
Macrophage Lysosomal Alkalinization Drives Invasive Aspergillosis in a Mouse Cystic Fibrosis Model of Airway Transplantation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Aspergillus Culture
2.2. FLARE Conidia AF633 Tagging
2.3. Orthotopic Tracheal Transplantation Model
2.4. Tissue Preparation, Grading of Fungal Invasion
2.5. Lung Macrophages Isolation
2.6. Tracheal Tissue Staining
2.7. Conidial Ingestion and Clearance Detection Imaging
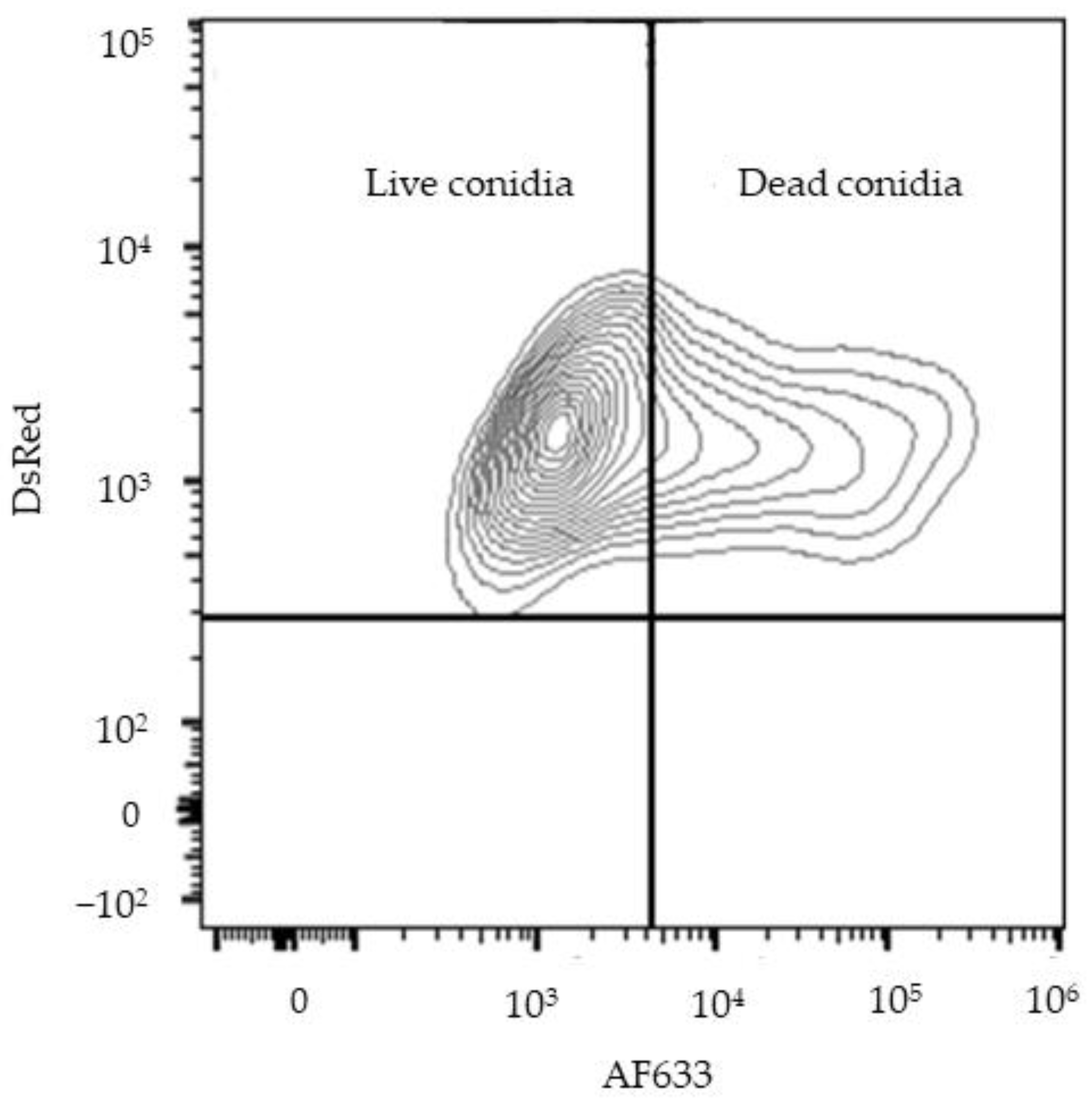
2.8. Flow Cytometry Methods
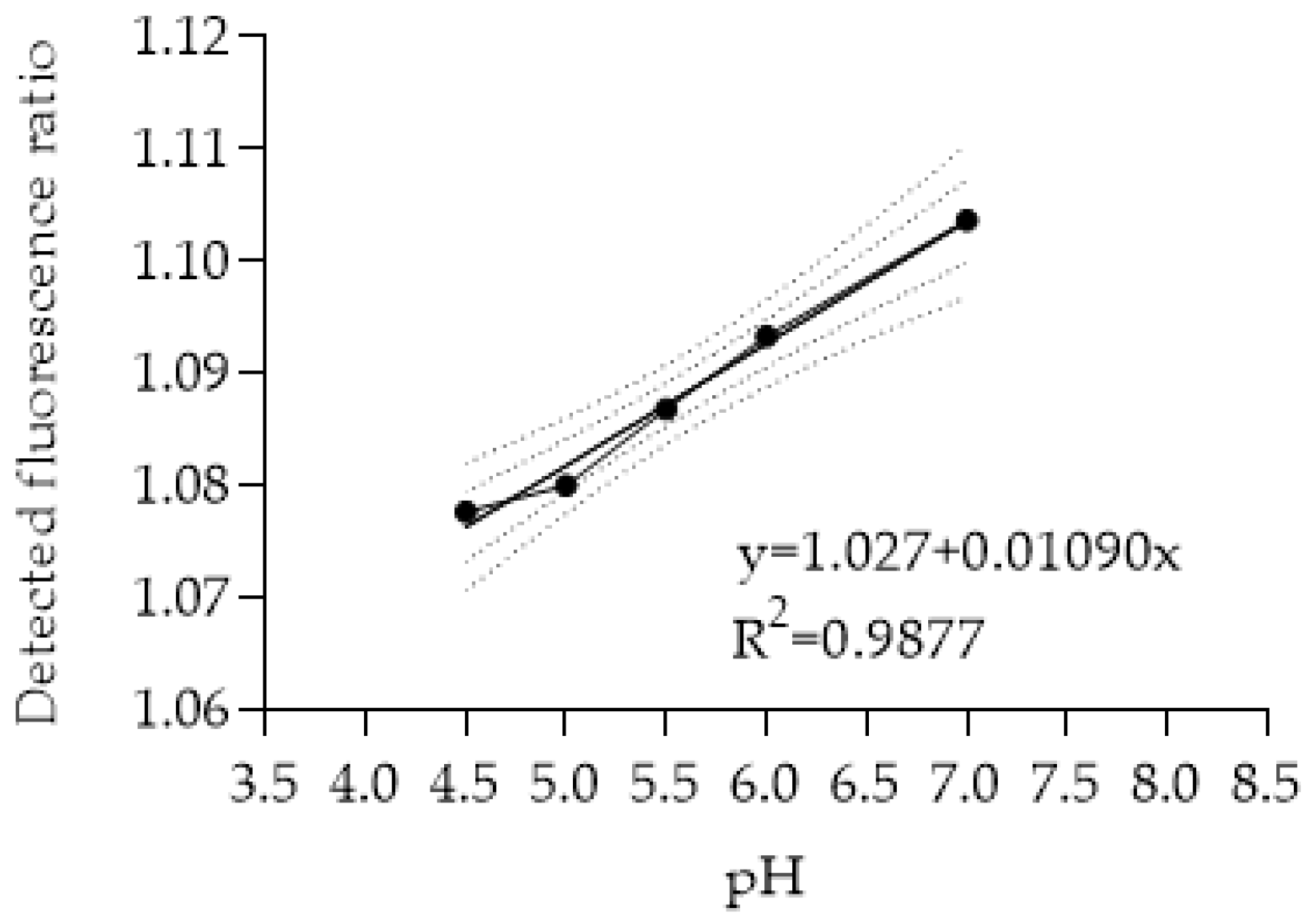
2.9. Determination of Lysosomal Alkalinization
2.10. Cellular pH Measurement
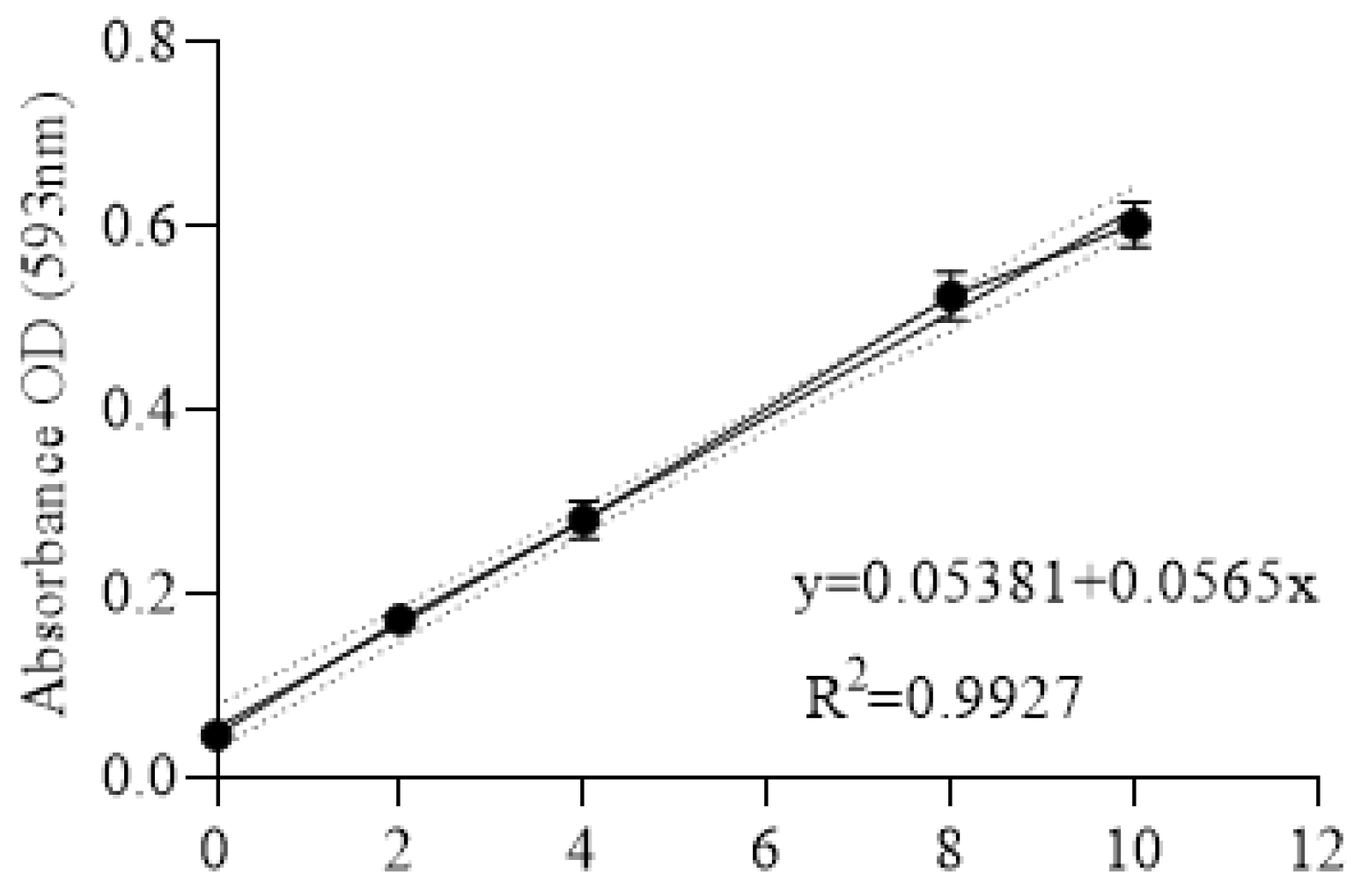
2.11. Iron Assay
2.12. pH Modulation Treatments
2.13. Statistics
3. Results
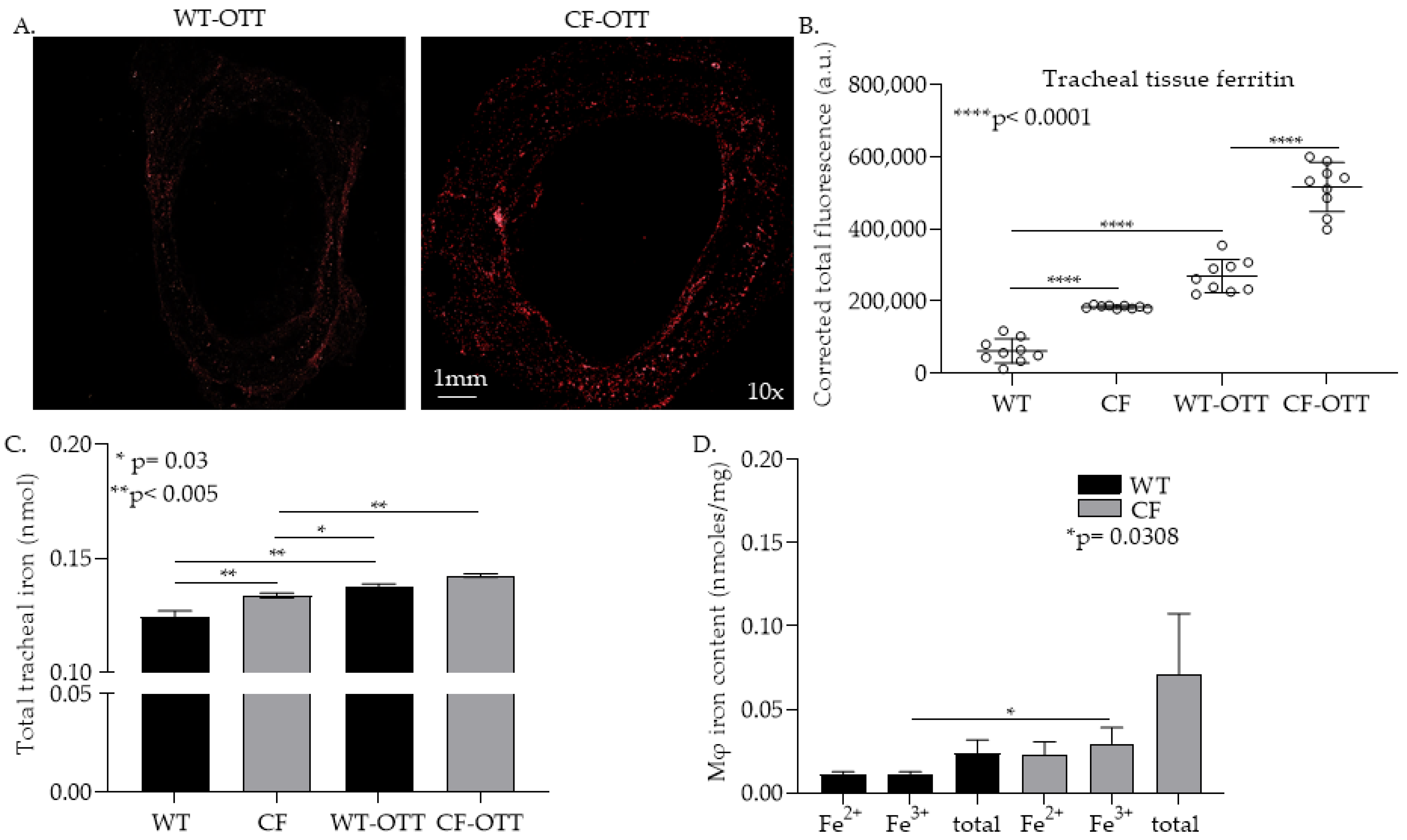
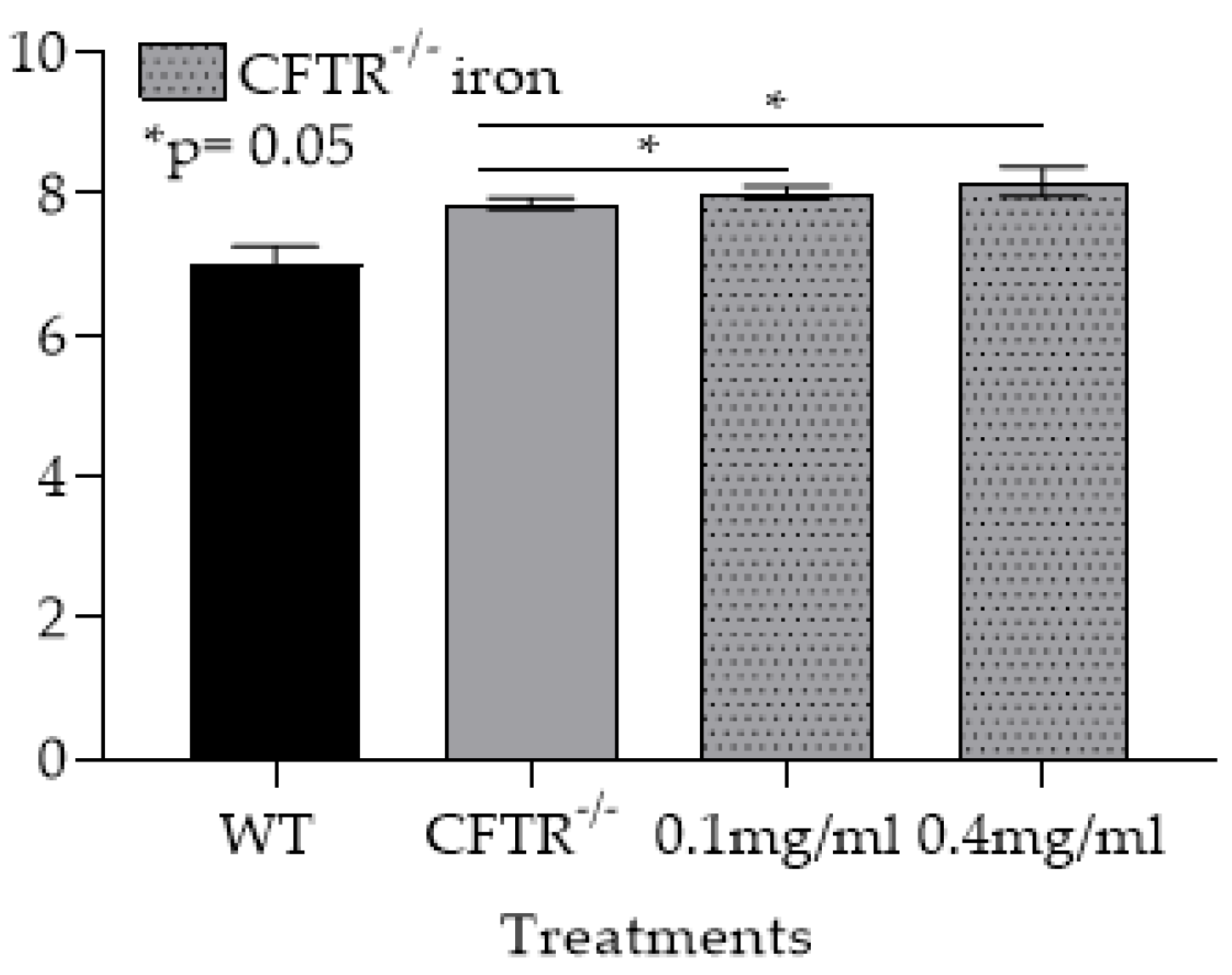
3.1. In the CF-OTT Model Both Tracheas and CFTR-/- mφs Had Higher Levels of Iron Compared to WT Control
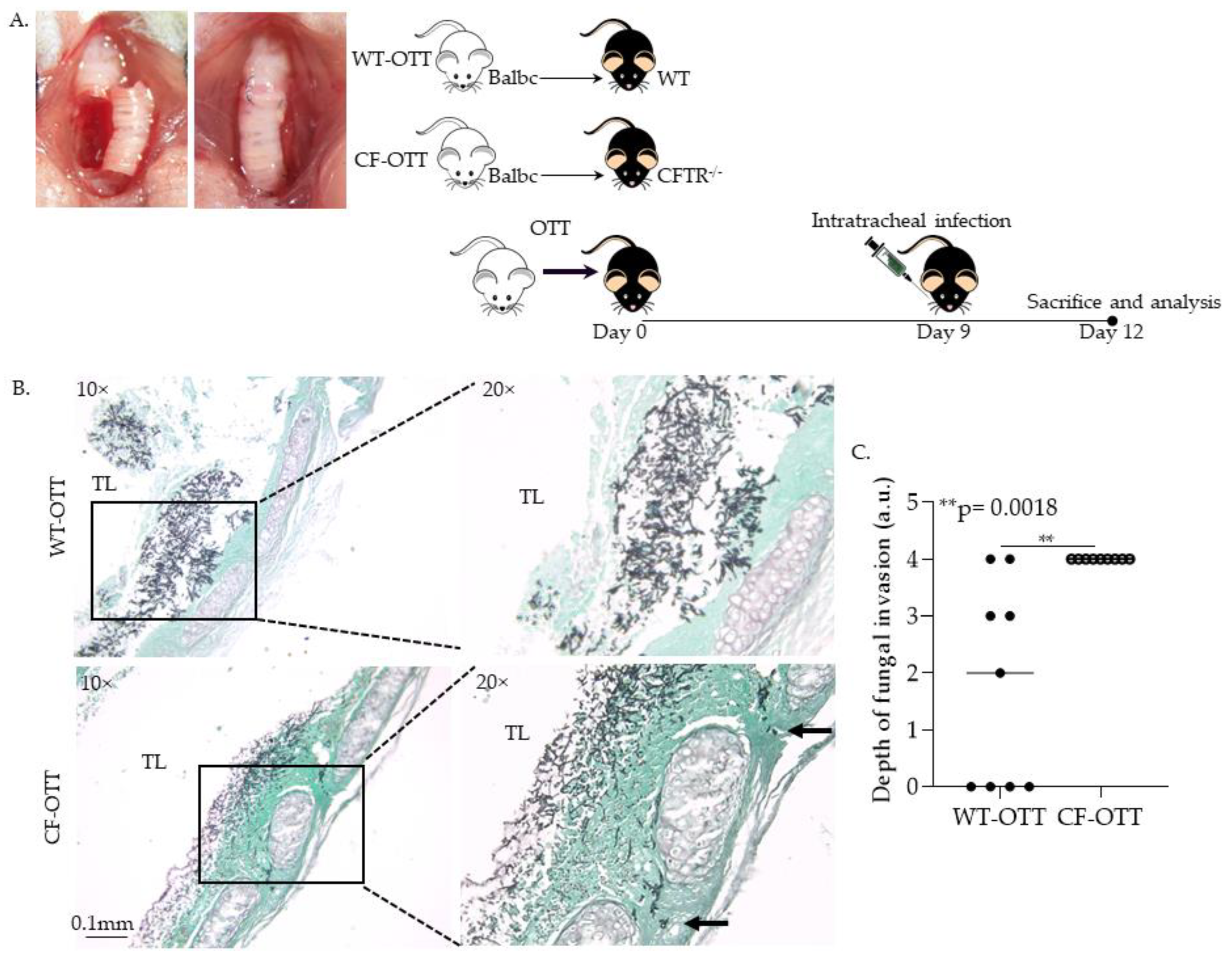
3.1.1. Af Was Significantly More Invasive in CF-OTT Recipients
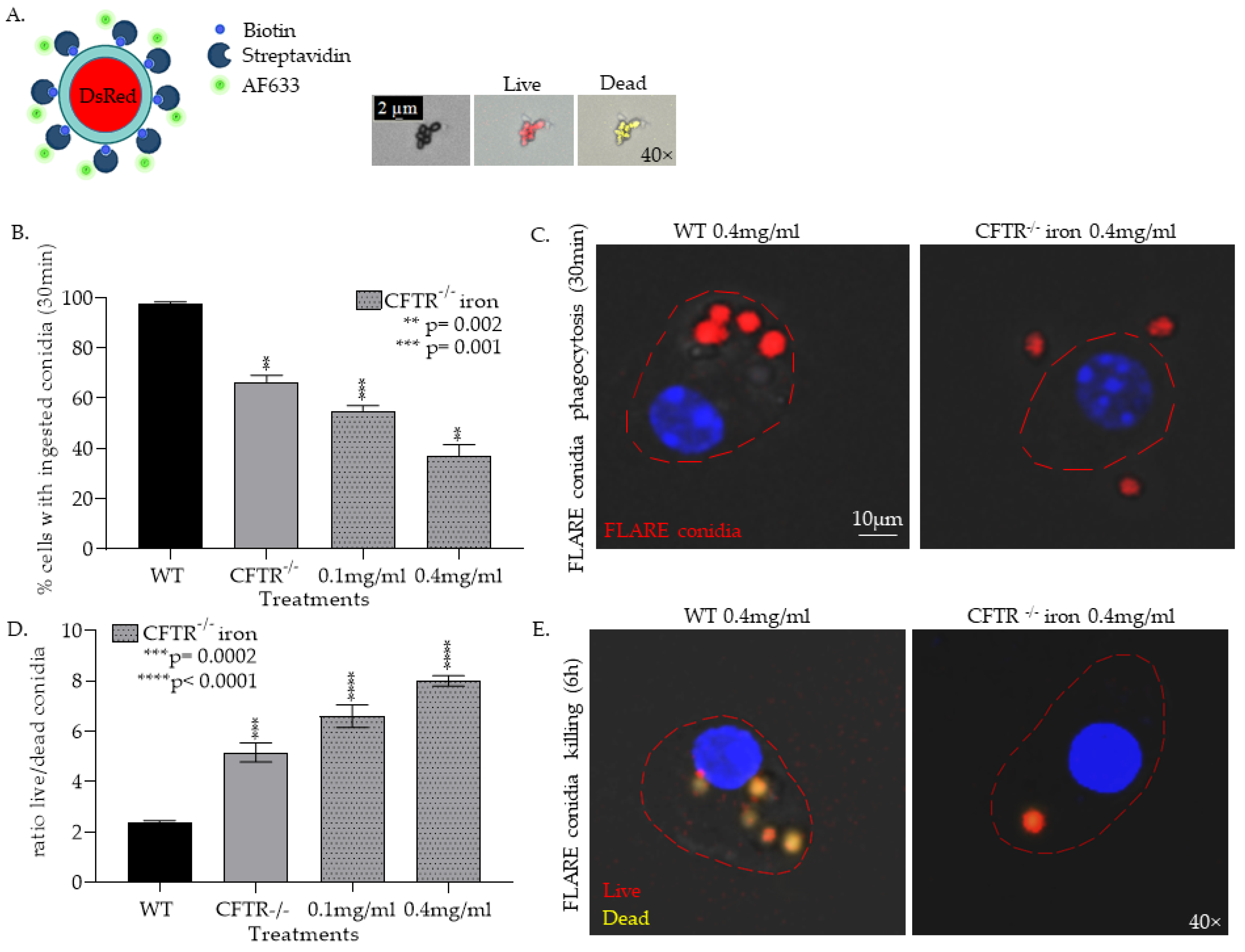
3.1.2. CFTR−/− mφs Have Impaired Ability to Phagocytose and Kill Af Conidia and Are Further Impaired by the Addition of Iron
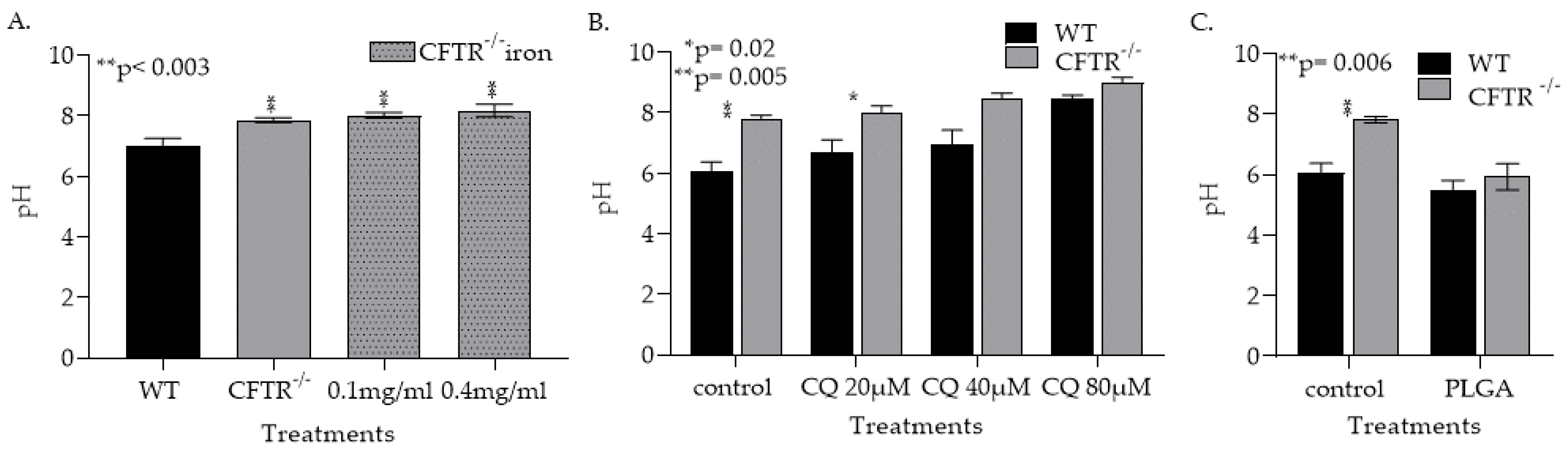
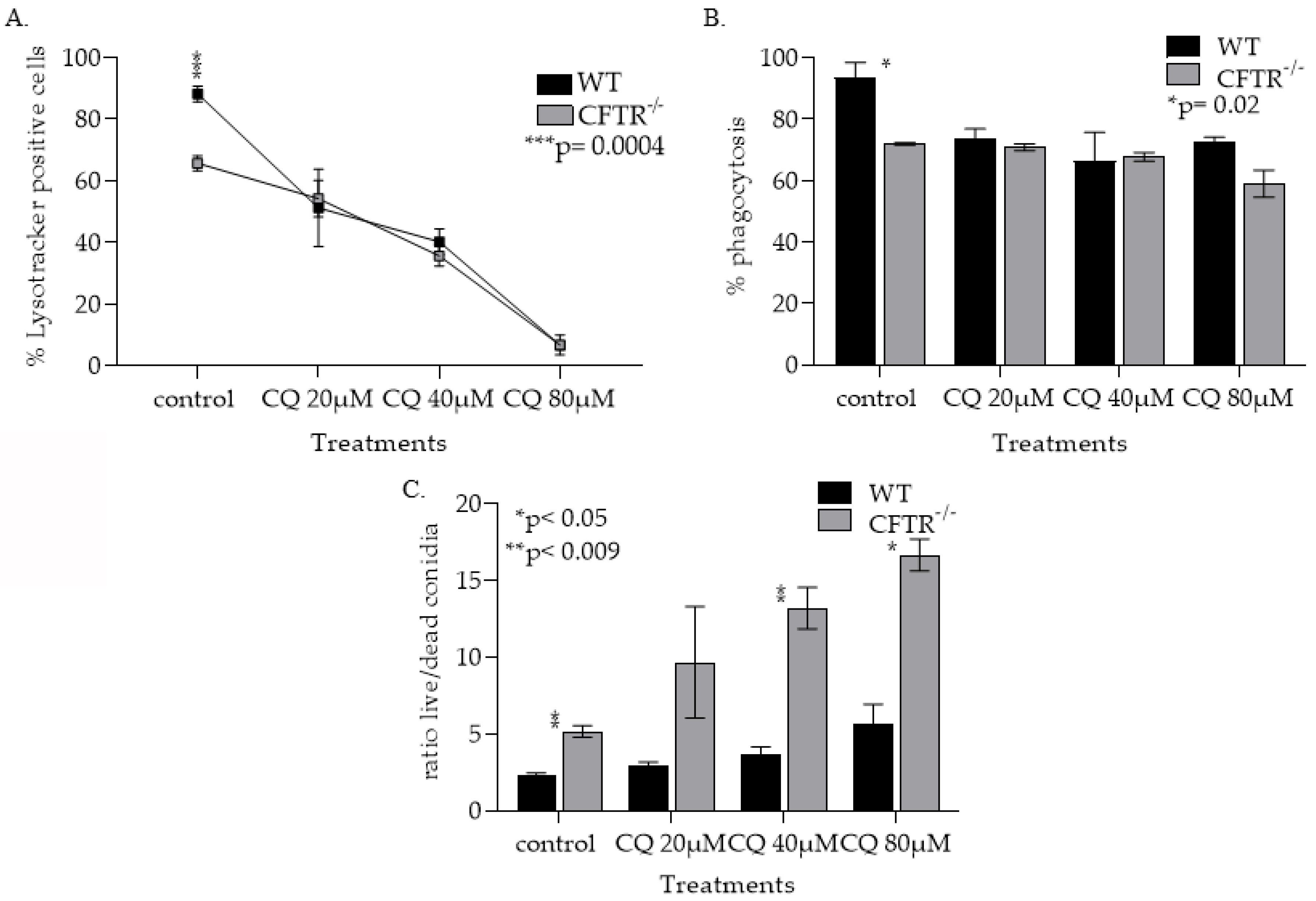
3.1.3. CFTR Deficiency, Iron and Treatment with Chloroquin Increase mφ Lysosomal pH
3.1.4. CQ Treatment Increased Lysosomal pH in Both WT and CFT−/− mφs and Lowering Their Ability to Phagocytose Kill Af Conidia
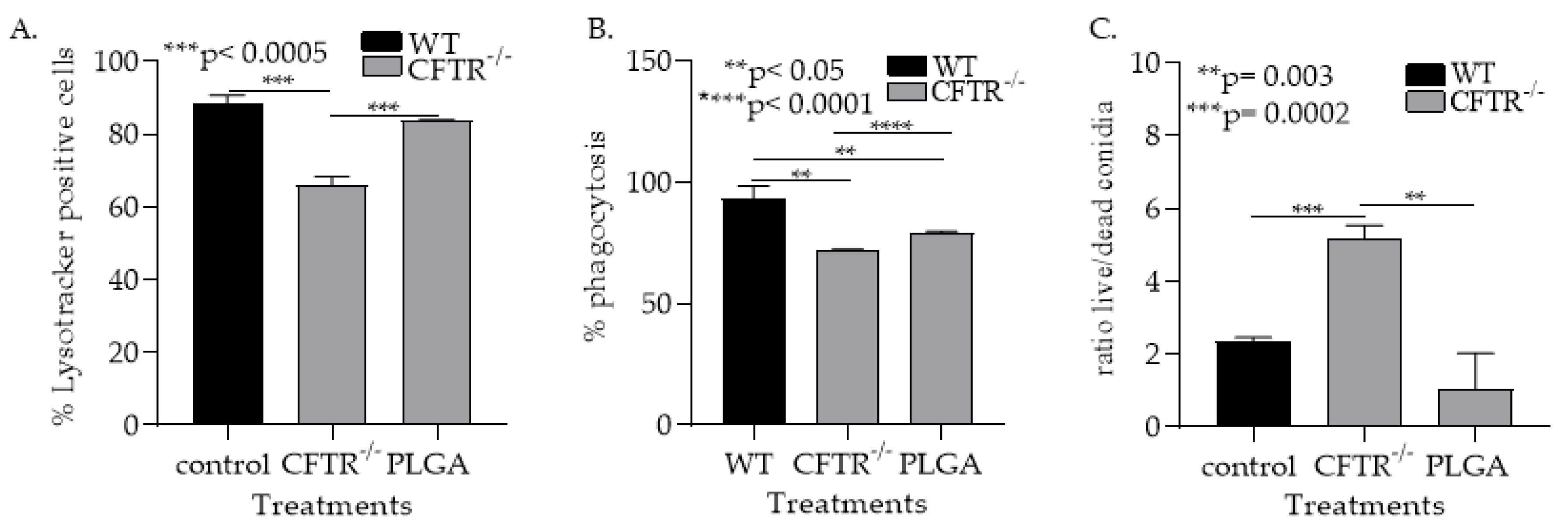
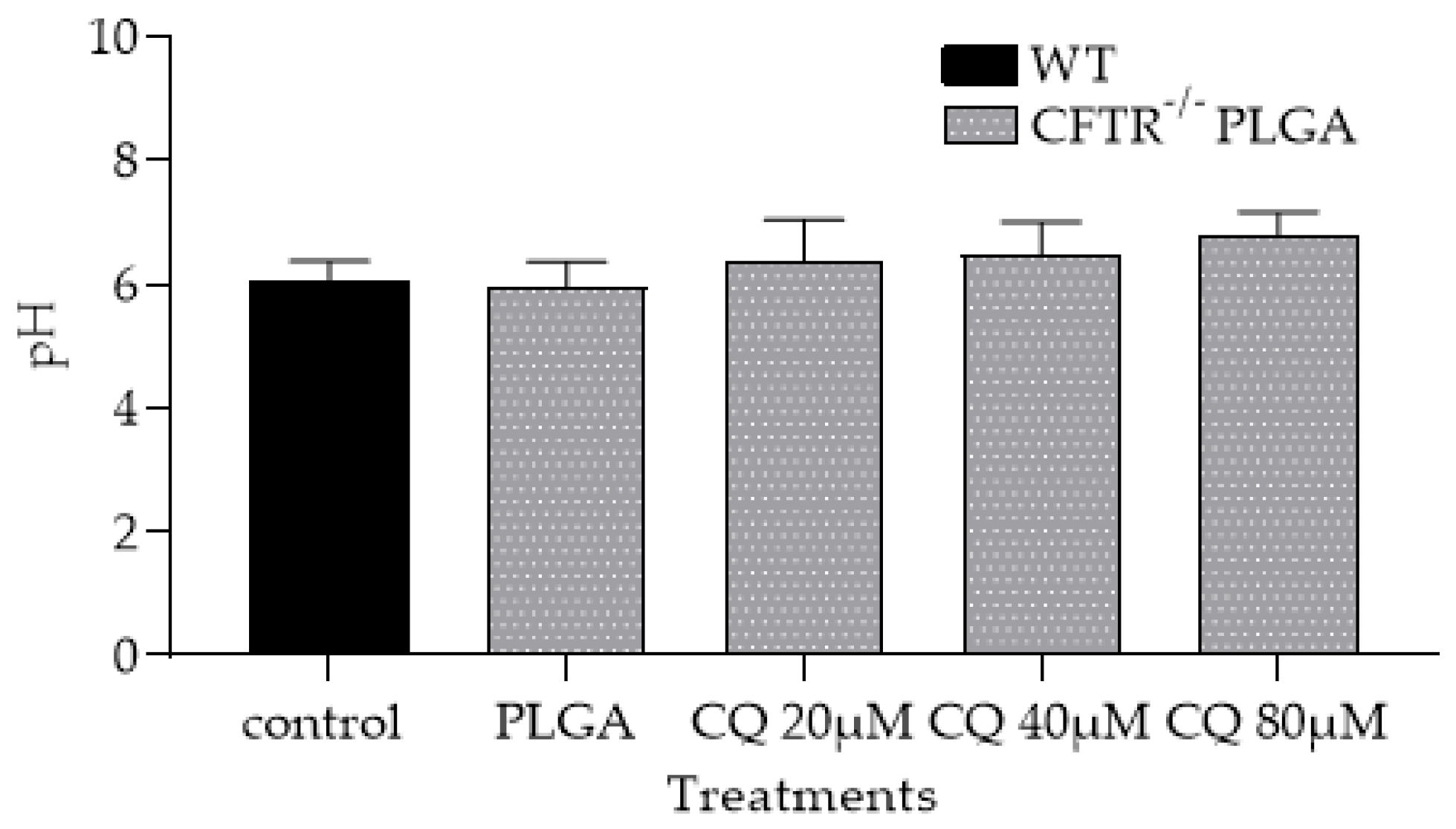
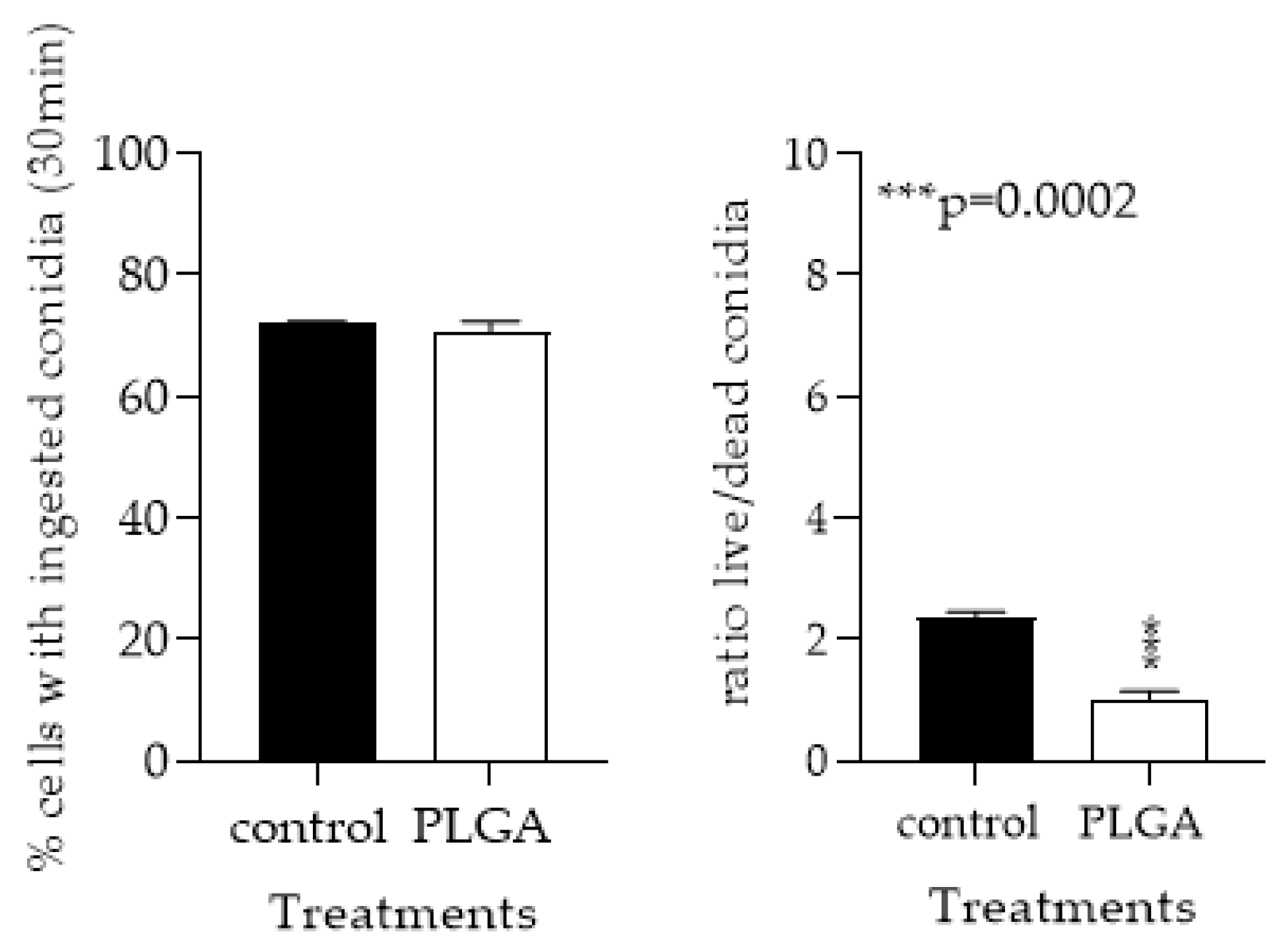
3.1.5. PLGA NPs Restore CFTR−/− mφ Lysosomal Acidity and Ability to Kill Af Conidia
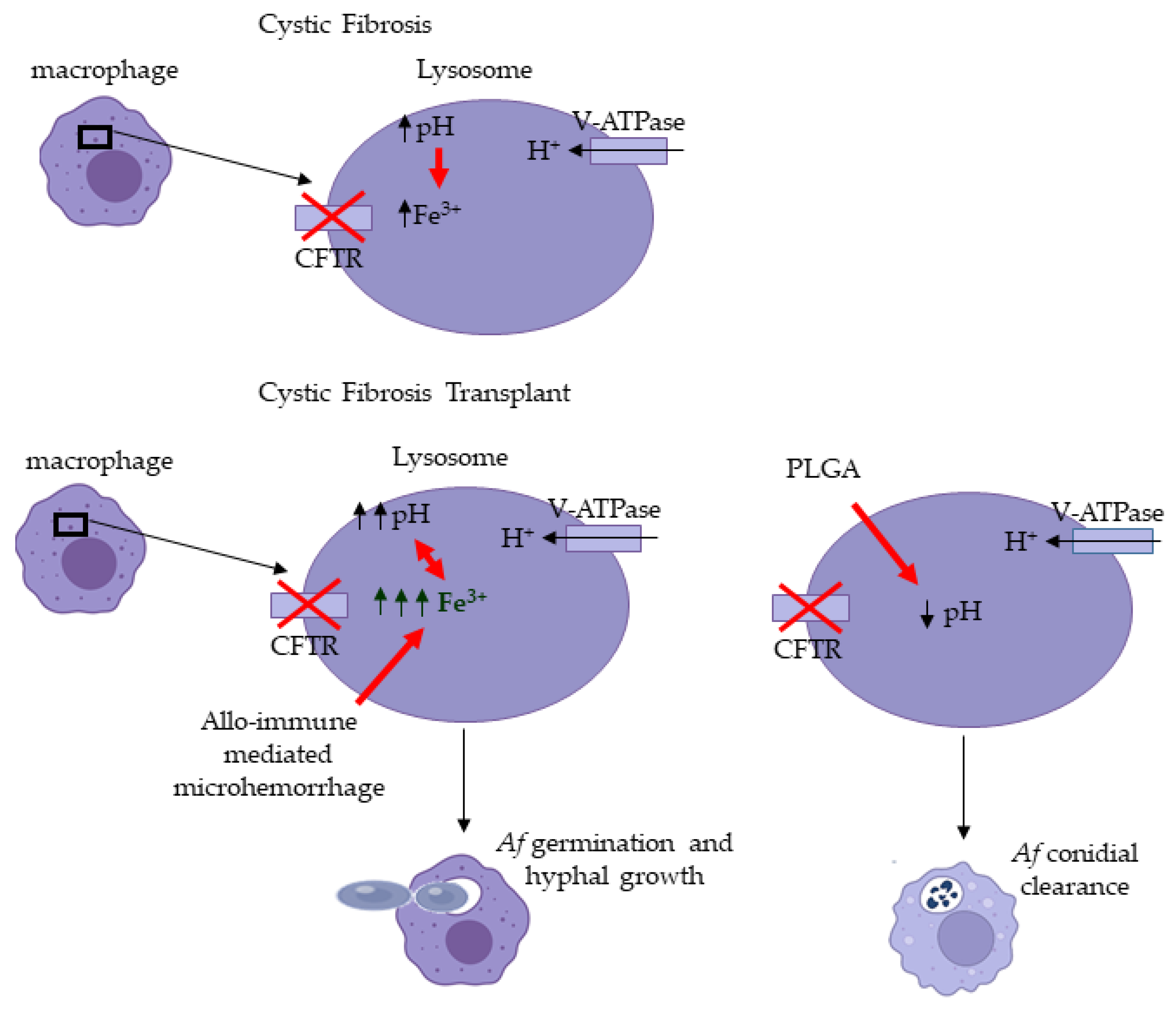
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [Green Version]
- Chabi, M.L.; Goracci, A.; Roche, N.; Paugam, A.; Lupo, A.; Revel, M.P. Pulmonary aspergillosis. Diagn. Interv. Imaging 2015, 96, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Alawi, A.; Ryan, C.F.; Flint, J.D.; Müller, N.L. Aspergillus-Related Lung Disease. Can. Respir. J. 2005, 12, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, M.; Silva, J.T.; San-Juan, R.; de Dios, B.; García-Luján, R.; López-Medrano, F.; Lizasoain, M.; Aguado, J.M. Aspergillus Tracheobronchitis: Report of 8 Cases and Review of the Literature. Medicine 2012, 91, 261–273. [Google Scholar] [CrossRef]
