
An Integrative View of the Phyllosphere Mycobiome of Native Rubber Trees in the Brazilian Amazon

 , ,
, ,  , , , , ,
, , , , ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Knowledge Discovery in Database (KDD) Approach
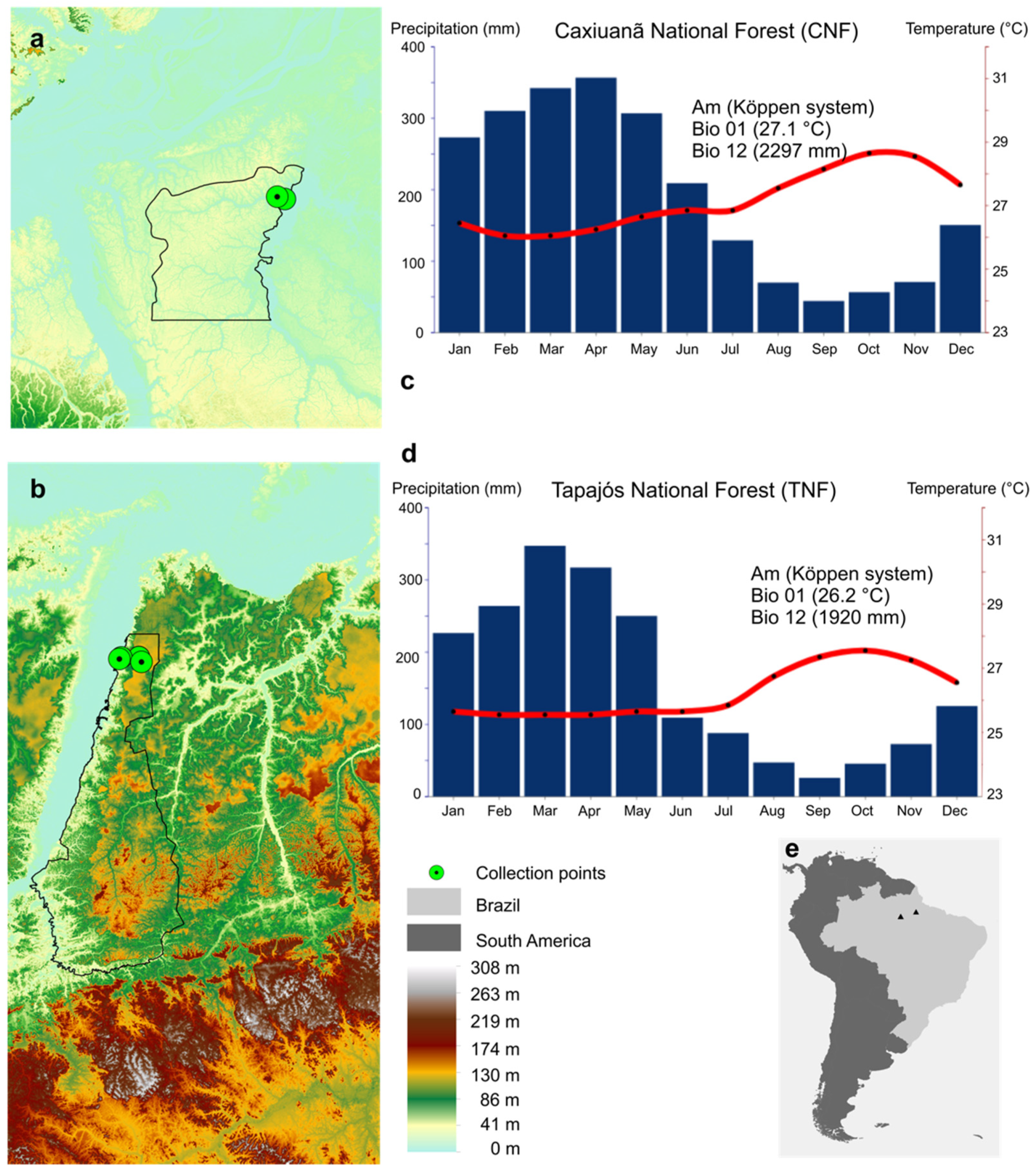
2.2. Study Areas
2.3. Field Sampling
2.4. Metabarcoding
2.4.1. DNA Extraction, PCR, Metagenomics Sequencing, Bioinformatic Analyses, and Community Ecology Analyses
2.4.2. Predicted Ecological Traits (Trophic Modes and Guilds)
2.5. Complex Networks Analyses
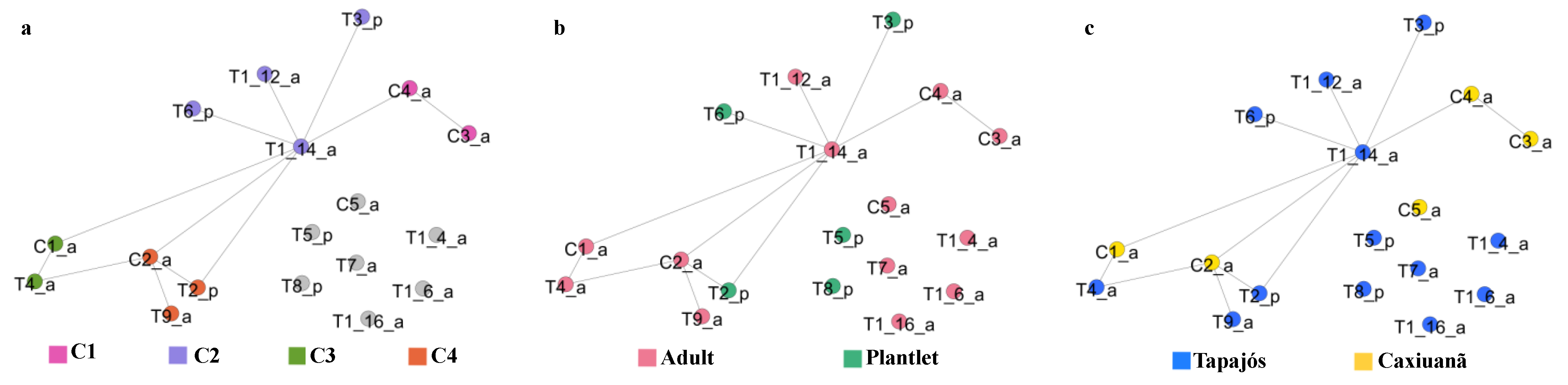
2.5.1. Tree Similarity Network
2.5.2. Co-Occurrence Networks
2.6. Case Study of a Selected Sampling Unit (C2 Tree)
2.6.1. Shotgun Metagenomics
2.6.2. Small RNA Metatranscriptomics
3. Results
3.1. In Silico KDD Approach
3.1.1. Literature Database
3.1.2. Host-Fungus Database
3.1.3. Fungal DNA Sequences Associated with H. brasiliensis
3.2. Field and Experimental Approach
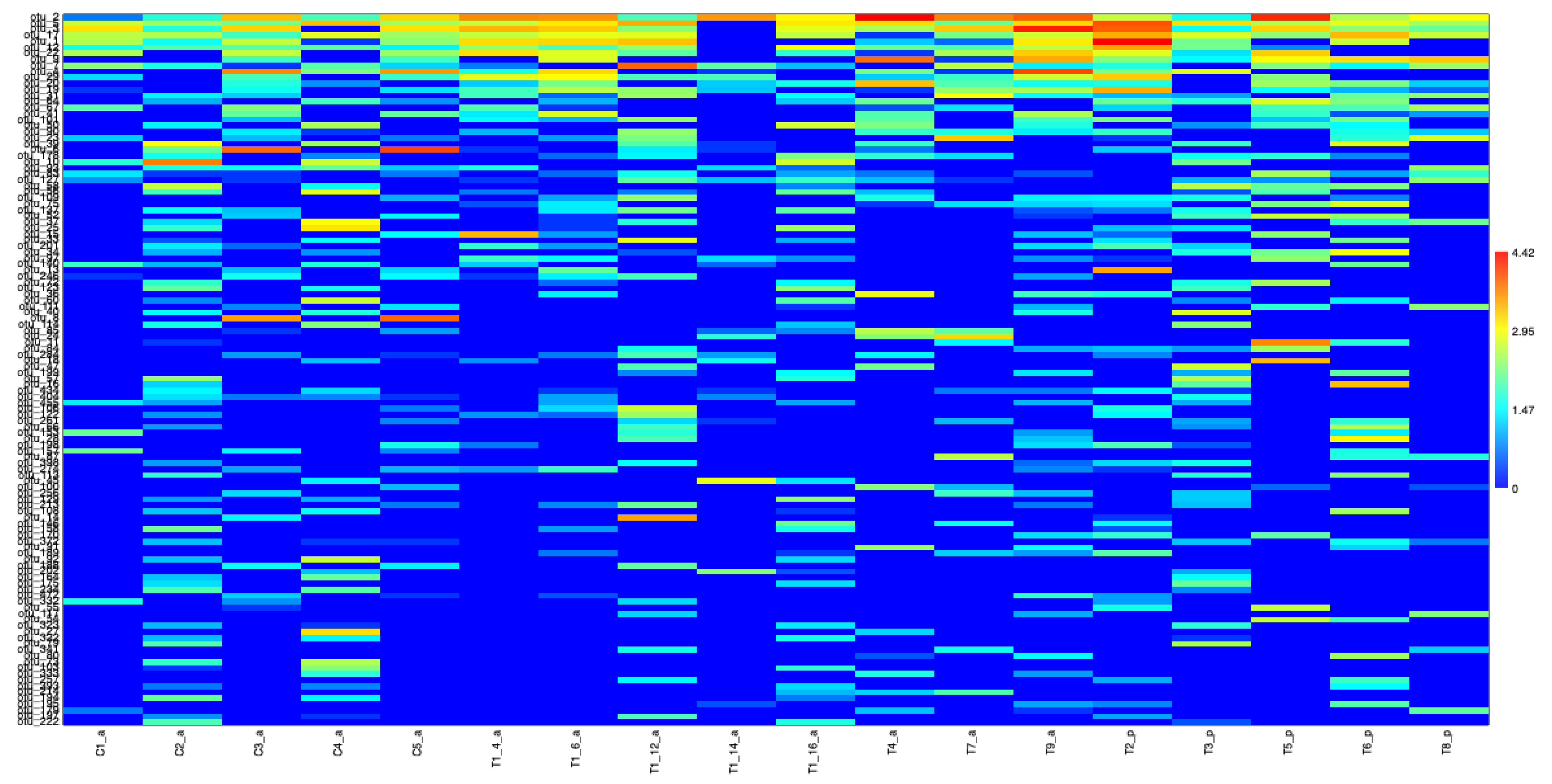
