
Enhancing Genome-Scale Model by Integrative Exometabolome and Transcriptome: Unveiling Carbon Assimilation towards Sphingolipid Biosynthetic Capability of Cordyceps militaris

 ,
,  , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation for Dry Weight and Metabolic Footprint Analysis
2.2. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
2.3. Metabolite Acquisition and Identification towards Differentially Accumulated Metabolites (DAMs) Analysis
2.4. Improvement of GSMM of C. militaris by Incorporating Metabolic Footprint and Transcriptome Data
3. Results and Discussion
3.1. Quantitative Metabolic Footprinting of C. militaris
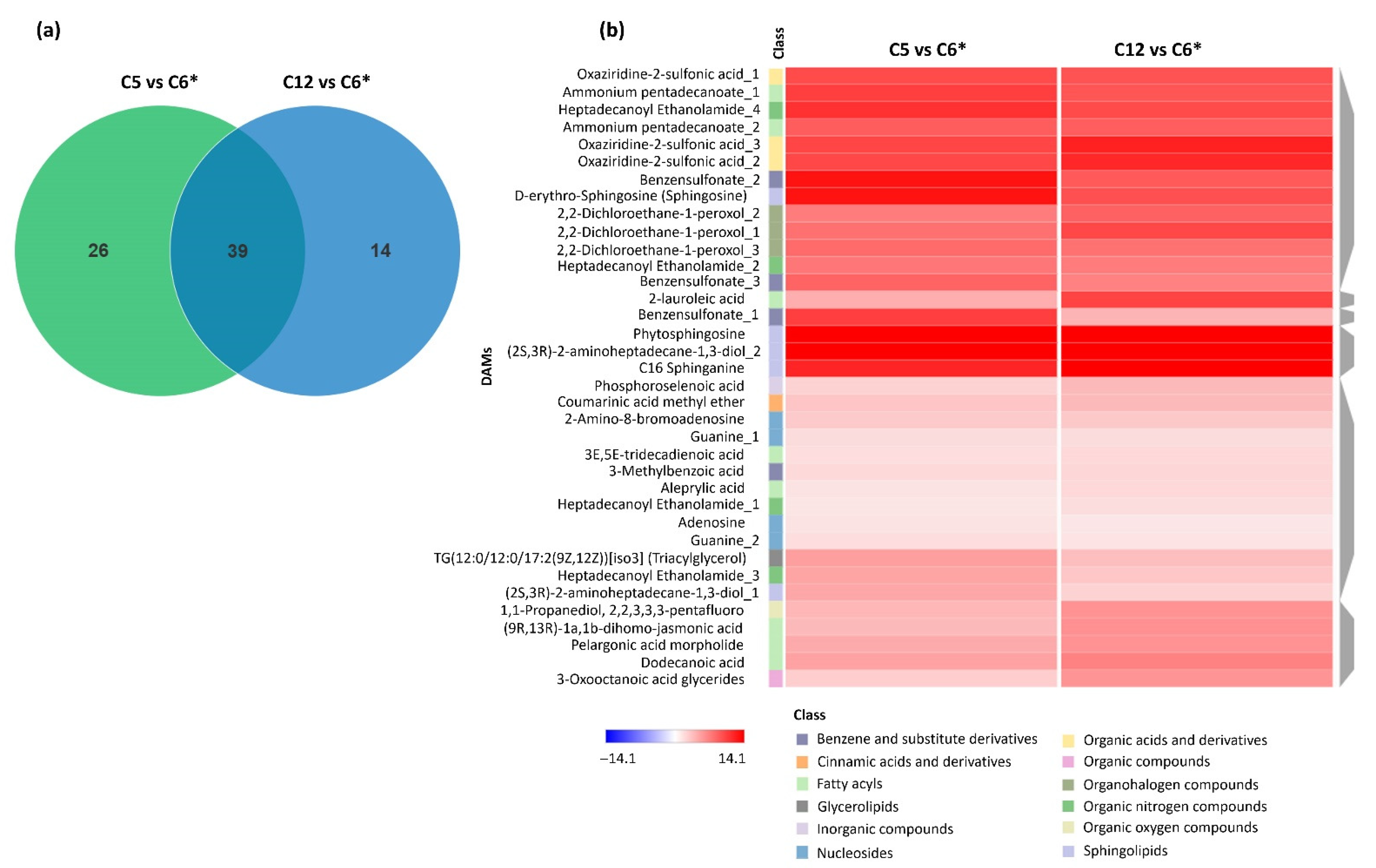
3.2. Identification of DAMs across Pairwise Comparisons of the Cultures using Different Carbon Sources
3.3. Identification of Significant Abundant DAMs in the Cultures Using Different Carbon Sources
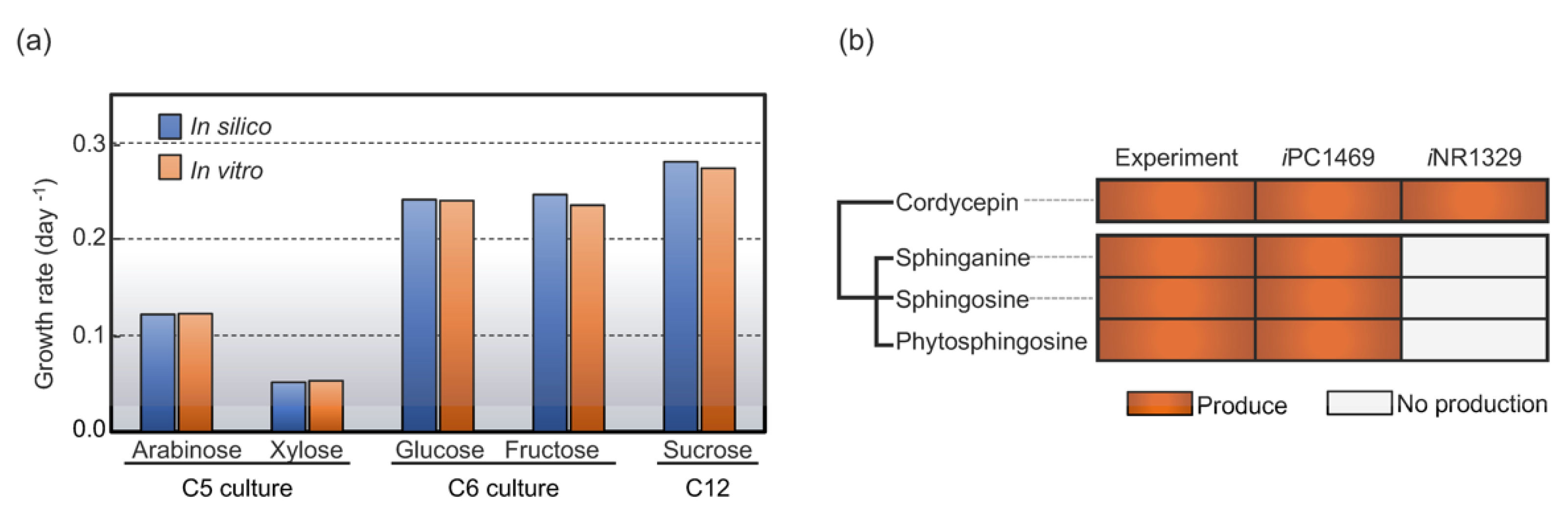
3.4. Enhancing Genome-Scale Metabolic Model (GSMM) Using Significant Metabolic Footprint Profiles
3.5. Integrative Transcriptome Analysis Using The Enhanced GSMM Revealed Metabolic Response in Lipid Biosynthetic Capability
3.5.1. Glycerolipid Biosynthetic Pathway
3.5.2. Sphingolipid Biosynthetic Capability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, M.; Guo, S.; Huaijun, Y.; Bu, N.; Dong, C. Comparison of major bioactive compounds of the caterpillar medicinal mushroom, Cordyceps militaris (Ascomycetes), fruiting bodies cultured on wheat substrate and pupae. Int. J. Med. Mushrooms. 2016, 18, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Tuli, H.S.; Sharma, A.K.; Sandhu, S.S.; Kashyap, D. Cordycepin: A bioactive metabolite with therapeutic potential. Life Sci. 2013, 93, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yan, H.; Zeng, B.; Hu, Z. Research progress on cordycepin synthesis and methods for enhancement of cordycepin production in Cordyceps militaris. Bioengineering 2022, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Zhang, M.; Bhandri, B. 3D printing of Cordyceps flower powder. J. Food Process Eng. 2019, 42, e13179. [Google Scholar] [CrossRef]
- Kunhorm, P.; Chaicharoenaudomrung, N.; Noisa, P. Enrichment of cordycepin for cosmeceutical applications: Culture systems and strategies. Appl. Microbiol. Biotechnol. 2019, 103, 1681–1691. [Google Scholar] [CrossRef]
- Das, S.K.; Masuda, M.; Hatashita, M.; Sakurai, A.; Sakakibara, M. Optimization of culture medium for cordycepin production using Cordyceps militaris mutant obtained by ion beam irradiation. Process Biochem. 2010, 45, 129–132. [Google Scholar] [CrossRef]
- Cui, J.D. Biotechnological production and applications of Cordyceps militaris, a valued traditional Chinese medicine. Crit. Rev. Biotechnol. 2015, 35, 475–484. [Google Scholar] [CrossRef]