- Agarwal, R.; Sehgal, I.S.; Dhooria, S.; Aggarwal, A.N. Challenging cases in fungal asthma. Med. Mycol. 2019, 57, S110–S117. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, A.P. Allergic bronchopulmonary aspergillosis in asthma. Expert Rev. Clin. Immunol. 2017, 13, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takazono, T.; Sheppard, D.C. Aspergillus in chronic lung disease: Modeling what goes on in the airways. Med. Mycol. 2016, 55, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassetti, M.; Bouza, E. Invasive mould infections in the ICU setting: Complexities and solutions. J. Antimicrob. Chemother. 2017, 72, i39–i47. [Google Scholar] [CrossRef] [Green Version]
- Lamoth, F.; Calandra, T. Early diagnosis of invasive mould infections and disease. J. Antimicrob. Chemother. 2017, 72, i19–i28. [Google Scholar] [CrossRef] [Green Version]
- Samanta, P.; Clancy, C.J.; Nguyen, M.H. Fungal infections in lung transplantation. J. Thorac. Dis. 2021, 13, 6695–6707. [Google Scholar] [CrossRef]
- Kosmidis, C.; Denning, D.W. The clinical spectrum of pulmonary aspergillosis. Thorax 2015, 70, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carsin, A.; Romain, T.; Ranque, S.; Reynaud-Gaubert, M.; Dubus, J.-C.; Mège, J.-L.; Vitte, J. Aspergillus fumigatus in cystic fibrosis: An update on immune interactions and molecular diagnostics in allergic bronchopulmonary aspergillosis. Allergy 2017, 72, 1632–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.; Brunel, S.F.; Warris, A. Aspergillus infections in cystic fibrosis. J. Infect. 2016, 72, S50–S55. [Google Scholar] [CrossRef] [PubMed]
- Geltner, C.; Lass-Flörl, C. Invasive pulmonary Aspergillosis in organ transplants—Focus on lung transplants. Respir. Investig. 2016, 54, 76–84. [Google Scholar] [CrossRef]
- Pasupneti, S.; Manouvakhova, O.; Nicolls, M.R.; Hsu, J.L. Aspergillus-related pulmonary diseases in lung transplantation. Med. Mycol. 2017, 55, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.R.; Denning, D.W.; Marshall, S.E.; Ross, D.J.; Berry, G.; Lewiston, N.J.; Stevens, D.A.; Theodore, J. Ulcerative Tracheobronchitis after Lung Transplantation: A New Form of Invasive Aspergillosis. Am. Rev. Respir Dis. 1991, 144, 552–556. [Google Scholar] [CrossRef]