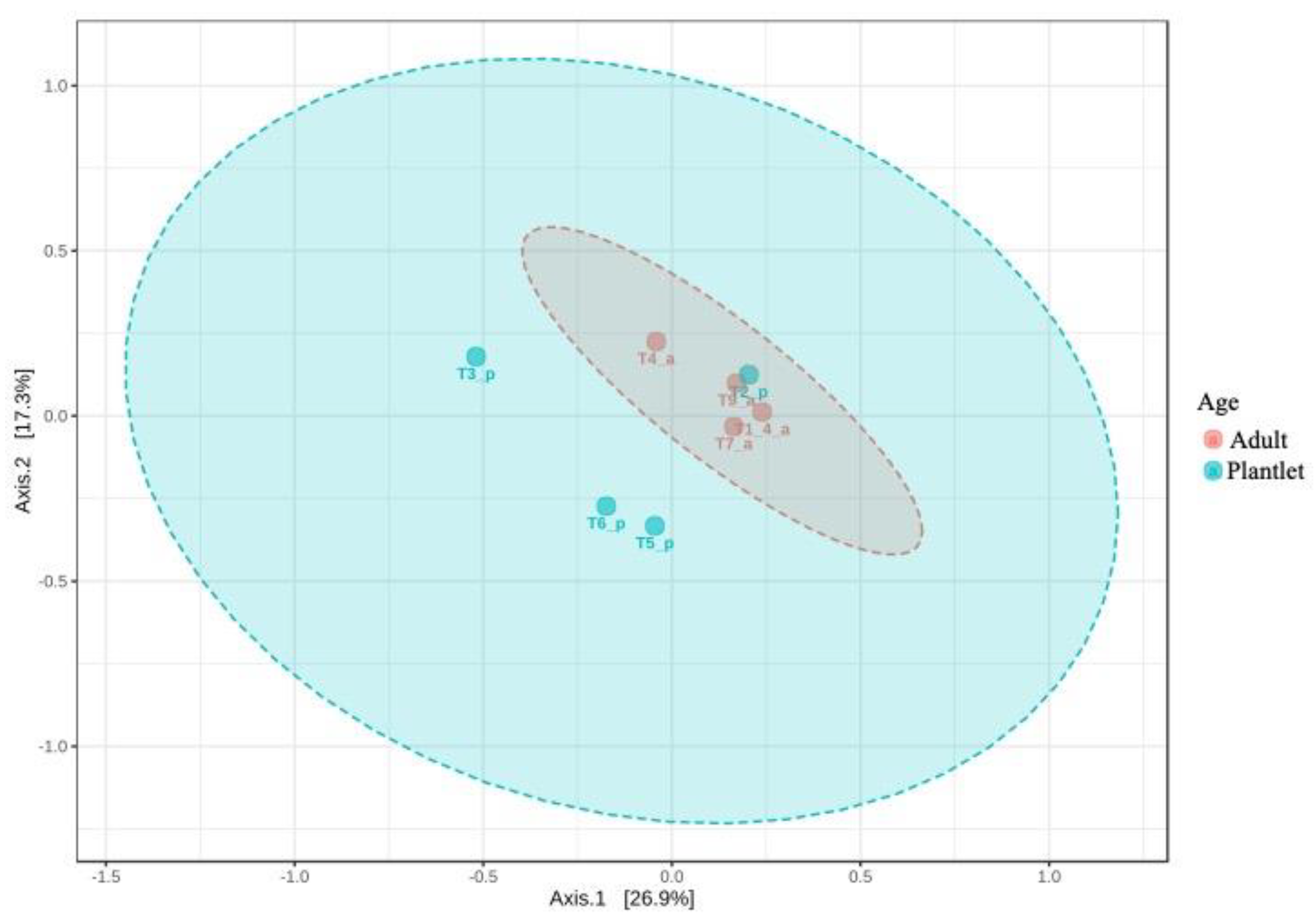
3.2.1. Metabarcoding
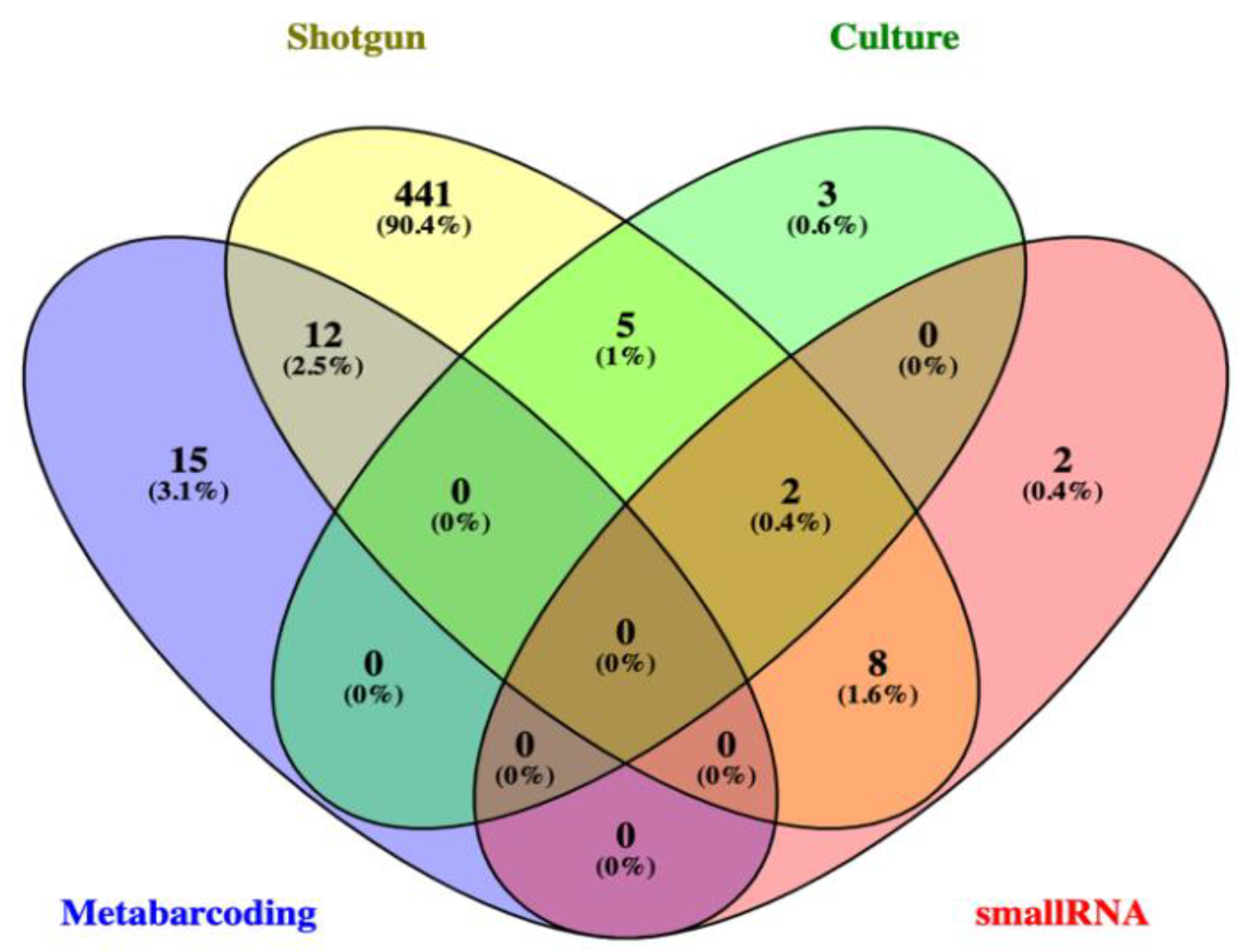
3.2.2. Shotgun Metagenomics and Small RNA Metatranscriptomics of a Selected Sampling Unit (C2)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, G.; Rybakova, D.; Grube, M.; Köberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2016, 67, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Vacher, C.; Hampe, A.; Porté, A.J.; Sauer, U.; Compant, S.; Morris, C.E. The Phyllosphere: Microbial Jungle at the Plant–Climate Interface. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 1–24. [Google Scholar] [CrossRef]
- Cordier, T.; Robin, C.; Capdevielle, X.; Desprez-Loustau, M.L.; Vacher, C. Spatial variability of phyllosphere fungal assemblages: Genetic distance predominates over geographic distance in a European beech stand (Fagus sylvatica). Fungal Ecol. 2012, 5, 509–520. [Google Scholar] [CrossRef]
