
Genome Assembly and Genetic Traits of the Pleuromutilin-Producer Clitopilus passeckerianus DSM1602

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clitopilus passeckerianus Growth and DNA Isolation
2.2. Intron/Exon Analysis of Ple Genes in C. passeckerianus
2.3. Nanopore Library Preparation and GridION Sequencing
2.4. Illumina Library Preparation and MiSeq Sequencing
2.5. Base Calling, Reads Processing, and Assembly
2.6. Gene Prediction and Genome Annotation
2.7. Comparative Genome Analyses and Phylogenetic Analysis
2.8. Identification and Analysis of the Mitogenome
2.9. Construction of the Yeast-PleBGCs
3. Results and Discussion
3.1. Generation of a High-Qualitiy Draft Genome of Clitopilus passeckerianus DSM1602
3.2. The C. passeckerianus Genome Has a Diploid Character
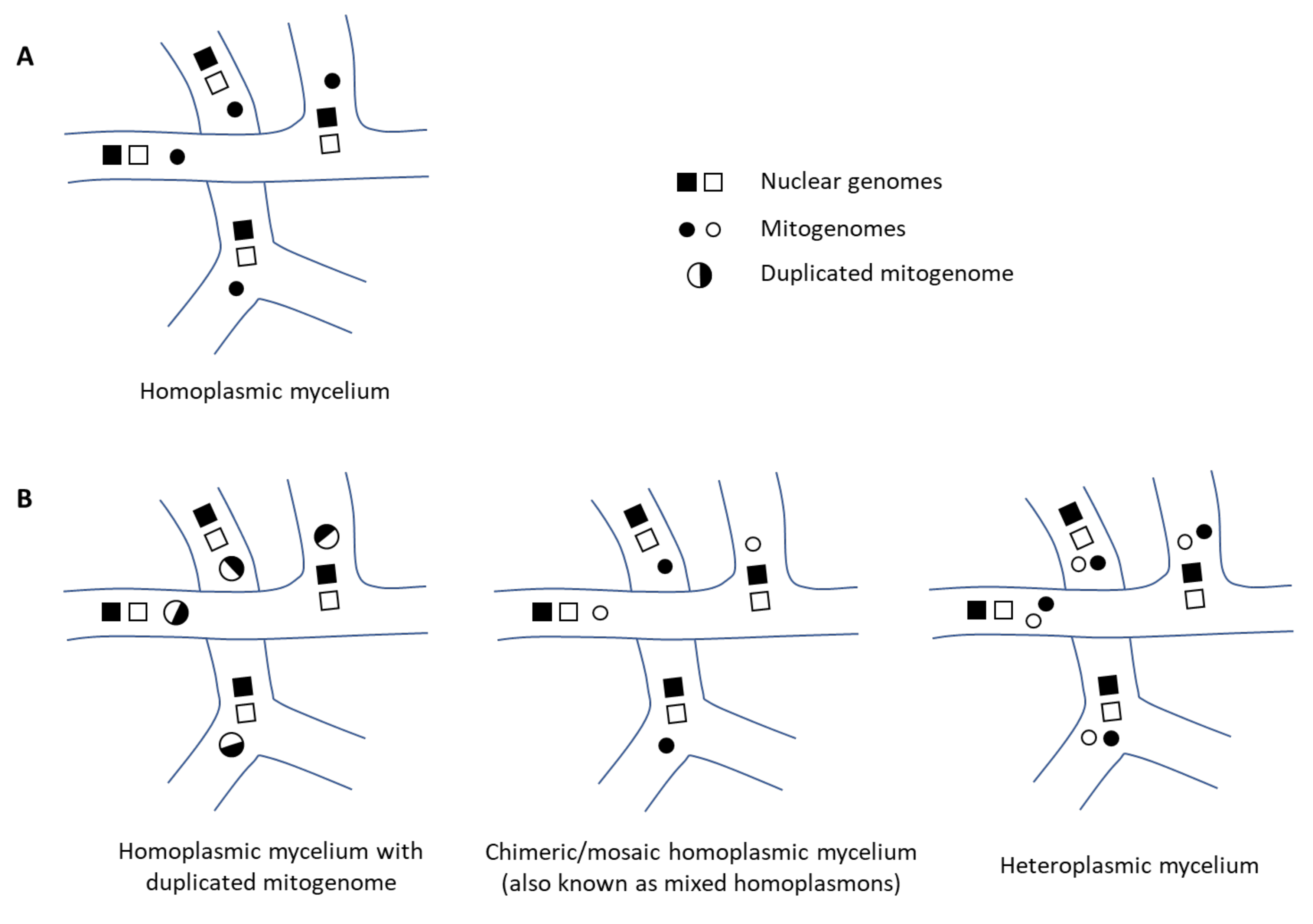
3.3. The Mitogenome Appears in Duplicate
3.4. Phylogeny and Comparative Genome Analysis Confirms Genetic Proximity of Clitopilus spp.
3.5. Clitopilus passeckerianus Harbors a Huge Genetic Potential for Secondary Metabolites
3.6. Two Pleuromutilin Biosynthesis Gene Clusters Present in C. passeckerianus
3.7. Refactoring the pleBGC for Heterolgous Expression in Saccharomyces cerevisiae
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumla, J.; Suwannarach, N.; Sungpalee, W.; Sri-Ngernyuang, K.; Lumyong, S. Clitopilus lampangensis (Agaricales, Entolomataceae), a new species from northern Thailand. MycoKeys 2019, 58, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Passecker, F. Ein neuer Unkrautpilz auf Champignonbeeten. (Pleurotus passeckerianus Pilát). Z. Pflanzenkrankh. (Pflanzenpathol.) Pflanzenschutz 1936, 46, 271–277. [Google Scholar]
- Lohrmann, F. Katzenohrräsling—Clitopilus passeckerianus. In Pilzbestimmer. Available online: https://pilzbestimmer.de/Detailed/10833.html (accessed on 14 April 2022).
- Bailey, A.M.; Alberti, F.; Kilaru, S.; Collins, C.M.; de Mattos-Shipley, K.; Hartley, A.J.; Hayes, P.; Griffin, A.; Lazarus, C.M.; Cox, R.J.; et al. Identification and manipulation of the pleuromutilin gene cluster from Clitopilus passeckerianus for increased rapid antibiotic production. Sci. Rep. 2016, 6, 25202. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.J.; de Mattos-Shipley, K.; Collins, C.M.; Kilaru, S.; Foster, G.D.; Bailey, A.M. Investigating pleuromutilin-producing Clitopilus species and related basidiomycetes. FEMS Microbiol. Lett. 2009, 297, 24–30. [Google Scholar] [CrossRef]
