1. Introduction
The nanotechnology field is dedicated to designing and producing materials with nanometric dimensions by using different devices, structures, and systems that are inspired constantly by nature. The rapid progress of nanotechnology involved the development of new materials with different proprieties, such as biodegradability, biocompatibility, versatility, stimuli-sensitive properties, and the ability to deliver therapeutic biomolecules (drugs, proteins, genes, and photosensitizer) to a target in a controlled manner. Nanotechnology is already used in biomedicine applications through the use of liposomes, nanoparticles, and nanotubes, which are currently interesting and promising alternatives. Moreover, the chemical functionalization of natural/synthetic polymer nanomaterials offers a variety of opportunities such as tolerability of the polymer proprieties such as solubility, bioadhesivity, antibacterial properties, and antifungal activity [
1].
Advanced nanomedicine systems require the combination of targeted delivery, imaging, and therapeutics in a single nanocarrier system. Hydrogel nanoparticles are promising nanomaterials due to advantages such as targeting, decreasing drug dose, the possibility of maintaining a desired therapeutic concentration, and low toxicity effects [
2]. Moreover, a hydrogel is a polymer three-dimensional network, capable of capturing large amounts of water or active principles due to its hydrophilic structure, presenting excellent properties such as a porous structure, sensitivity to different environmental factors (pH, temperature, light), biocompatibility, and the similarity of their physical properties to natural tissue, which led to its use as support material in tissue engineering [
3,
4,
5,
6,
7,
8].
Chitosan (CS) is a cationic polysaccharide obtained via deacetylation and partial depolymerization of chitin under alkaline conditions or enzymatic hydrolysis, and is the second most common natural polysaccharide after cellulose”.
Although chitin is insoluble in most organic solvents, CS is soluble in dilute acidic solutions with pH < 6 due to the presence of primary amino groups (with pKa = 6.3) which become protonated. An increase in pH above 6, leads to the deprotonation of amino groups and CS loses its soluble character [
9]. Chitosan represents a natural polymer with outstanding properties such as bioadhesivity, biocompatibility, biodegradability, non-toxicity, bioavailability, etc., which justify its attractiveness, especially for bio-related applications [
2,
9]. Moreover, CS shows important biological activity, including antiviral, antifungal, and antibacterial properties; antihyperglycemic, hypocholesterolemic, and antitumoral effects; genetic material transfection; and wound healing promotion [
2].
The highly reactive amine moieties may be employed in a wide range of physical/chemical reactions for CS modification to gain a specific effect. One example is ionotropic gelation through ionic interactions between chitosan’s amine groups and negatively charged crosslinking agents, which results in a simple and flexible materials preparation procedure [
10].
The diversity and efficiency of chitosan-based micro/nano-particulate hydrogel systems for drug targeting and controlled drug release applications have been extensively recorded in the literature [
9,
11,
12,
13]. Thus, physical crosslinking using small molecular weight anionic crosslinkers, such as sodium tripolyphosphate (TPP), has been employed in many studies [
14]. However, the crosslinking process is reversible, and a pH increase leads to fast destructuring of the hydrogels through dissociation of the TPP/CS complexes into free chitosan chains [
15]. Such phenomena are creating problems for applications that involve high pH variations, e.g., the passage through the gastrointestinal tract. A solution to alleviate the problems associated with the reversibility of ionic crosslinking in chitosan-based systems employs a supplementary covalent crosslinking step [
13,
15,
16,
17]. Chitosan’s particular chemistry favors crosslinking processes using aldehydes which form imine bonds with the amine functions [
18]. Chitosan double ionic and covalent crosslinking has rarely been performed [
18,
19].
CS nanoparticles prepared via emulsification and crosslinking using the amino group of CS and the aldehyde group of glutaraldehyde (GA) were first obtained and characterized by Ohya et al. The nanoparticles were used as a system for circulatory delivery of 5-fluorouracil [
20]. Furthermore, as an anionic crosslinker for CS, TPP is widely used and has been extensively investigated for the preparation of micro/nanoparticles, hydrogels, and scaffold systems in biomedical applications. For increasing micro/nanoparticle systems stability, both types of crosslinkers (e.g., TPP and GA) are needed [
21]. Few studies are mentioned in the literature concerning the preparation of nanoparticle system-based CS or modified CS via the double ionic/covalent crosslinking method [
16,
17,
22,
23,
24,
25]. Adrenalin-loaded gelatin/CS particles for application in ocular drug administration were prepared using a two-step cross-linking process performed in an emulsion-phase separation system by Peptu et. al., (2010) [
17]. Another interesting study concerning the obtaining of nanoparticles-based CS and gelatin via double crosslinking for drug targeting was realized by Jatariu (Cadinoiu) et. al, (2012) [
22]. For the first time, polymer magnetic nanoparticles based on the chitosan-maltose derivative and magnetite double crosslinking technique (ionic and covalent) in reverse emulsion for antitumor drug delivery were prepared by Alupei et al., (2016) [
16].