- Masuda, M.; Urabe, E.; Sakurai, A.; Sakakibara, M. Production of cordycepin by surface culture using the medicinal mushroom Cordyceps militaris. Enzym. Microb. Technol. 2006, 39, 641–646. [Google Scholar] [CrossRef]
- Mao, X.-B.; Eksriwong, T.; Chauvatcharin, S.; Zhong, J.-J. Optimization of carbon source and carbon/nitrogen ratio for cordycepin production by submerged cultivation of medicinal mushroom Cordyceps militaris. Process Biochem. 2005, 40, 1667–1672. [Google Scholar] [CrossRef]
- Wongsa, B.; Raethong, N.; Chumnanpuen, P.; Wong-ekkabut, J.; Laoteng, K.; Vongsangnak, W. Alternative metabolic routes in channeling xylose to cordycepin production of Cordyceps militaris identified by comparative transcriptome analysis. Genomics 2020, 112, 629–636. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.; Liu, L.; Feng, J.; Zhang, T.; Qin, S.; Zhao, X.; Wang, C.; Li, D.; Han, W.; et al. Study of the whole genome, methylome and transcriptome of Cordyceps militaris. Sci. Rep. 2019, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Raethong, N.; Wang, H.; Nielsen, J.; Vongsangnak, W. Optimizing cultivation of Cordyceps militaris for fast growth and cordycepin overproduction using rational design of synthetic media. Comput. Struct. Biotechnol. J. 2020, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, Q.; Zhang, J.; Liu, K.; Li, K.; Liu, G.; Dong, C. Comparative transcriptome analysis between a spontaneous albino mutant and its sibling strain of Cordyceps militaris in response to light stress. Front. Microbiol. 2018, 9, 1237. [Google Scholar] [CrossRef]
- Raethong, N.; Laoteng, K.; Vongsangnak, W. Uncovering global metabolic response to cordycepin production in Cordyceps militaris through transcriptome and genome-scale network-driven analysis. Sci. Rep. 2018, 8, 9250. [Google Scholar] [CrossRef]
- Lin, M.; Guo, S.; Xie, D.; Li, S.; Hu, H. Lipidomic profiling of wild cordyceps and its substituents by liquid chromatography-electrospray ionization-tandem mass spectrometry. LWT 2022, 163, 113497. [Google Scholar] [CrossRef]
- Mi, J.-N.; Han, Y.; Xu, Y.; Kou, J.; Wang, J.-R.; Jiang, Z.-H. New immunosuppressive sphingoid base and ceramide analogues in wild Cordyceps. Sci. Rep. 2016, 6, 38641. [Google Scholar] [CrossRef]
- Mi, J.; Han, Y.; Xu, Y.; Kou, J.; Li, W.-J.; Wang, J.-R.; Jiang, Z.-H. Deep profiling of immunosuppressive glycosphingolipids and sphingomyelins in wild Cordyceps. J. Agric. Food Chem. 2018, 66, 8991–8998. [Google Scholar] [CrossRef]
- Maru, M.; Haraguchi, M.; Higashi, H.; Kato, S.; Kurimura, T.; Naiki, M.; Wakamiya, N. Anti-tumor activity of ceramides and glycosphingolipids in a murine tumor system. Int. J. Cancer 1993, 53, 645–650. [Google Scholar] [CrossRef]
