. Basic Microbiol. 2009, 49, 231–241. [Google Scholar] [CrossRef]
- Przybyl, K.; Dahm, H.; Ciesielska, A.; Molinski, K. Cellulolytic activity and virulence of Ophiostoma ulmi and O. novo-ulmi isolates. For. Pathol. 2006, 36, 58–67. [Google Scholar] [CrossRef]
- Svaldi, R.; Elgersma, D.M. Further studies on the activity of cell wall degrading enzymes of aggressive and non-aggressive isolates of Ophiostoma ulmi. Eur. J. For. Pathol. 1982, 12, 29–36. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Valley Forge | Susceptible | |||
|---|---|---|---|---|
| 0 h | 96 h | 0 h | 96 h | |
| Total reads | 75,742,361 (±0.8%) | 73,144,879 (±3.9%) | 66,441,814 (±8.6%) | 75,518,322 (±4.6%) |
| Alignment (%) | 73.7 (±0.7%) | 71.5 (±1.5%) | 70.1 (±0.9%) | 69.8 (±0.4%) |
| Treatment | GO ID | GO Description | p-Value | FDR |
|---|---|---|---|---|
| V0 vs. S0 | GO:0008152 | Metabolic process | 2.3 × 10−110 | 1.1 × 10−106 |
| GO:0009987 | Cellular process | 4 × 10−94 | 1 × 10−90 | |
| GO:0044237 | Cellular metabolic process | 4.5 × 10−83 | 7.5 × 10−80 | |
| GO:0044238 | Primary metabolic process | 1.2 × 10−55 | .5 × 10−52 | |
| GO:0050896 | Response to stimulus | 4.4 × 10−46 | 4.4 × 10−43 | |
| V96 vs. V0 | GO:0050896 | Response to stimulus | 4.5 × 10−14 | 4.20 × 10−11 |
| GO:0006950 | Response to stress | 2.4 × 10−12 | 1.10 × 10−9 | |
| GO:0042221 | Response to chemical stimulus | 1.9 × 10−11 | 5.90 × 10−9 | |
| GO:0019748 | Secondary metabolic process | 2.5 × 10−7 | 5.80 × 10−5 | |
| GO:0006952 | Defense response | 7.1 × 10−7 | 1.3 × 10−4 | |
| S96 vs. S0 | GO:0006950 | Response to stress | 7.5 × 10−14 | 9.2 × 10−11 |
| GO:0050896 | Response to stimulus | 6.2 × 10−13 | 3.8 × 10−10 | |
| GO:0008152 | Metabolic process | 6.9 × 10−12 | 2.8 × 10−9 | |
| GO:0019748 | Secondary metabolic process | 4.6 × 10−10 | 1.4 × 10−7 | |
| GO:0009628 | Response to abiotic stimulus | 2.4 × 10−8 | 5.8 × 10−6 | |
| V96 vs. S96 | GO:0008152 | Metabolic process | 3.50 × 10−107 | 1.50 × 10−103 |
| GO:0009987 | Cellular process | 3.10 × 10−79 | 6.40 × 10−76 | |
| GO:0044237 | Cellular metabolic process | 7.00 × 10−77 | 9.70 × 10−74 | |
| GO:0044238 | Primary metabolic process | 3.50 × 10−53 | 3.60 × 10−50 | |
| GO:0019538 | Protein metabolic process | 1.10 × 10−42 | 7.80 × 10−40 |
| Treatment | Pathway ID | Pathway Name | p-Value | FDR |
|---|---|---|---|---|
| V0 vs. S0 | ko00195 | Photosynthesis | 1.7 × 10−19 | 1.1 × 10−17 |
| ko01200 | Carbon metabolism | 8.4 × 10−12 | 2.1 × 10−10 | |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 1 × 10−8 | 1.8 × 10−7 | |
| ko00020 | Citrate cycle (TCA cycle) | 9.2 × 10−8 | 1.5 × 10−6 | |
| ko00620 | Pyruvate metabolism | 9.8 × 10−6 | 1.4 × 10−4 | |
| V96 vs. V0 | ko09110 | Biosynthesis of secondary metabolites | 7.9 × 10−5 | 1.9 × 10−3 |
| ko00940 | Phenylpropanoid biosynthesis | 4.5 × 10−4 | 8.2 × 10−3 | |
| ko00350 | Tyrosine metabolism | 5.4 × 10−3 | 1.1 × 10−2 | |
| ko00270 | Cysteine and methionine metabolism | 8.4 × 10−3 | 1.9 × 10−2 | |
| ko00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 2.4 × 10−2 | 2.2 × 10−2 | |
| S96 vs. S0 | ko00630 | Glyoxylate and dicarboxylate metabolism | 2.9 × 10−4 | 8.5 × 10−3 |
| ko09110 | Biosynthesis of secondary metabolites | 3.6 × 10−4 | 8.5 × 10−3 | |
| ko00071 | Fatty acid degradation | 5.7 × 10−4 | 1.0 × 10−2 | |
| ko00910 | Nitrogen metabolism | 6.6 × 10−4 | 1.0 × 10−2 | |
| ko00940 | Phenylpropanoid biosynthesis | 3.1 × 10−3 | 4.1 × 10−2 | |
| V96 vs. S96 | ko00195 | Photosynthesis | 1.8 × 10−17 | 1.1 × 10−15 |
| Ko01200 | Carbon metabolism | 1.4 × 10−15 | 5.8 × 10−14 | |
| ko00190 | Oxidative phosphorylation | 1 × 10−12 | 2.6 × 10−11 | |
| ko00020 | Citrate cycle (TCA cycle) | 7.3 × 10−10 | 1.5 × 10−8 | |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 3.5 × 10−9 | 5.4 × 10−8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.T.; Coutin, J.F.; Shukla, M.; Dhaliwal, A.K.; Nigg, M.; Bernier, L.; Sherif, S.M.; Saxena, P.K. Deciphering the Genome-Wide Transcriptomic Changes during Interactions of Resistant and Susceptible Genotypes of American Elm with Ophiostoma novo-ulmi. J. Fungi 2022, 8, 120. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020120
Islam MT, Coutin JF, Shukla M, Dhaliwal AK, Nigg M, Bernier L, Sherif SM, Saxena PK. Deciphering the Genome-Wide Transcriptomic Changes during Interactions of Resistant and Susceptible Genotypes of American Elm with Ophiostoma novo-ulmi. Journal of Fungi. 2022; 8(2):120. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020120
Chicago/Turabian StyleIslam, Md Tabibul, Jose Freixas Coutin, Mukund Shukla, Amandeep Kaur Dhaliwal, Martha Nigg, Louis Bernier, Sherif M. Sherif, and Praveen K. Saxena. 2022. "Deciphering the Genome-Wide Transcriptomic Changes during Interactions of Resistant and Susceptible Genotypes of American Elm with Ophiostoma novo-ulmi" Journal of Fungi 8, no. 2: 120. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8020120