- Brown, S.P.; Leopold, D.R.; Busby, P.E. Protocols for investigating the leaf mycobiome using high-throughput DNA sequencing. Methods Mol. Biol. 2018, 1848, 39–51. [Google Scholar] [CrossRef]
- Harrison, J.G.; Griffin, E.A. The diversity and distribution of endophytes across biomes, plant phylogeny and host tissues: How far have we come and where do we go from here? Environ. Microbiol. 2020, 22, 2107–2123. [Google Scholar] [CrossRef] [Green Version]
- Atsatt, P.R.; Whiteside, M.D. Novel symbiotic protoplasts formed by endophytic fungi explain their hidden existence, lifestyle switching, and diversity within the plant kingdom. PLoS ONE 2014, 9, e95266. [Google Scholar] [CrossRef]
- Nelson, A.; Vandegrift, R.; Carroll, G.C.; Roy, B.A. Double lives: Transfer of fungal endophytes from leaves to woody substrates. PeerJ 2020, 8, e9341. [Google Scholar] [CrossRef]
- Thomas, D.C.; Vandegrift, R.; Roy, B.A. An agent-based model of the foraging ascomycete hypothesis. Fungal Ecol. 2020, 47, 100963. [Google Scholar] [CrossRef]
- Vaz, A.B.M.; Fonseca, P.L.C.; Silva, F.F.; Quintanilha-Peixoto, G.; Sampedro, I.; Siles, J.A.; Carmo, A.; Kato, R.B.; Azevedo, V.; Badotti, F.; et al. Foliar mycoendophytome of an endemic plant of the Mediterranean biome (Myrtus communis) reveals the dominance of basidiomycete woody saprotrophs. PeerJ 2020, 8, e10487. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, M.; Kushveer, J.S.; Sarma, V.V. A worldwide list of endophytic fungi with notes on ecology and diversity. Mycosphere 2019, 10, 798–1079. [Google Scholar] [CrossRef]
- IPNI. International Plant Names Index. Available online: http://www.ipni.org (accessed on 1 February 2022).
- Ter Steege, H.; Pitman, N.C.; Sabatier, D.; Baraloto, C.; Salomão, R.P.; Guevara, J.E.; Silman, M.R. Hyperdominance in the Amazonian tree flora. Science 2013, 342, 6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, K.P. Rubber (Hevea brasiliensis). In Tree Crops; Nair, K.P., Ed.; Springer: New York, NY, USA, 2021; pp. 287–332. [Google Scholar] [CrossRef]
- Vaz, A.B.M.; Fonseca, P.L.C.; Badotti, F.; Skaltsas, D.; Tomé, L.M.R.; Silva, A.C.; Cunha, M.C.; Soares, M.A.; Santos, V.L.; Oliveira, G.; et al. A multiscale study of fungal endophyte communities of the foliar endosphere of native rubber trees in Eastern Amazon. Sci. Rep. 2018, 8, 16151. [Google Scholar] [CrossRef]
- Araújo, K.S.; Brito, V.N.; Veloso, T.G.; de Leite, T.S.; Alves, J.L.; da Hora Junior, B.T.; Moreno, H.L.; Pereira, O.L.; Mizubuti, E.S.; de Queiroz, M.V. Diversity and distribution of endophytic fungi in different tissues of Hevea brasiliensis native to the Brazilian Amazon forest. Mycol. Prog. 2020, 19, 1057–1068. [Google Scholar] [CrossRef]
- Martelli, A. Python in a Nutshell: A Desktop Quick Reference, 2nd ed.; O’Reilly Media: Sebastopol, Ukraine, 2006; pp. 1–718. [Google Scholar]
- Venny. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 3 February 2022).