- Hsu, J.L.; Khan, M.A.; Sobel, R.A.; Jiang, X.; Clemons, K.V.; Nguyen, T.T.; Stevens, D.A.; Martinez, M.; Nicolls, M.R. Aspergillus fumigatus invasion increases with progressive airway ischemia. PLoS ONE 2013, 8, e77136. [Google Scholar] [CrossRef]
- Gregson, A.L. Infectious Triggers of Chronic Lung Allograft Dysfunction. Curr. Infect. Dis. Rep. 2016, 18, 21. [Google Scholar] [CrossRef]
- Robbins, N.; Wright, G.D.; Cowen, L.E. Antifungal Drugs: The Current Armamentarium and Development of New Agents. Microbiol. Spectr. 2016, 4, 4–5. [Google Scholar] [CrossRef]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef]
- Kriegl, L.; Boyer, J.; Egger, M.; Hoenigl, M. Antifungal stewardship in solid organ transplantation. Transpl. Infect. Dis. 2022. [Google Scholar] [CrossRef]
- Iversen, M.; Burton, C.M.; Vand, S.; Skovfoged, L.; Carlsen, J.; Milman, N.; Andersen, C.B.; Rasmussen, M.; Tvede, M. Aspergillus infection in lung transplant patients: Incidence and prognosis. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Warris, A.; Bercusson, A.; Armstrong-James, D. Aspergillus colonization and antifungal immunity in cystic fibrosis patients. Med. Mycol. 2019, 57, S118–S126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Worley, S.; Mallory, G.B., Jr.; Arrigain, S.; Robertson, J.; Schecter, M.G.; Elidemir, O.; Danziger-Isakov, L.A. Fungal infections in pediatric lung transplant recipients: Colonization and invasive disease. J. Heart Lung Transpl. 2009, 28, 1226–1230. [Google Scholar] [CrossRef] [Green Version]
- Salehi, S.; Reed, E.F. The divergent roles of macrophages in solid organ transplantation. Curr. Opin. Organ Transpl. 2015, 20, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Londino, J.D.; Matalon, S. Chloride secretion across adult alveolar epithelial cells contributes to cardiogenic edema. Proc. Natl. Acad. Sci. USA 2013, 110, 10055–10056. [Google Scholar] [CrossRef] [Green Version]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate Immun. 2016, 8, 550–563. [Google Scholar] [CrossRef]