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Maille, G.O.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS online: Simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’ayan, A. Clustergrammer, A web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Grego, T.; Pesquita, C.; Bastos, H.P.; Couto, F.M. Chemical entity recognition and resolution to ChEBI. ISRN Bioinform. 2012, 2012, 619427. [Google Scholar] [CrossRef]
- Moretti, S.; Tran, V.D.T.; Mehl, F.; Ibberson, M.; Pagni, M. MetaNetX/MNXref: Unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 2021, 49, D570–D574. [Google Scholar] [CrossRef]
- Nguyen, N.; Vongsangnak, W.; Shen, B.; Nguyen, P.-V.; Leong, H. Megafiller: A retrofitted protein function predictor for filling gaps in metabolic networks. J. Proteom. Bioinf. 2014, s9, 1–11. [Google Scholar] [CrossRef]
- Vongsangnak, W.; Raethong, N.; Mujchariyakul, W.; Nguyen, N.N.; Leong, H.W.; Laoteng, K. Genome-scale metabolic network of Cordyceps militaris useful for comparative analysis of entomopathogenic fungi. Gene 2017, 626, 132–139. [Google Scholar] [CrossRef]
- Thananusak, R.; Laoteng, K.; Raethong, N.; Zhang, Y.; Vongsangnak, W. Metabolic responses of carotenoid and cordycepin biosynthetic pathways in Cordyceps militaris under light-programming exposure through genome-wide transcriptional analysis. Biology 2020, 9, 242. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kusonmano, K.; Vongsangnak, W.; Chumnanpuen, P. Informatics for Metabolomics. In Translational Biomedical Informatics: A Precision Medicine Perspective; Shen, B., Tang, H., Jiang, X., Eds.; Springer: Singapore, 2016; pp. 91–115. [Google Scholar]
- Mi, J.-N.; Wang, J.-R.; Jiang, Z.-H. Quantitative profiling of sphingolipids in wild Cordyceps and its mycelia by using UHPLC-MS. Sci. Rep. 2016, 6, 20870. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Fujita, T.; Inoue, K.; Yamamoto, S.; Ikumoto, T.; Sasaki, S.; Toyama, R.; Chiba, K.; Hoshino, Y.; Okumoto, T. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J. Antibiot. 1994, 47, 208–215. [Google Scholar] [CrossRef]
- Pavicic, T.; Wollenweber, U.; Farwick, M.; Korting, H.C. Anti-microbial and -inflammatory activity and efficacy of phytosphingosine: An in vitro and in vivo study addressing acne vulgaris. Int. J. Cosmet. Sci. 2007, 29, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Maiti, A. The role of sphingosine-1-phosphate and ceramide-1-phosphate in inflammation and cancer. Mediat. Inflamm. 2017, 2017, 4806541. [Google Scholar] [CrossRef]