Damour I. and Muti H., (2021) succeeded in obtaining stable CPH based on lecithin/CS conjugates via a dual ionic/covalent crosslinking method. They found that the dual ionic/covalent crosslinking improved nanoparticles’ drug loading/releasing properties [
26].
On the other hand, the chemical modification of CS is a promising method for achieving desired polymer properties. CS solubility in acidic solutions, its polycationic character, and the presence of NH
2 groups in its structure, allow a variety of chemical modification strategies leading to new CS derivatives with new physical or biological properties [
27].
The aim of this work is the chemical modification of chitosan by Michael addition of poly (ethylene glycol) methyl ether acrylate (PEGA) to the amine groups to further prepare submicronic particulate hydrogels. In this investigation, the synthesis of new submicronic particulate hydrogels based on CS grafted with poly (ethylene glycol) methyl ether acrylate (CS-PEGA) through a two-step cross-linking technique (ionic followed by covalent), in a reverse emulsion, is described for the first time. CPH characterization was carried out in terms of structural, size, morphological, swelling, and drug loading/release properties which were controlled by varying the synthesis parameters (polymer concentration, dispersion speed, and amount of ionic cross-linking agent). In vitro tests were performed to demonstrate CPH capacity for drug loading and release, hemocompatibility, and cytotoxicity. The two-step cross-linking technique was revealed to be a valid method for the synthesis of nontoxic CPH-based materials.
2. Results and Discussion
The work presented herein is focused on the effect of CS modification with PEG-acrylate on polymer processability during the preparation of submicronic nanoparticulate hydrogel drug formulations. The use of PEGA to chemically modify CS may lead to the improvement of its solubility in water and diminish its pH-dependent water swelling character by adding hydrophilic segments to the CS linear chain. In principle, native chitosan may be used for emulsion double crosslinking using ionic gelation in the first step followed by network fixation via covalent crosslinking at the emulsion interface level in the second step. Amine groups of CS are involved in both crosslinking steps and partial grafting with PEG-acrylate may be the source of uncertainties concerning the double crosslinking process. Moreover, the pegylated chitosan will further affect pH-dependent water swelling and drug release properties.
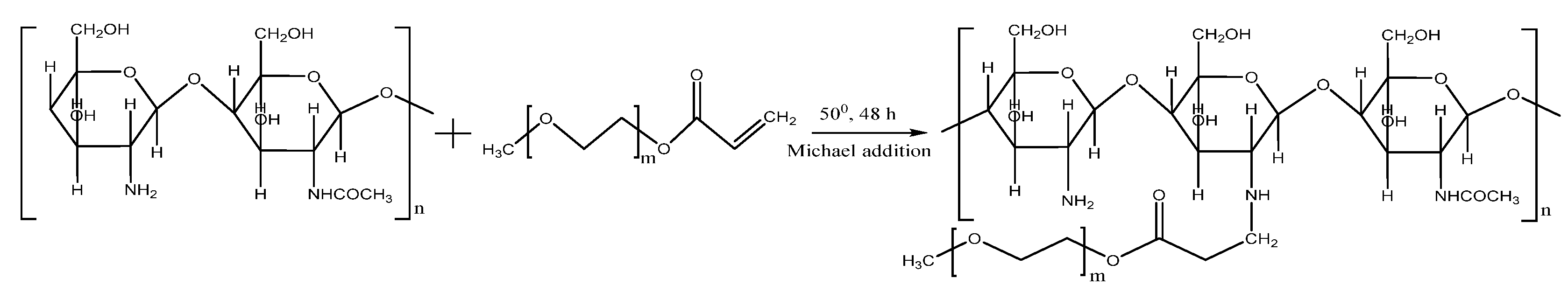
2.1. Synthesis and Characterization of CS-PEGA
The proposed pathway to synthesize CS-PEGA is a Michael addition reaction, which involves the addition of the acrylate PEGA end chains to the amino groups of CS, at 50 °C, as presented in
Scheme 1.
CS-PEGA structural characterization was performed via FTIR and
1H NMR aiming to have qualitative and quantitative proofs of CS grafting.