- Swahn, H.; Harris, A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes 2019, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Dagenais, T.R.T.; Keller, N.P. Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis. Clin. Microbiol. Rev. 2009, 22, 447–465. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Warris, A.; Van der Meer, J.W.M.; Fenton, M.J.; Verver-Janssen, T.J.G.; Jacobs, L.E.H.; Andresen, T.; Verweij, P.E.; Kullberg, B.J. Aspergillus fumigatus Evades Immune Recognition during Germination through Loss of Toll-Like Receptor-4-Mediated Signal Transduction. J. Infect. Dis. 2003, 188, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.L.; Metz, A.E.; Horn, D.; Schoeb, T.R.; Hewitt, M.M.; Schwiebert, L.M.; Faro-Trindade, I.; Brown, G.D.; Steele, C. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 2009, 182, 4938–4946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalçin, E.; Talim, B.; Ozçelik, U.; Doğru, D.; Cobanoğlu, N.; Pekcan, S.; Kiper, N. Does defective apoptosis play a role in cystic fibrosis lung disease? Arch. Med. Res. 2009, 40, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Haggie, P.M.; Verkman, A.S. Cystic fibrosis transmembrane conductance regulator-independent phagosomal acidification in macrophages. J. Biol. Chem. 2007, 282, 31422–31428. [Google Scholar] [CrossRef] [Green Version]
- Law, S.M.; Stanfield, S.J.; Hardisty, G.R.; Dransfield, I.; Campbell, C.J.; Gray, R.D. Human cystic fibrosis monocyte derived macrophages display no defect in acidification of phagolysosomes when measured by optical nanosensors. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2020, 19, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Grassmé, H.; Döring, G.; Gulbins, E. Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J. Immunol. 2010, 184, 5104–5111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Reyna, S.; Holbrook, J.; Jarosz-Griffiths, H.H.; Peckham, D.; McDermott, M.F. Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations. Cell Mol. Life Sci. 2020, 77, 4485–4503. [Google Scholar] [CrossRef]