- SediĆ, M.; GrbČIĆ, P.; PaveliĆ, S.K. Bioactive sphingolipids as biomarkers predictive of disease severity and treatment response in cancer: Current status and translational challenges. Anticancer Res. 2019, 39, 41. [Google Scholar] [CrossRef]
- Noda, T.; Ono, M.; Iimure, K.; Araki, T. Characterization of a germination-accelerating factor from the silkworm (Bombyx mori Linnaeus) of entomopathogenic fungus Nomuraea rileyi (Farlow) samson. Biosci. Biotechnol. Biochem. 2010, 74, 1226–1230. [Google Scholar] [CrossRef]
- Lennon, M.B.; Suhadolnik, R.J. Biosynthesis of 3′-deoxyadenosine by Cordyceps militaris: Mechanism of reduction. Biochim. Et Biophys. Acta (BBA)-Nucleic Acids Protein Synth. 1976, 425, 532–536. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pairwise Comparison Sets | Number of Significant DAMs | Up-Accumulated Metabolites | Down-Accumulated Metabolites |
|---|---|---|---|
| C5 vs. C6 cultures | 252 | 212 | 40 |
| C12 vs. C6 cultures | 188 | 187 | 1 |
| C5 vs. C12 cultures | 30 | 17 | 13 |
| Characteristics | iNR1329 * | iPC1469 (This Study) |
|---|---|---|
| Number of Genes | 1329 | 1469 |
| Number of Metabolites | 1171 | 1229 |
| Number of Reactions | 1821 | 1904 |
| Enzymatic reactions | 1391 | 1404 |
| Transport reactions | 271 | 339 |
| Exchange reactions | 137 | 140 |
| Spontaneous reactions | 21 | 21 |
| Biomass synthesis reaction | 1 | 1 |
| Biosynthetic capacities of GSMM | Cordycepin | Cordycepin, sphinganine, phytosphingosine, and sphingosine |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheawchanlertfa, P.; Chitcharoen, S.; Raethong, N.; Liu, Q.; Chumnanpuen, P.; Soommat, P.; Song, Y.; Koffas, M.; Laoteng, K.; Vongsangnak, W. Enhancing Genome-Scale Model by Integrative Exometabolome and Transcriptome: Unveiling Carbon Assimilation towards Sphingolipid Biosynthetic Capability of Cordyceps militaris. J. Fungi 2022, 8, 887. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080887
Cheawchanlertfa P, Chitcharoen S, Raethong N, Liu Q, Chumnanpuen P, Soommat P, Song Y, Koffas M, Laoteng K, Vongsangnak W. Enhancing Genome-Scale Model by Integrative Exometabolome and Transcriptome: Unveiling Carbon Assimilation towards Sphingolipid Biosynthetic Capability of Cordyceps militaris. Journal of Fungi. 2022; 8(8):887. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080887
Chicago/Turabian StyleCheawchanlertfa, Pattsarun, Suwalak Chitcharoen, Nachon Raethong, Qing Liu, Pramote Chumnanpuen, Panyawarin Soommat, Yuanda Song, Mattheos Koffas, Kobkul Laoteng, and Wanwipa Vongsangnak. 2022. "Enhancing Genome-Scale Model by Integrative Exometabolome and Transcriptome: Unveiling Carbon Assimilation towards Sphingolipid Biosynthetic Capability of Cordyceps militaris" Journal of Fungi 8, no. 8: 887. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8080887