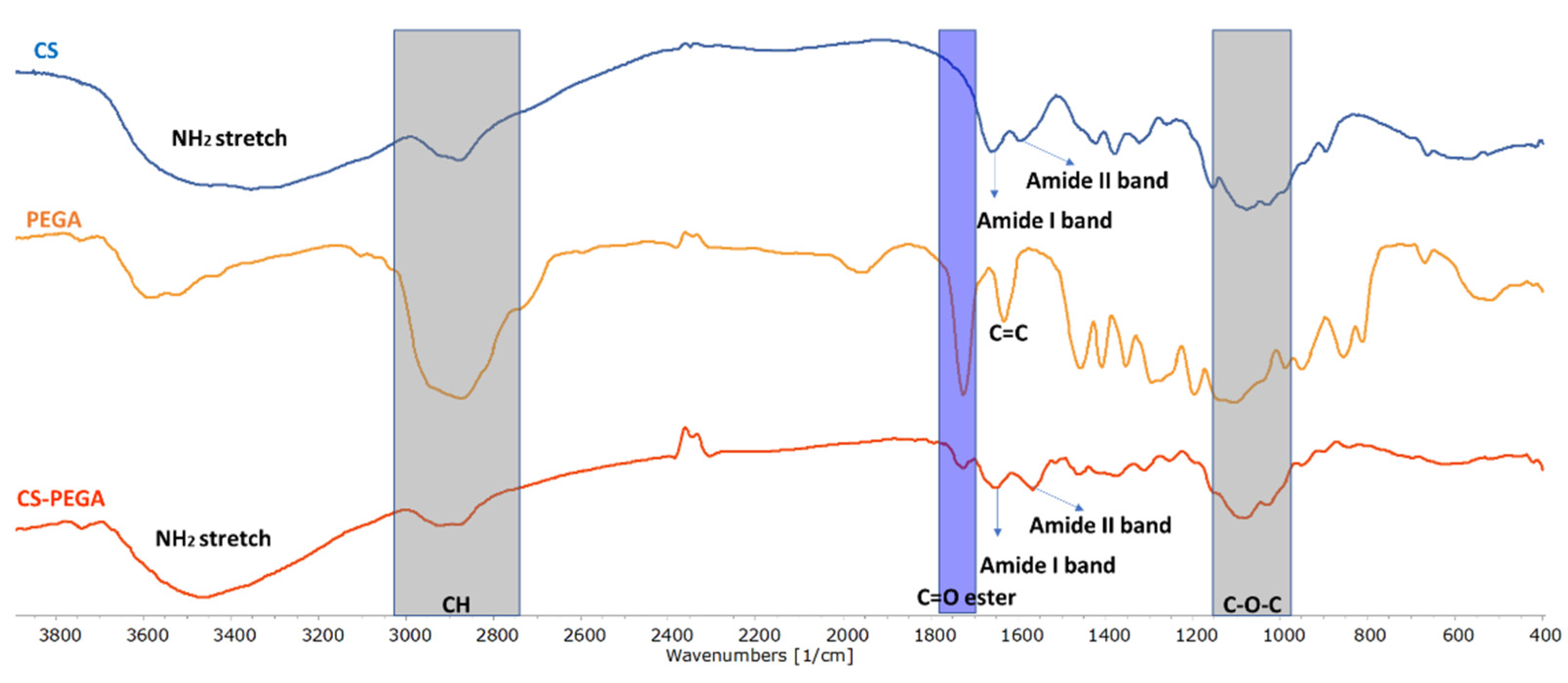
Figure 1 illustrates the overlapped FTIR spectra of the starting precursors (CS and PEGA) and the synthesized polymer (CS-PEGA obtained using a molar ratio of NH
2:PEGA = 1:0.5). The assigned characteristic bands from the obtained spectra are presented in
Table S1.
FTIR CS spectra (
Figure 1) showed absorption bands at 1076 cm
−1 (C-O-C stretching vibration) [
28], 1659 cm
−1, 1423 cm
−1, and 1378 cm
−1 (NHAc units, amide I, NH
2 bending, and amide III, respectively) [
29]; and a strong peak at 3355 cm
−1 attributed to the axial stretching vibration of (OH) overlapped with the (NH
2) stretching band of CS molecules. The PEGA spectra show characteristic signals at 1631 cm
−1 (C=C starching vibration), 1724 cm
−1 (C=O asymmetrical and symmetrical stretching), and intense peaks at 2874 cm
−1 (CH) and 3522 cm
−1 (OH stretching vibration). The FTIR spectrum of CS-PEGA proves PEGA grafting onto CS through the appearance of the specific signal of (C=O) at 1733 cm
−1, as highlighted.