- Orlando, V.; Morin, G.; Laffont, A.; Lénart, D.; Solórzano Barrera, C.; Mustafy, T.; Sankhe, S.; Villemure, I.; Mailhot, G. CFTR deletion affects mouse osteoblasts in a gender-specific manner. J. Cell. Physiol. 2020, 235, 6736–6753. [Google Scholar] [CrossRef]
- Hazlett, H.F.; Hampton, T.H.; Aridgides, D.S.; Armstrong, D.A.; Dessaint, J.A.; Mellinger, D.L.; Nymon, A.B.; Ashare, A. Altered iron metabolism in cystic fibrosis macrophages: The impact of CFTR modulators and implications for Pseudomonas aeruginosa survival. Sci. Rep. 2020, 10, 10935. [Google Scholar] [CrossRef]
- Di Pietro, C.; Zhang, P.-X.; O’Rourke, T.K.; Murray, T.S.; Wang, L.; Britto, C.J.; Koff, J.L.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Ezrin links CFTR to TLR4 signaling to orchestrate anti-bacterial immune response in macrophages. Sci. Rep. 2017, 7, 10882. [Google Scholar] [CrossRef]
- Schupp, J.C.; Khanal, S.; Gomez, J.L.; Sauler, M.; Adams, T.S.; Chupp, G.L.; Yan, X.; Poli, S.; Zhao, Y.; Montgomery, R.R.; et al. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 202, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell. Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggie, P.M.; Verkman, A.S. Unimpaired lysosomal acidification in respiratory epithelial cells in cystic fibrosis. J. Biol. Chem. 2009, 284, 7681–7686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.L.; Manouvakhova, O.V.; Clemons, K.V.; Inayathullah, M.; Tu, A.B.; Sobel, R.A.; Tian, A.; Nazik, H.; Pothineni, V.R.; Pasupneti, S.; et al. Microhemorrhage-associated tissue iron enhances the risk for Aspergillus fumigatus invasion in a mouse model of airway transplantation. Sci. Transl. Med. 2018, 10, eaag2616. [Google Scholar] [CrossRef] [Green Version]
- Baz, M.A.; Ghio, A.J.; Roggli, V.L.; Tapson, V.F.; Piantadosi, C.A. Iron accumulation in lung allografts after transplantation. Chest 1997, 112, 435–439. [Google Scholar] [CrossRef]
- Saha, B.K.; Chong, W.H. Lung transplant to manage end-stage lung disease due to idiopathic pulmonary hemosiderosis: A review of the literature. Respir. Investig. 2022, 60, 82–89. [Google Scholar] [CrossRef]
- Meng, F.; Fleming, B.A.; Jia, X.; Rousek, A.A.; Mulvey, M.A.; Ward, D.M. Lysosomal iron recycling in mouse macrophages is dependent upon both LcytB and Steap3 reductases. Blood Adv. 2022, 6, 1692–1707. [Google Scholar] [CrossRef]