- Kilaru, S.; Collins, C.M.; Hartley, A.J.; Bailey, A.M.; Foster, G.D. Establishing Molecular Tools for Genetic Manipulation of the Pleuromutilin-Producing Fungus Clitopilus passeckerianus. Appl. Environ. Microbiol. 2009, 75, 7196–7204. [Google Scholar] [CrossRef]
- de Mattos-Shipley, K.M.J.; Foster, G.D.; Bailey, A.M. Insights into the Classical Genetics of Clitopilus passeckerianus—The Pleuromutilin Producing Mushroom. Front. Microbiol. 2017, 8, 1056. [Google Scholar] [CrossRef]
- Smith, M.L.; Anderson, J.B. Mitochondrial DNAs of the fungus Armillaria ostoyae: Restriction map and length variation. Curr. Genet. 1994, 25, 545–553. [Google Scholar] [CrossRef]
- Smith, M.L.; Duchesne, L.C.; Bruhn, J.N.; Anderson, J.B. Mitochondrial genetics in a natural population of the plant pathogen armillaria. Genetics 1990, 126, 575–582. [Google Scholar] [CrossRef]
- Saville, B.J.; Yoell, H.; Anderson, J.B. Genetic exchange and recombination in populations of the root-infecting fungus Armillaria gallica. Mol. Ecol. 1996, 5, 485–497. [Google Scholar] [CrossRef]
- Kavanagh, F.; Hervey, A.; Robbins, W.J. Antibiotic Substances from Basidiomycetes. Proc. Natl. Acad. Sci. USA 1951, 37, 570–574. [Google Scholar] [CrossRef]
- Paukner, S.; Riedl, R. Pleuromutilins: Potent Drugs for Resistant Bugs—Mode of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 7, a027110. [Google Scholar] [CrossRef]
- Van Duijkeren, E.; Greko, C.; Pringle, M.; Baptiste, K.E.; Catry, B.; Jukes, H.; Moreno, M.; Pomba, M.C.M.F.; Pyörälä, S.; Rantala, M.; et al. Pleuromutilins: Use in food-producing animals in the European Union, development of resistance and impact on human and animal health. J. Antimicrob. Chemother. 2014, 69, 2022–2031. [Google Scholar] [CrossRef]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2008, 41, 1091–1099. [Google Scholar] [CrossRef]
- Yang, L.P.H.; Keam, S.J. Retapamulin. Drugs 2008, 68, 855–873. [Google Scholar] [CrossRef]
- Alberti, F.; Khairudin, K.; Venegas, E.R.; Davies, J.; Hayes, P.M.; Willis, C.L.; Bailey, A.M.; Foster, G.D. Heterologous expression reveals the biosynthesis of the antibiotic pleuromutilin and generates bioactive semi-synthetic derivatives. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Yamane, M.; Minami, A.; Liu, C.; Ozaki, T.; Takeuchi, I.; Tsukagoshi, T.; Tokiwano, T.; Gomi, K.; Oikawa, H. Biosynthetic Machinery of Diterpene Pleuromutilin Isolated from Basidiomycete Fungi. ChemBioChem 2017, 18, 2317–2322. [Google Scholar] [CrossRef]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2013, 42, D26–D31. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.A.; Otillar, R.; Riley, R.; Salamov, A.A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2013, 42, D699–D704. [Google Scholar] [CrossRef]