- Brasil. Plano de Manejo da Floresta Nacional de Caxiuanã, Volume I—Diagnóstico; Instituto Chico Mendes: Brasília, Brazil, 2012; 214p. [Google Scholar]
- Brasil. Plano de Manejo da Floresta Nacional de Tapajós, Volume 1—Informações Gerais; IBAMA: Brasília, Brazil, 2004; 580p. [Google Scholar]
- Hora-Júnior, B.T.; de Macedo, D.M.; Barreto, R.W.; Evans, H.C.; Mattos, C.R.R.; Maffia, L.A.; Mizubuti, E.S.G. Erasing the Past: A New Identity for the Damoclean Pathogen Causing South American Leaf Blight of Rubber. PLoS ONE 2014, 9, e104750. [Google Scholar] [CrossRef] [Green Version]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.J.W.T.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: Cambridge, UK, 1990; pp. 315–322. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mah, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- McIlroy, M.D. A Research UNIX Reader: Annotated Excerpts from the Programmer’s Manual, 1971–1986; AT&T Bell Laboratories: Murray Hill, NY, USA, 1987; pp. 1–115. [Google Scholar]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Hiland, K.; Kjller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi–recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Andrade, R.F.S.; Rocha-Neto, I.C.; Santos, L.B.L.; De Santana, C.N.; Diniz, M.V.C.; Lobo, T.P.; Goés-Neto, A.; Pinho, S.T.R.; El-Hani, C.N. 2011. Detecting network communities: An application to phylogenetic analysis. PLoS Comput. Biol. 2011, 7, e1001131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, M.; Heymann, S. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International ICWSM Conference, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 January 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Opens external link in new window. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- De Wood Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.; Clements, J.; Arndt, W.; Miller, B.; Wheeler, T.; Schreiber, F.; Bateman, A.; Eddy, S. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef]
- Hanski, I. Dynamics of Regional Distribution: The Core and Satellite Species Hypothesis. Oikos 1982, 38, 210. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Hernandez-Agreda, A.; Gates, R.D.; Ainsworth, T.D. Defining the Core Microbiome in Corals’ Microbial Soup. Trends Microbiol. 2017, 25, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Denchev, C.M.; Denchev, T.T. Validation of the generic names Meira and Acaromyces and nineteen species names of basidiomycetous yeasts. Mycobiota 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Boekhout, T.; Theelen, B.; Houbraken, J.; Robert, V.; Scorzetti, G.; Gafni, A.; Gerson, U.; Sztejnberg, A. Novel anamorphic mite associated fungi belonging to the Ustilaginomycetes: Meira geulakonigii gen. nov., sp. nov., Meira argovae sp. nov. and Acaromyces ingoldii gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, T.A.; Aime, M.C. The genus Meira: Phylogenetic placement and description of a new species. Antonie Leeuwenhoek 2013, 103, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, F.; Yamagishi, D.; Akamatsu, H.; Izawa, H.; Kodama, M.; Otani, H. Meira nashicola sp. nov., a novel basidiomycetous, anamorphic yeastlike fungus isolated from Japanese pear fruit with reddish stain. Mycoscience 2006, 47, 36–40. [Google Scholar] [CrossRef]
- Cao, Y.; Li, P.-D.; Zhao, J.; Wang, H.-K.; Jeewon, R.; Bhoyroo, V.; Aruna, B.; Lin, F.-C.; Wang, Q. Morph-molecular characterization of Meira nicotianae sp. nov., a novel basidiomycetous, anamorphic yeast-like fungus associated with growth improvement in tobacco plant. Phytotaxa 2018, 365, 169–181. [Google Scholar] [CrossRef]