- Nairz, M.; Theurl, I.; Swirski, F.K.; Weiss, G. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflug. Arch. 2017, 469, 397–418. [Google Scholar] [CrossRef] [Green Version]
- Barriere, H.; Bagdany, M.; Bossard, F.; Okiyoneda, T.; Wojewodka, G.; Gruenert, D.; Radzioch, D.; Lukacs, G.L. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol. Biol. Cell. 2009, 20, 3125–3141. [Google Scholar] [CrossRef] [Green Version]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 29. [Google Scholar] [CrossRef]
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaëc, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Massip-Copiz, M.M.; Santa-Coloma, T.A. Extracellular pH and lung infections in cystic fibrosis. Eur. J. Cell. Biol. 2018, 97, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Massey, M.K.; Reiterman, M.J.; Mourad, J.; Luckie, D.B. Is CFTR an exchanger?: Regulation of HCO(3) (-)Transport and extracellular pH by CFTR. Biochem. Biophys Rep. 2021, 25, 100863. [Google Scholar] [CrossRef] [PubMed]
- Jhingran, A.; Mar, K.B.; Kumasaka, D.K.; Knoblaugh, S.E.; Ngo, L.Y.; Segal, B.H.; Iwakura, Y.; Lowell, C.A.; Hamerman, J.A.; Lin, X.; et al. Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Rep. 2012, 2, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.K.; Mendez, O.; Bowen, S.; Mohanakumar, T. Isolation and In Vitro Culture of Murine and Human Alveolar Macrophages. JoVE 2018, 20, 57287. [Google Scholar] [CrossRef]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14.1.1–14.1.14. [Google Scholar] [CrossRef]
- Ibrahim-Granet, O.; Philippe, B.; Boleti, H.; Boisvieux-Ulrich, E.; Grenet, D.; Stern, M.; Latgé, J.P. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 2003, 71, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Thywißen, A.; Heinekamp, T.; Dahse, H.-M.; Schmaler-Ripcke, J.; Nietzsche, S.; Zipfel, P.F.; Brakhage, A.A. Conidial Dihydroxynaphthalene Melanin of the Human Pathogenic Fungus Aspergillus fumigatus Interferes with the Host Endocytosis Pathway. Front. Microbiol. 2011, 2, 96. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.V.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H.; et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol. Cell 2020, 77, 645–655. [Google Scholar] [CrossRef]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef]