- Peng, L.; Shan, X.; Wang, Y.; Martin, F.; Vilgalys, R.; Yuan, Z. Hybrid Genome Assembly and Gene Repertoire of the Root Endophyte Clitopilus hobsonii QYL-10 (Entolomataceae, Agaricales, Basidiomycetes). Mol. Plant-Microbe Interactions 2021, 34, 711–714. [Google Scholar] [CrossRef]
- Ullmann, L.; Wibberg, D.; Busche, T.; Rückert, C.; Müsgens, A.; Kalinowski, J.; Blank, L.M. Seventeen Ustilaginaceae High-Quality Genome Sequences Allow Phylogenomic Analysis and Provide Insights into Secondary Metabolite Synthesis. J. Fungi 2022, 8, 269. [Google Scholar] [CrossRef]
- Wibberg, D.; Stadler, M.; Lambert, C.; Bunk, B.; Spröer, C.; Rückert, C.; Kalinowski, J.; Cox, R.J.; Kuhnert, E. High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers. 2020, 106, 7–28. [Google Scholar] [CrossRef]
- Wibberg, D.; Andersson, L.; Tzelepis, G.; Rupp, O.; Blom, J.; Jelonek, L.; Pühler, A.; Fogelqvist, J.; Varrelmann, M.; Schlüter, A.; et al. Genome analysis of the sugar beet pathogen Rhizoctonia solani AG2-2IIIB revealed high numbers in secreted proteins and cell wall degrading enzymes. BMC Genom. 2016, 17, 245. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Borodovsky, M.; Lomsadze, A. Eukaryotic Gene Prediction Using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinform. 2011, 35, 4.6.1–4.6.10. [Google Scholar] [CrossRef]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef]
- Rupp, O.; Becker, J.; Brinkrolf, K.; Timmermann, C.; Borth, N.; Pühler, A.; Noll, T.; Goesmann, A. Construction of a Public CHO Cell Line Transcript Database Using Versatile Bioinformatics Analysis Pipelines. PLoS ONE 2014, 9, e85568. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Dieckmann, M.A.; Beyvers, S.; Nkouamedjo-Fankep, R.C.; Hanel, P.H.G.; Jelonek, L.; Blom, J.; Goesmann, A. EDGAR3.0: Comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 2021, 49, W185–W192. [Google Scholar] [CrossRef]
- Wibberg, D.; Genzel, F.; Verwaaijen, B.; Blom, J.; Rupp, O.; Goesmann, A.; Zrenner, R.; Grosch, R.; Pühler, A.; Schlüter, A. Draft genome sequence of the potato pathogen Rhizoctonia solani AG3-PT isolate Ben3. Arch. Microbiol. 2017, 199, 1065–1068. [Google Scholar] [CrossRef]
- Wibberg, D.; Genzel, F.; Verwaaijen, B.; Blom, J.; Rupp, O.; Goesmann, A.; Zrenner, R.; Grosch, R.; Pühler, A.; Schlüter, A. Genome Analyses of the Less Aggressive Rhizoctonia solani AG1-IB Isolates 1/2/21 and O8/2 Compared to the Reference AG1-IB Isolate 7/3/14. J. Fungi 2021, 7, 832. [Google Scholar] [CrossRef]