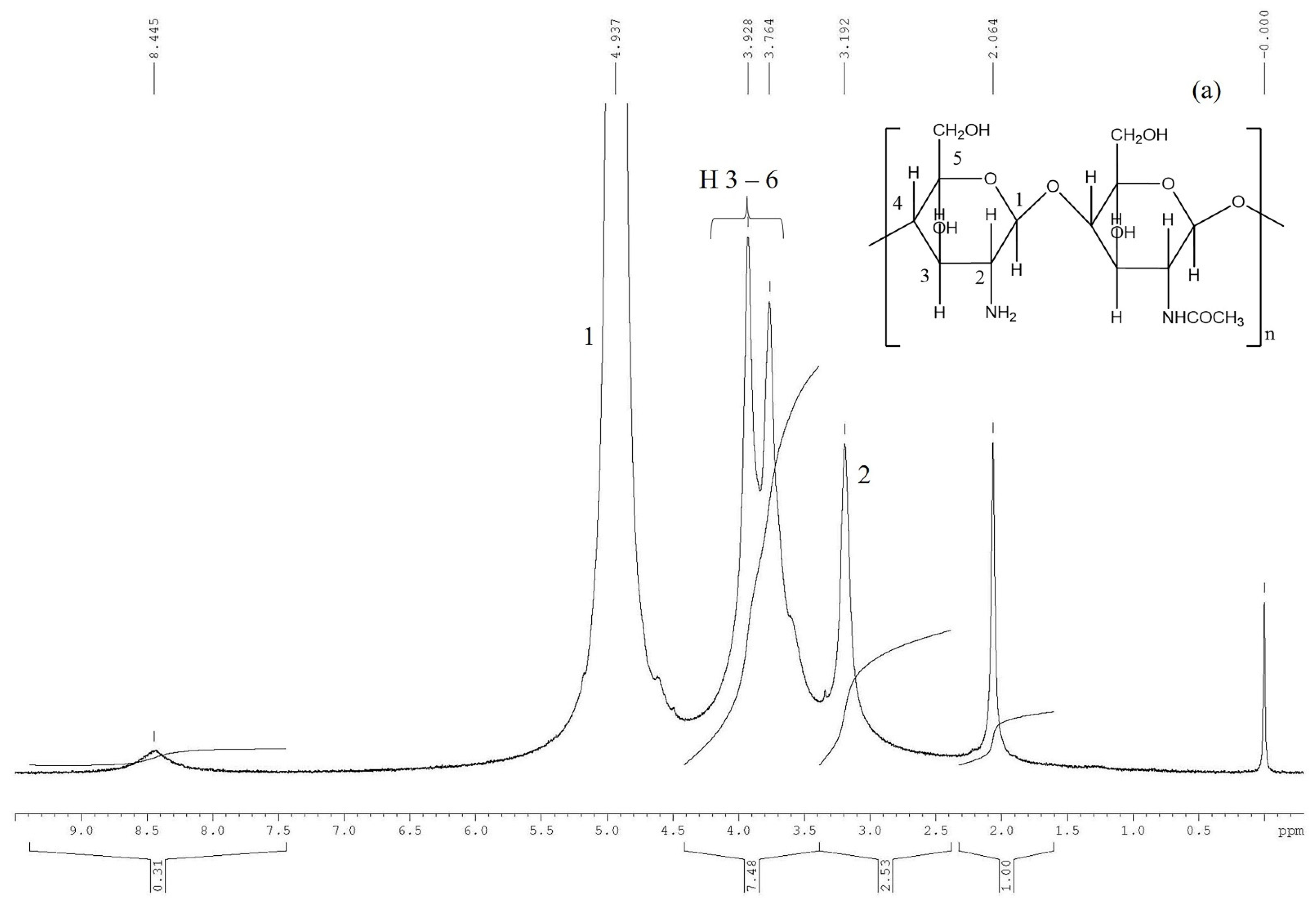
1H NMR spectroscopy was employed to obtain more information regarding the chemical composition of the synthesized polymer.
Figure 2 displays the
1H-NMR spectra of CS, PEGA, and CS-PEGA (molar ratio NH
2:PEGA = 1:0.5 in PEGA synthesis feed).
The
1H NMR spectrum recorded for CS (
Figure 2a) reveals the characteristic signal of methyl protons from the chitin residue, the resonance peaks of the acetylated group found at 1.90 ppm, and the peak corresponding to the H
2 methine protons of the deacetylated groups at 3.01 ppm. H
3, H
4, H
5, and H
6 protons of the deacetylated and acetylated groups were observed in the 3.5–3.8 ppm range. Based on the spectrum, the hydrolysis degree of the CS was determined to be D = 88%.