- Zeng, J.; Shirihai, O.S.; Grinstaff, M.W. Modulating lysosomal pH: A molecular and nanoscale materials design perspective. J. Life Sci. 2020, 2, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Martin, A.; Han, X.; Shirihai, O.S.; Grinstaff, M.W. Biodegradable PLGA Nanoparticles Restore Lysosomal Acidity and Protect Neural PC-12 Cells against Mitochondrial Toxicity. Ind. Eng. Chem. Res. 2019, 58, 13910–13917. [Google Scholar] [CrossRef]
- Nunley, D.R.; Gal, A.A.; Vega, J.D.; Perlino, C.; Smith, P.; Lawrence, E.C. Saprophytic fungal infections and complications involving the bronchial anastomosis following human lung transplantation. Chest 2002, 122, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunley, D.R.; Ohori, P.; Grgurich, W.F.; Iacono, A.T.; Williams, P.A.; Keenan, R.J.; Dauber, J.H. Pulmonary aspergillosis in cystic fibrosis lung transplant recipients. Chest 1998, 114, 1321–1329. [Google Scholar] [CrossRef] [Green Version]
- Helmi, M.; Love, R.B.; Welter, D.; Cornwell, R.D.; Meyer, K.C. Aspergillus infection in lung transplant recipients with cystic fibrosis: Risk factors and outcomes comparison to other types of transplant recipients. Chest 2003, 123, 800–808. [Google Scholar] [CrossRef]
- Stites, S.W.; Walters, B.; O’Brien-Ladner, A.R.; Bailey, K.; Wesselius, L.J. Increased iron and ferritin content of sputum from patients with cystic fibrosis or chronic bronchitis. Chest 1998, 114, 814–819. [Google Scholar] [CrossRef]
- Stites, S.W.; Plautz, M.W.; Bailey, K.; O’Brien-Ladner, A.R.; Wesselius, L.J. Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am. J. Respir. Crit. Care Med. 1999, 160, 796–801. [Google Scholar] [CrossRef]
- Reid, D.W.; Carroll, V.; O’May, C.; Champion, A.; Kirov, S.M. Increased airway iron as a potential factor in the persistence of Pseudomonas aeruginosa infection in cystic fibrosis. Eur. Respir J. 2007, 30, 286–292. [Google Scholar] [CrossRef]
- Swanson, J. CFTR: Helping to acidify macrophage lysosomes. Nat. Cell Biol. 2006, 8, 908–909. [Google Scholar] [CrossRef]