- Villa, V.D.; Storck, R. Nucleotide Composition of Nuclear and Mitochondrial Deoxyribonucleic Acid of Fungi. J. Bacteriol. 1968, 96, 184–190. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Eikmeyer, F.; Hadiati, A.; Szczepanowski, R.; Wibberg, D.; Schneiker-Bekel, S.; Rogers, L.M.; Brown, C.J.; Top, E.M.; Pühler, A.; Schlüter, A. The complete genome sequences of four new IncN plasmids from wastewater treatment plant effluent provide new insights into IncN plasmid diversity and evolution. Plasmid 2012, 68, 13–24. [Google Scholar] [CrossRef]
- n.d. N50, L50, and Related Statistics. In Wikipedia. Available online: https://en.wikipedia.org/wiki/N50,_L50,_and_related_statistics (accessed on 30 May 2022).
- Wibberg, D.; Jelonek, L.; Rupp, O.; Hennig, M.; Eikmeyer, F.; Goesmann, A.; Hartmann, A.; Borriss, R.; Grosch, R.; Pühler, A.; et al. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J. Biotechnol. 2013, 167, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Hintz, W.; Anderson, J.B.; Horgen, P.A. Nuclear migration and mitochondrial inheritance in the mushroom Agaricus bitorquis. Genetics 1988, 119, 35–41. [Google Scholar] [CrossRef]
- Jin, T.; Sonnenberg, A.S.M.; Van Griensven, L.J.L.D.; Horgen, P.A. Investigation of Mitochondrial Transmission in Selected Matings between Homokaryons from Commercial and Wild-Collected Isolates of Agaricus bisporus (= Agaricus brunnescens). Appl. Environ. Microbiol. 1992, 58, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Horgen, P.A. Uniparental Mitochondrial Transmission in the Cultivated Button Mushroom, Agaricus bisporus. Appl. Environ. Microbiol. 1994, 60, 4456–4460. [Google Scholar] [CrossRef] [PubMed]
- Baptista-Ferreira, J.L.C.; Economou, A.; Casselton, L.A. Mitochondrial genetics of Coprinus: Recombination of mitochondrial genomes. Curr. Genet. 1983, 7, 405–407. [Google Scholar] [CrossRef] [PubMed]
- May, G.; Taylor, J.W. Patterns of mating and mitochondrial DNA inheritance in the agaric Basidiomycete Coprinus cinereus. Genetics 1988, 118, 213–220. [Google Scholar] [CrossRef]
- Barroso, G.; Labarère, J. Genetic evidence for nonrandom sorting of mitochondria in the basidiomycete Agrocybe aegerita. Appl. Environ. Microbiol. 1997, 63, 4686–4691. [Google Scholar] [CrossRef]
- Aanen, D.K.; Kuyper, T.W.; Debets, A.J.M.; Hoekstra, R.F. The evolution of non–reciprocal nuclear exchange in mushrooms as a consequence of genomic conflict. Proc. R. Soc. B Boil. Sci. 2004, 271, 1235–1241. [Google Scholar] [CrossRef]
- Ye, L.-Y.; Deng, Y.-J.; Mukhtar, I.; Meng, G.-L.; Song, Y.-J.; Cheng, B.; Hao, J.-B.; Wu, X.-P. Mitochondrial genome and diverse inheritance patterns in Pleurotus pulmonarius. J. Microbiol. 2020, 58, 142–152. [Google Scholar] [CrossRef]