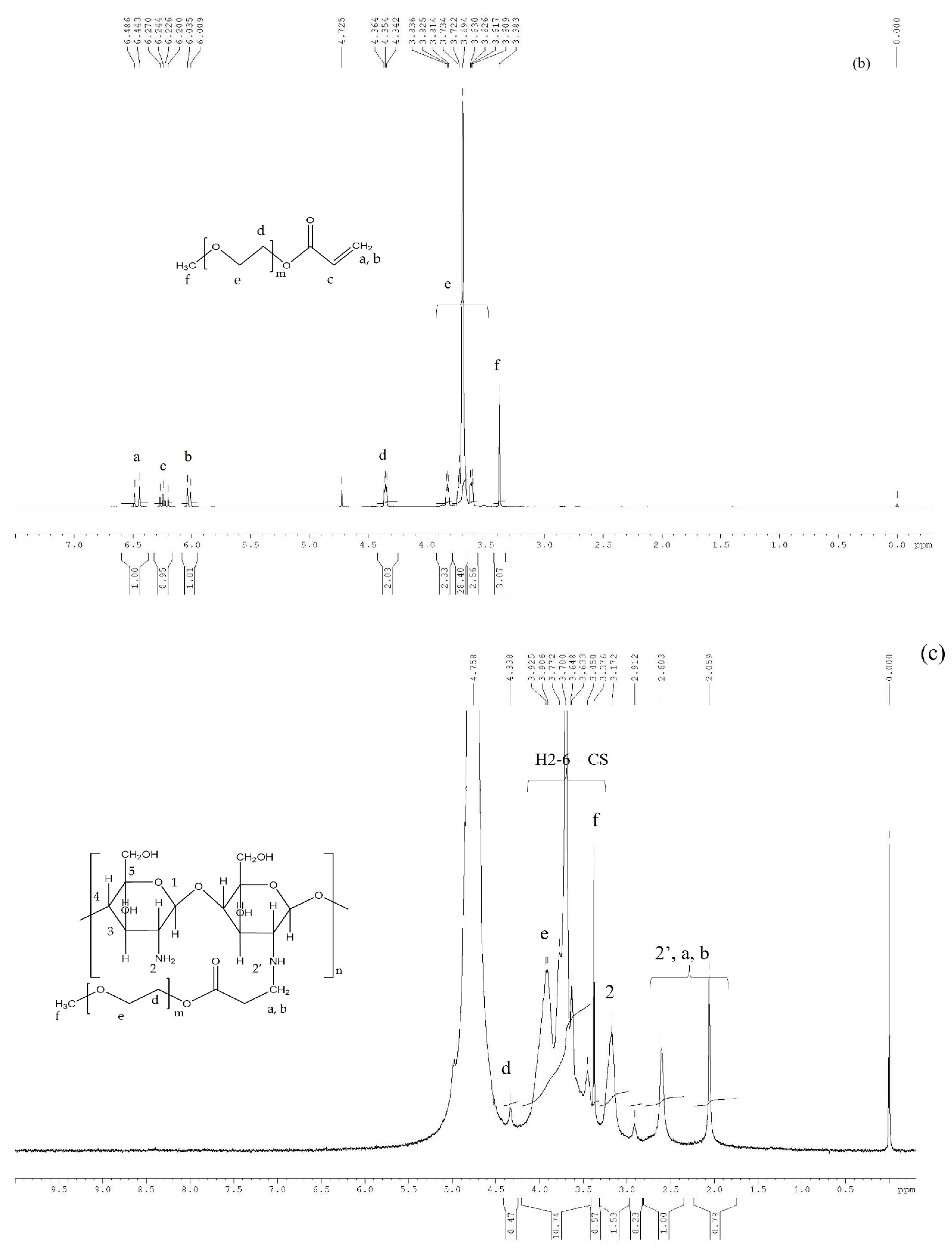
The PEGA
1H NMR spectrum (
Figure 2b) was recorded in D
2O and the characteristic observed peaks were as follows: 6.44 ppm (CH
2 = CHCOO), 6.24 ppm (CH
2 = CHCOO), 6.005 ppm (CH
2 = CHCOO), 4.35 ppm (–COOCH
2–), 3.6–3.83 ppm (–COOCH
2CH
2–, –OCH
2CH
2–, –CH
2CH
2OCH
3), and 3.38 ppm (–OCH
3).
In the case of CS-PEGA, compared to CS, the peaks corresponding to the (–COOCH
2CH
2–) and (–NH-CH
2CH
2COO–) appear at 4.33 ppm and 2.60 ppm, respectively. A clear signal at 3.37 ppm corresponds to the (–OCH
3) group of the PEGA structural unit. The signals of PEGA and in-chain methylene groups overlap with the signals of H
3, H
4, H
5, and H
6 protons of CS. The CS-PEGA spectrum (
Figure 2c) was similar to that obtained by Han et al., also confirming the successful synthesis of CS-PEGA [
30].
The obtained spectrum was further used to calculate the CS substitution yield. Previously, the calculation of DS used the peak intensity of the PEG graft (–COOCH
2–, 4.33 ppm) and (–CH
3, 2.00 ppm) specific to chitin residues. Thus, DS calculated using Equation (1) was around 16%.
However, because of the influence of neighboring peaks, such calculations may be highly biased, and a different protocol was used for DS calculation taking into consideration only the CS backbone peaks:
where I
2 is the integration of the peak found at 3.17 ppm, corresponding to the unmodified chitosan rings, and I
2′ represents the integration of the modified chitosan, probably situated at 2.6 ppm. However, the NMR spectrum previously reported was slightly different [
30] and in our case, the 2′ peak is overlapping with the peaks corresponding to the PEG methylene protons, a and b. Therefore, the 2′ integration value was indirectly obtained using the calculated CS hydrolysis degree, through Equation (3):
where D represents the calculated hydrolysis degree.
Finally, the obtained functionalization degree using Equation (2) was DS = 20%, much lower than that previously obtained by Han et al. In principle, higher substitution degrees may be reached (e.g., a 1:1 NH2:PEG molar ratio led to a DS of 51%) but our goal was to leave a certain amount of amine groups available while achieving complete water solubilization of CS-PEGA.