- Limtong, S.; Polburee, P.; Chamnanpa, T.; Khunnamwong, P.; Limtong, P. Meira siamensis sp. nov., a novel anamorphic ustilaginomycetous yeast species isolated from the vetiver grass phylloplane. Int. J. Syst. Evol. Microbiol. 2017, 67, 2418–2422. [Google Scholar] [CrossRef]
- Gerson, U.; Gafni, A.; Paz, Z.; Sztejnberg, A. A tale of three acaropathogenic fungi in Israel: Hirsutella, Meira, and Acaromyces. Exp. Appl. Acarol. 2008, 46, 183–194. [Google Scholar] [CrossRef]
- Hikawa, M.; Endoh, R.; Takeuchi, Y.; Futai, K. Yeast-like fungus Meira nashicola isolated from a bog bean Menyanthes trifoliata. Jpn. J. Mycol. 2014, 55, 5–11. [Google Scholar]
- Tanaka, E.; Shimizu, K.; Imanishi, Y.; Yasuda, F.; Tanaka, C. Isolation of basidiomycetous anamorphic yeast-like fungus Meira argovae found on Japanese bamboo. Mycoscience 2008, 49, 329–333. [Google Scholar] [CrossRef]
- De Vis, R.M.; Moraes, G.J.D.; Bellini, M.R. Mites (Acari) of rubber trees (Hevea brasiliensis Muell. Arg., Euphorbiaceae) in Piracicaba, State of São Paulo, Brazil. Neotrop. Entomol. 2006, 35, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, W.S.; Daud, R.D. Insights on plant mite occurrence in natural vegetation remnants from Brazil. Syst. Appl. Acarol. 2017, 22, 302–322. [Google Scholar] [CrossRef]
- Rezende, J.M.; Lofego, A.C.; Nuvoloni, F.M.; Navia, D. Mites from Cerrado fragments and adjacent soybean crops: Does the native vegetation help or harm the plantation? Exp. Appl. Acarol. 2014, 64, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Paz, Z.; Burdman, S.; Gerson, U.; Sztejnberg, A. Antagonistic effects of the endophytic fungus Meira geulakonigii on the citrus rust mite Phyloocoptruta oleivora. J. Appl. Microbiol. 2007, 103, 2570–2579. [Google Scholar] [CrossRef] [PubMed]
- Paz, Z.; Gerson, U.; Sztejnberg, A. Assaying three new fungi against citrus mites in the laboratory, and a field trial. Biocontrol 2007, 52, 855–862. [Google Scholar] [CrossRef]
- Paz, Z.; Bilkis, I.; Gerson, U.; Kerem, Z.; Sztejnberg, A. Argovin, a novel natural product secreted by the fungus Meira argovae, is antagonistic to mites. Entomol. Exp. Appl. 2011, 140, 247–253. [Google Scholar] [CrossRef]
- Gleason, M.L.; Zhang, R.; Batzer, J.C.; Sun, G. Stealth pathogens: The sooty blotch and flyspeck fungal complex. Annu. Rev. Phytopathol. 2019, 57, 135–164. [Google Scholar] [CrossRef]
- Rosli, H.; Batzer, J.C.; Harrington, T.C.; Gleason, M.L. Peltaster gemmifer: A new species in the sooty blotch and flyspeck species complex from the United States. Mycologia 2018, 110, 822–834. [Google Scholar] [CrossRef]
- Aguiar, I.D.J.A. Fungos do herbário do Instituto Nacional de Pesquisas da Amazônia. I. Tipos nomenclaturais da coleção Chaves Batista. Acta Amazon. 1988, 18, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.S.; Kozlowski, T.T. Stomatal characteristics, leaf waxes, and transpiration rates of Theobroma cacao and Hevea brasiliensis seedlings. Ann. Bot. 1988, 61, 425–432. [Google Scholar] [CrossRef]
- Kulakovskaya, T.V.; Shashkov, A.S.; Kulakovskaya, E.V.; Golubev, W.I. Characterization of an antifungal glycolipid secreted by the yeast Sympodiomycopsis paphiopedili. FEMS Yeast Res. 2004, 5, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezierska, S.; Claus, S.; Van Bogaert, I. Yeast glycolipid biosurfactants. FEBS Lett. 2018, 592, 1312–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, H.-X.; Li, J.; Chen, H.; Wang, W. The insights into the evolutionary history of Translucidithyrium: Based on a newly-discovered species. MycoKeys 2020, 76, 1–16. [Google Scholar] [CrossRef]