- Wang, P.; Sha, T.; Zhang, Y.; Cao, Y.; Mi, F.; Liu, C.; Yang, D.; Tang, X.; He, X.; Dong, J.; et al. Frequent heteroplasmy and recombination in the mitochondrial genomes of the basidiomycete mushroom Thelephora ganbajun. Sci. Rep. 2017, 7, 1626. [Google Scholar] [CrossRef]
- Choi, J.-H.; Suzuki, T.; Ono, A.; Kotajima, M.; Tanaka, Y.; Suzuki, T.; Kawagishi, H.; Dohra, H. The complete mitochondrial genome sequence of the fairy ring-forming fungus Lepista sordida. Mitochondrial DNA Part B 2022, 7, 712–714. [Google Scholar] [CrossRef]
- Wibberg, D.; Rupp, O.; Blom, J.; Jelonek, L.; Kröber, M.; Verwaaijen, B.; Goesmann, A.; Albaum, S.; Grosch, R.; Pühler, A.; et al. Development of a Rhizoctonia solani AG1-IB Specific Gene Model Enables Comparative Genome Analyses between Phytopathogenic R. solani AG1-IA, AG1-IB, AG3 and AG8 Isolates. PLoS ONE 2015, 10, e0144769. [Google Scholar] [CrossRef]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Peng, L.; Shan, X.; Yang, Y.; Wang, Y.; Druzhinina, I.S.; Pan, X.; Jin, W.; He, X.; Wang, X.; Zhang, X.; et al. Facultative symbiosis with a saprotrophic soil fungus promotes potassium uptake in American sweetgum trees. Plant Cell Environ. 2021, 44, 2793–2809. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Wendt, K.U.; Schulz, G.E. Isoprenoid biosynthesis: Manifold chemistry catalyzed by similar enzymes. Structure 1998, 6, 127–133. [Google Scholar] [CrossRef]
- Dickschat, J.S. Isoprenoids in three-dimensional space: The stereochemistry of terpene biosynthesis. Nat. Prod. Rep. 2011, 28, 1917–1936. [Google Scholar] [CrossRef]
- Chen, H.-P.; Liu, J.-K. Secondary Metabolites from Higher Fungi. In Progress in the Chemistry of Organic Natural Products; Springer: Cham, Switzerland, 2017; Volume 106, pp. 1–201. [Google Scholar] [CrossRef]
- Koczyk, G.; Pawłowska, J.; Muszewska, A. Terpenoid Biosynthesis Dominates among Secondary Metabolite Clusters in Mucoromycotina Genomes. J. Fungi 2021, 7, 285. [Google Scholar] [CrossRef]
- Gressler, M.; Löhr, N.A.; Schäfer, T.; Lawrinowitz, S.; Seibold, P.S.; Hoffmeister, D. Mind the mushroom: Natural product biosynthetic genes and enzymes of Basidiomycota. Nat. Prod. Rep. 2021, 38, 702–722. [Google Scholar] [CrossRef]
- Schmidt-Dannert, C. Biosynthesis of Terpenoid Natural Products in Fungi. Adv. Biochem. Eng. Biotechnol. 2015, 148, 19–61. [Google Scholar] [CrossRef]