2.2. Preparation and Characterization of CPH
The proposed pathway to obtain CPH is a double crosslinking technique in reverse emulsion (
w/
o) [
10,
11,
16,
17,
18,
19]. The main problem related to the crosslinking procedure consists of the CS chemical modification impact on amine groups’ availability for further reactions. Furthermore, the improved water solubility of CS-PEGA should provide better preparation conditions for the particulate hydrogel’s formulation. The prepared systems were evaluated according to the following parameters: size and polydispersity, morphology, and zeta potential FTIR.
The selected method for particle preparation was the double crosslinking (ionic and covalent) technique in a water-in-oil emulsion, the advantage being the use of a lower amount of covalent crosslinker, which leads to a decrease in final product toxicity. The fundamental principle of the process is the formation of an interconnected/interpenetrated network in the first stage through the majority ionic crosslinking process, followed by the covalent crosslinking step. The experimental program concerning the preparation of CHP is presented in
Table 1.
The parameters taken into consideration for the preparation process are CS-PEGA initial concentration, NH2/TPP molar ratio, and stirring speed in the emulsion formation phase. The initial water/oil phase ratio, surfactants concentration, and GA content in the organic phase were kept constant throughout the study.
Samples from A1 to A4 were obtained in the first experiments of the study and the molar ratio NH2/Na5P3O10 was varied at 1:0.5 (A1) and 1:1 (A2), the time of the ionic cross-linking process was varied at 20 min (A3) and 40 min (A4), while the water/oil phase ratio, surfactants concentration, and GA content in the organic phase were kept constant. SEM microphotographs showed a nonuniform spherical shape, which suggests that the amount of the ionic crosslinker (A1 and A2) or ionic reaction time (A3 and A4) were insufficient for their favorable formation. In addition, the formation of aggregates with average sizes of 2–3 μm and a fairly wide dimensional polydispersity, proves that they couldn’t be used in further investigations. After the first stages of optimization, which involved increasing the time of the ionic cross-linking/polymer ratio and molar ratio NH2/Na5P3O10, the prepared CPH presented a spherical shape, were individualized, and the diameter and polydispersity were much reduced compared to the first attempts.
The preparation yields (
Table 1) revealed that the initial CS-PEGA amount was diminished during thorough purification procedures. Final yields were around 80% (samples A5, A6) at lower speeds and decreased at higher speeds to 76% (sample A7). Although yield variation is almost negligible, it could be considered that the centrifugation-washing cycles led to fractionation through the loss of the particle populations having the smallest size. Thus, after correlating the preparation yields with the preparation parameters, it may be observed that the highest yields are obtained at the lowest emulsification speeds because the emulsification conditions (stirring speed) favor the formation of larger size particles. The formation of larger size particles may lead to the encapsulation of CS-PEGA material with low network connectivity as compared with lower size particles. In principle, the polymers which do not benefit from crosslinking during the preparation process are eliminated during the purification step, contributing to lower yields. Thus, larger-sized particles may encapsulate free chains that are not eliminated by purification, increasing the final yields.
Particle size measurements confirm that preparations performed at lower emulsification speeds result in larger particle size. Furthermore, the initial concentration of the CS-PEGA affected the preparation yields. Thus, switching the initial concentration from 0.5% to 0.35% (wt) led to a decrease in the preparation yields from 78% to 56% together with a drastic reduction in particle average size from A8 to A9. On the other hand, increasing the polymer concentration to 0.75% (wt) also led to diminished yields associated with the loss of free polymer. Higher polymer density may affect the crosslinking processes, both ionic and covalent. Finally, the increase in TPP amount is correlated with a particle size reduction, from 562 nm (A8) to 141 nm (A10) and, consequently, with diminished yields from 78 to 59%.
CPH samples were also analyzed by zeta potential measurements and
Table 1 summarizes the arithmetic average of zeta potentials. The analyzed samples were dispersed in distilled water at a mass concentration of 1.25 g/L and sonicated for 15 min to avoid agglomeration. In principle, a large negative or positive value of zeta potential for suspended particles in an aqueous medium indicates that the system is stable against flocculation or coagulation. As shown in
Table 1, the obtained results for samples A8, A9, and A10 indicate that CPH have good stability in aqueous suspension. The measured zeta potential values are explained by the lower availability of the amine groups due to PEG grafting and covalent crosslinking.