- Skaltsas, D.N.; Badotti, F.; Vaz, A.B.M.; Silva, F.F.; Gazis, R.; Wurdack, K.; Castlebury, L.; Góes-Neto, A.; Chaverri, P. Exploration of stem endophytic communities revealed developmental stage as one of the drivers of fungal endophytic community assemblages in two Amazonian hardwood genera. Sci. Rep. 2019, 9, 12685. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Lan, G.; Wu, Z.; Chen, B.; Quan, F.; Li, M.; Sun, S.; Du, H. Phyllosphere fungal communities of rubber trees exhibited biogeographical patterns, but not bacteria. Environ. Microbiol. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Carroll, G.C. The foraging ascomycete. In Proceedings of the 16th International Botanical Congress, St. Louis, MO, USA, 1–7 August 1999. [Google Scholar]
- Thomas, D.C.; Vandegrift, R.; Ludden, A.; Carroll, G.C.; Roy, B.A. Spatial Ecology of the Fungal Genus Xylaria in a Tropical Cloud Forest. Biotropica 2016, 48, 381–393. [Google Scholar] [CrossRef]
- Liu, Y.X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315. [Google Scholar] [CrossRef]
- Wang, Z.; Li, M.; Ju, W.-B.; Ye, W.; Xue, L.; Boufford, D.; Gao, X.; Yue, B.; Liu, Y.; Pierce, N. The Entomophagous Caterpillar Fungus Ophiocordyceps Sinensis is Consumed by Its Lepidopteran Host as a Plant Endophyte. Fungal Ecol. 2020, 47. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef]
- Liu, L.; Lu, L.; Li, H.; Meng, Z.; Dong, T.; Peng, C.; Xu, X. Divergence of Phyllosphere Microbial Communities Between Females and Males of the Dioecious Populus cathayana. MPMI 2021, 34, 351–361. [Google Scholar] [CrossRef]
- Murphy, C.L.; Youssef, N.H.; Hanafy, R.A.; Couger, M.B.; Stajich, J.E.; Wang, Y.; Baker, K.; Dagar, S.S.; Griffith, G.W.; Farag, I.F.; et al. Horizontal gene transfer as an indispensable driver for evolution of Neocallimastigomycota into a distinct gut-dwelling fungal lineage. Appl. Environ. Microbiol. 2019, 85, e00988-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazis, R.; Chaverri, P. Diversity of fungal endophytes in leaves and stems of wild rubber trees (Hevea brasiliensis) in Peru. Fungal Ecol. 2010, 3, 240–254. [Google Scholar] [CrossRef]
- Gazis, R.; Chaverri, P. Wild trees in the Amazon basin harbor a great diversity of beneficial endosymbiotic fungi: Is this evidenceof protective mutualism? Fungal Ecol. 2015, 17, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.K.; Mendoza-Mendoza, A.; Zeilinger, S.; Horwitz, B.A. Mycoparasitism as a mechanism of Trichoderma-mediated suppression of plant diseases. Fungal Biol. Rev. 2022, 39, 15–33. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, P.L.C.; Skaltsas, D.; da Silva, F.F.; Kato, R.B.; de Castro, G.M.; García, G.J.Y.; Quintanilha-Peixoto, G.; Mendes-Pereira, T.; do Carmo, A.O.; Aguiar, E.R.G.R.; et al. An Integrative View of the Phyllosphere Mycobiome of Native Rubber Trees in the Brazilian Amazon. J. Fungi 2022, 8, 373. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8040373
Fonseca PLC, Skaltsas D, da Silva FF, Kato RB, de Castro GM, García GJY, Quintanilha-Peixoto G, Mendes-Pereira T, do Carmo AO, Aguiar ERGR, et al. An Integrative View of the Phyllosphere Mycobiome of Native Rubber Trees in the Brazilian Amazon. Journal of Fungi. 2022; 8(4):373. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8040373
Chicago/Turabian StyleFonseca, Paula Luize Camargos, Demetra Skaltsas, Felipe Ferreira da Silva, Rodrigo Bentes Kato, Giovanni Marques de Castro, Glen Jasper Yupanqui García, Gabriel Quintanilha-Peixoto, Thairine Mendes-Pereira, Anderson Oliveira do Carmo, Eric Roberto Guimarães Rocha Aguiar, and et al. 2022. "An Integrative View of the Phyllosphere Mycobiome of Native Rubber Trees in the Brazilian Amazon" Journal of Fungi 8, no. 4: 373. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8040373