- Yap, H.-Y.Y.; Muria-Gonzalez, M.J.; Kong, B.-H.; Stubbs, K.A.; Tan, C.-S.; Ng, S.-T.; Tan, N.-H.; Solomon, P.S.; Fung, S.-Y.; Chooi, Y.-H. Heterologous expression of cytotoxic sesquiterpenoids from the medicinal mushroom Lignosus rhinocerotis in yeast. Microb. Cell Factories 2017, 16, 103. [Google Scholar] [CrossRef]
- Harvey, C.J.B.; Tang, M.; Schlecht, U.; Horecka, J.; Fischer, C.R.; Lin, H.-C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M.; et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, eaar5459. [Google Scholar] [CrossRef]
- Maury, J.; Kannan, S.; Jensen, N.B.; Öberg, F.K.; Kildegaard, K.R.; Forster, J.; Nielsen, J.; Workman, C.T.; Borodina, I. Glucose-Dependent Promoters for Dynamic Regulation of Metabolic Pathways. Front. Bioeng. Biotechnol. 2018, 6, 63. [Google Scholar] [CrossRef]
- Curran, K.A.; Karim, A.S.; Gupta, A.; Alper, H.S. Use of expression-enhancing terminators in Saccharomyces cerevisiae to increase mRNA half-life and improve gene expression control for metabolic engineering applications. Metab. Eng. 2013, 19, 88–97. [Google Scholar] [CrossRef]
- Sakai, A.; Shimizu, Y.; Hishinuma, F. Integration of heterologous genes into the chromosome of Saccharomyces cerevisiae using a delta sequence of yeast retrotransposon Ty. Appl. Microbiol. Biotechnol. 1990, 33, 302–306. [Google Scholar] [CrossRef]
- Botas, A.; Eitel, M.; Schwarz, P.N.; Buchmann, A.; Costales, P.; Núñez, L.E.; Cortés, J.; Morís, F.; Krawiec, M.; Wolański, M.; et al. Genetic Engineering in Combination with Semi-Synthesis Leads to a New Route for Gram-Scale Production of the Immunosuppressive Natural Product Brasilicardin A. Angew. Chem. Int. Ed. 2021, 60, 13536–13541. [Google Scholar] [CrossRef]
- Schwarz, P.N.; Roller, L.; Kulik, A.; Wohlleben, W.; Stegmann, E. Engineering metabolic pathways in Amycolatopsis japonicum for the optimization of the precursor supply for heterologous brasilicardin congeners production. Synth. Syst. Biotechnol. 2018, 3, 56–63. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Total number of raw bases (ONT) | 22.81 Gb |
| Number of reads (ONT) | 4.59 million |
| Total number of raw bases (Illumina) | 4.70 Gb |
| Number of reads (Illumina) | 15.7 million |
| Number of contigs | 167 |
| Largest contig | 3,100,797 bp |
| Total length | 65,616,532 bp |
| GC content | 49.43% |
| N50 (shortest contig length at ½ genome length) 1 | 673,524 bp |
| Number of predicted genes | 23,566 |
| Feature | MtDNA-1 | MtDNA-2 |
|---|---|---|
| Length | 40.291 bp | 40.382 bp |
| Number of CDS | 32 | 32 |
| Number of encoded tRNA | 26 | 26 |
| Number of encoded rRNA clusters | 3 | 3 |
| GC content | 27.1% | 27.1% |
| Sequence Identity | 99.63% |