2.2.1. CPH Characterization
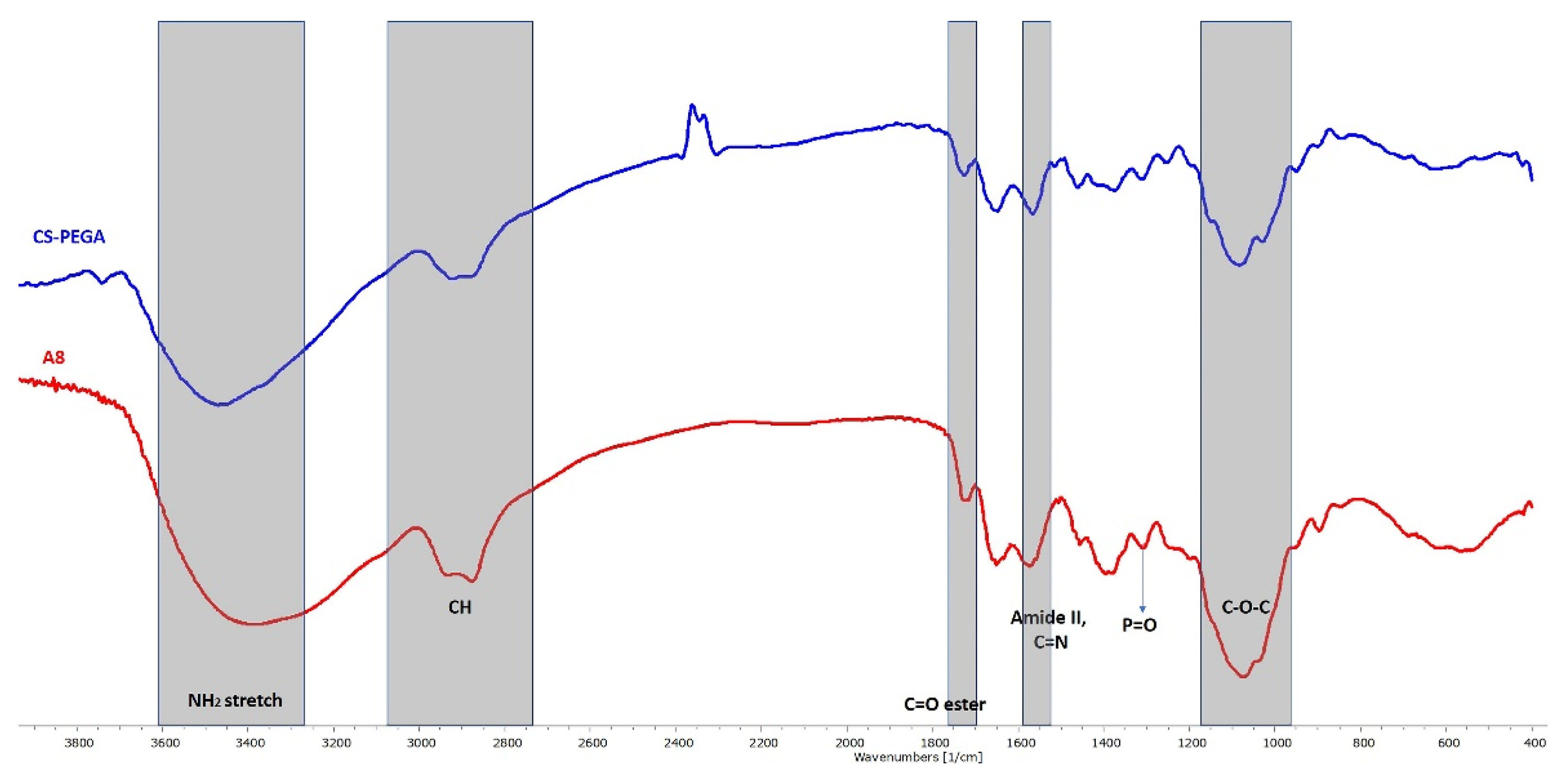
To better understand the polymer network formation after the double crosslinking process, CPH were first investigated by FTIR spectroscopy. The recorded spectra revealed a similar profile for all CPH and confirmed the success of both crosslinking processes, thus only the FTIR spectra for sample A8 are displayed (
Figure 3).
Table S2 presents in detail the characteristic absorption bands. The first crosslinking process confirmed was the ionic one, the spectra revealing at 846 cm
−1 the presence of a new bond formed between P
3O
105− anions of TPP and NH
3+ cations from CS-PEGA. The characteristic absorption bands corresponding to the covalent crosslinking process, namely C=N, were observed at 1573 cm
−1 [
13,
16,
31,
32,
33,
34].