- Haggie, P.M.; Verkman, A.S. Defective organellar acidification as a cause of cystic fibrosis lung disease: Reexamination of a recurring hypothesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L859–L867. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Barasch, J.; Kiss, B.; Prince, A.; Saiman, L.; Gruenert, D.; AI-Awqati, Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature 1991, 352, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Botelho, R.J.; Grinstein, S. Phagosome maturation: Aging gracefully. Biochem. J. 2002, 366, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Delves, P.J.; Roitt, I.M. The Immune System. N. Engl. J. Med. 2000, 343, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Gombart, Z.J.; Chen, J.W. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: Possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasylnka, J.A.; Moore, M.M. Aspergillus fumigatus conidia survive and germinate in acidic organelles of A549 epithelial cells. J. Cell Sci. 2003, 116, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Rammaert, B.; Jouvion, G.; de Chaumont, F.; Garcia-Hermoso, D.; Szczepaniak, C.; Renaudat, C.; Olivo-Marin, J.C.; Chrétien, F.; Dromer, F.; Bretagne, S. Absence of Fungal Spore Internalization by Bronchial Epithelium in Mouse Models Evidenced by a New Bioimaging Approach and Transmission Electronic Microscopy. Am. J. Pathol. 2015, 185, 2421–2430. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthaiou, E.I.; Chiu, W.; Conrad, C.; Hsu, J. Macrophage Lysosomal Alkalinization Drives Invasive Aspergillosis in a Mouse Cystic Fibrosis Model of Airway Transplantation. J. Fungi 2022, 8, 751. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070751
Matthaiou EI, Chiu W, Conrad C, Hsu J. Macrophage Lysosomal Alkalinization Drives Invasive Aspergillosis in a Mouse Cystic Fibrosis Model of Airway Transplantation. Journal of Fungi. 2022; 8(7):751. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070751
Chicago/Turabian StyleMatthaiou, Efthymia Iliana, Wayland Chiu, Carol Conrad, and Joe Hsu. 2022. "Macrophage Lysosomal Alkalinization Drives Invasive Aspergillosis in a Mouse Cystic Fibrosis Model of Airway Transplantation" Journal of Fungi 8, no. 7: 751. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070751