| Alignment Length | 40,405 bp |
| Identical Positions | 40,259 bp |
| Gaps | 137 |
| Gene Name | Alternative Name | Function | pleBGC | Length (bp) | Number of Introns | Protein Length (aa) | Nucleotide Identity | Amino Acid Identity (Similarity) |
|---|---|---|---|---|---|---|---|---|
| ple1 | Pl-p450-3 | Cytochrome P450 | 1 | 2107 | 10 | 523 | 93.9% | 98.1% (98.9%) |
| 2 | 2108 | 10 | 522 | |||||
| ple2 | Pl-atf | Acetyltransferase | 1 | 1301 | 3 | 377 | 93.7% | 96.6% (98.1%) |
| 2 | 1304 | 3 | 377 | |||||
| ple3 | Pl-cyc | Terpene synthase | 1 | 3041 | 3 | 959 | 96.7% | 98.6% (99.3%) |
| 2 | 3040 | 3 | 959 | |||||
| ple4 | Pl-gps | GGPP synthase | 1 | 1291 | 4 | 350 | 95.0% | 98.6% (99.4%) |
| 2 | 1290 | 4 | 350 | |||||
| ple5 | Pl-p450-1 | Cytochrome P450 | 1 | 2286 | 13 | 523 | 96.3% | 98.5% (99.4%) |
| 2 | 2284 | 13 | 523 | |||||
| ple6 | Pl-p450-2 | Cytochrome P450 | 1 | 2166 | 11 | 525 | 90.4% | 97.9% (99.2%) |
| 2 | 2166 | 11 | 525 | |||||
| ple7 | Pl-sdr | Dehydrogenase | 1 | 945 | 3 | 253 | 89.7% | 97.2% (98.4%) |
| 2 | 945 | 3 | 253 |
| Promoters 1 | Genes 2 | Terminators 3 | |||
|---|---|---|---|---|---|
| Yeast-pleBGC-A | Yeast-pleBGC-B | Yeast-pleBGC-C | All Yeast-pleBGCs | All Yeast-pleBGCs | |
| pSPG4 | pYIG1 | pPGK1 | ple1 | (P450-3) | tCYC* |
| pCTA1 | pYEF3 | pTEF1 | ple2 | (acetyltransferase) | tVPS13 |
| pACS1 | pTDH3 | pADH2 | ple3 | (terpene synthase) | tCPS1 |
| pICL1 | pADH1 | pADY2 | ple4 | (GGPP synthase) | tPRM9 |
| pADH2 | pPGK1 | pFBA1 | ple5 | (P450-1) | tHIS5 |
| pATO1 | pTEF1 | pATO1 | ple6 | (P450-2) | tSPG5 |
| pADY2 | pFBA1 | pCTA1 | ple7 | (dehydrogenase) | tUBX6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schafhauser, T.; Wibberg, D.; Binder, A.; Rückert, C.; Busche, T.; Wohlleben, W.; Kalinowski, J. Genome Assembly and Genetic Traits of the Pleuromutilin-Producer Clitopilus passeckerianus DSM1602. J. Fungi 2022, 8, 862. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080862
Schafhauser T, Wibberg D, Binder A, Rückert C, Busche T, Wohlleben W, Kalinowski J. Genome Assembly and Genetic Traits of the Pleuromutilin-Producer Clitopilus passeckerianus DSM1602. Journal of Fungi. 2022; 8(8):862. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080862
Chicago/Turabian StyleSchafhauser, Thomas, Daniel Wibberg, Antonia Binder, Christian Rückert, Tobias Busche, Wolfgang Wohlleben, and Jörn Kalinowski. 2022. "Genome Assembly and Genetic Traits of the Pleuromutilin-Producer Clitopilus passeckerianus DSM1602" Journal of Fungi 8, no. 8: 862. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080862