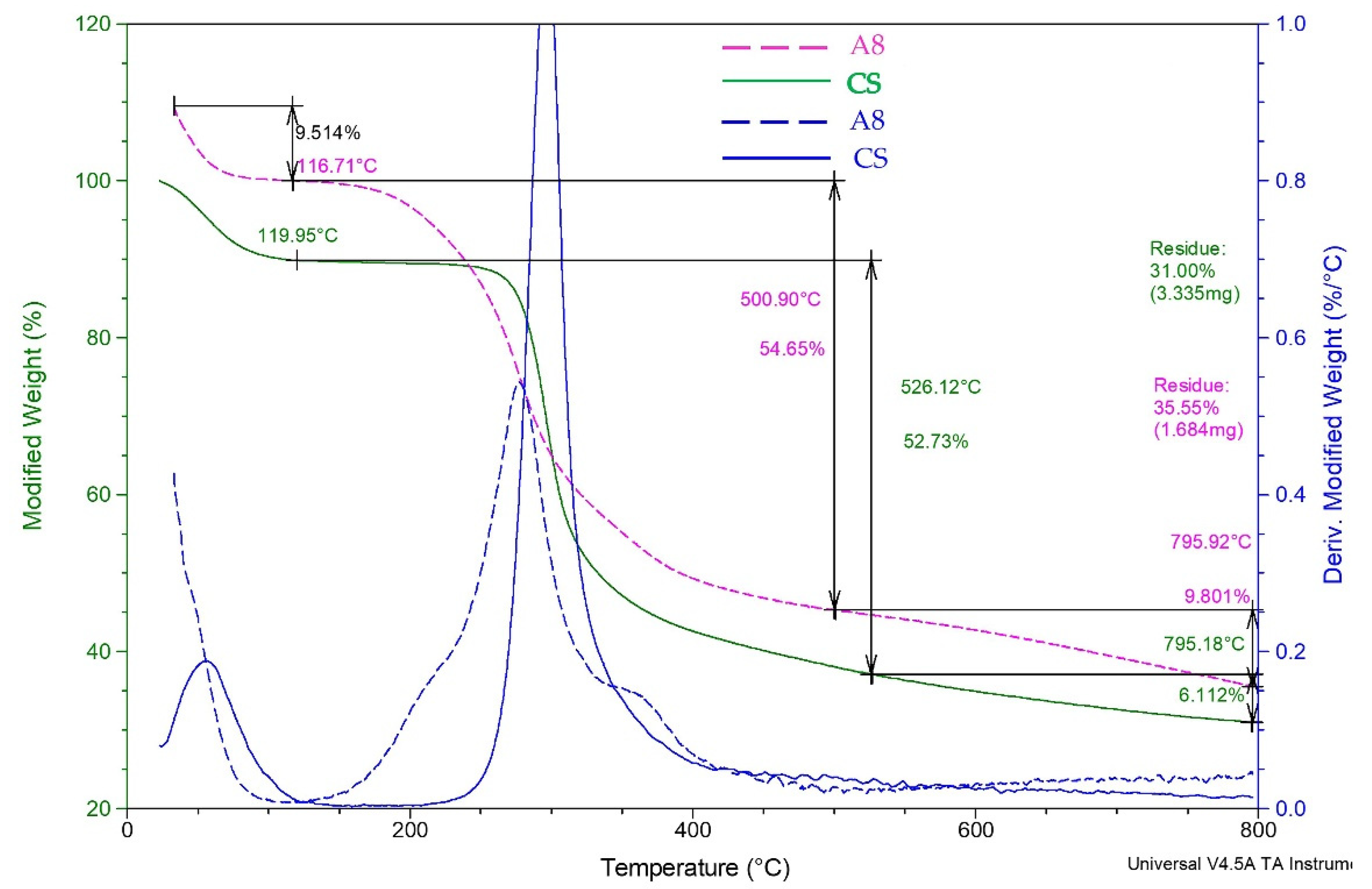
Furthermore, the CPH were also subjected to TGA analysis (
Figure 4). The data results from the TGA analysis indicated that CPH decomposition proceeds in three steps as shown in
Figure 4. TGA thermograms (
Figure 4) showed a steady weight loss in the temperature range of 100–500 °C. The slight weight losses (9.514% and 10.13%) up to 116 °C, and 119 °C respectively, correspond to adsorbed water and moisture loss by the CS and CPH. The second decomposition step (A8—54.65%, CS—52.73%) in the temperature range of 116/119–500 °C is mainly due to the degradation of oligosaccharides and PEG. The main difference between CS and A8 samples is related to the early onset of A8 degradation due to the chemical modification in correlation with the native CS. Thus, the presence of covalent GA bridges is probably the reason for the decrease in thermal stability. Moreover, the A8 sample presents a distinct extra shoulder in DTG corresponding to the additional PEG, between 350 and 400 °C. Overall, thermal analysis confirms the structural composition of A8 concerning CS-PEGA content and covalent crosslinking. Supplementary TGA analysis performed on A9 and A10 samples (
Figure S1, Supplementry Material) showed that different preparation conditions, such as concentration and TPP amount, do not significantly influence the TGA profile.
2.2.2. CPH Morphology
SEM technique was used to inspect the morphology of the prepared submicronic gel systems,
Figure 5. Results indicate that they have a spherical shape, are well individualized, generally have a submicronic diameter, and reduced dimensional polydispersity. The adapted emulsion double crosslinking procedure allows the obtaining of clear particulate systems with relatively low dimensional dispersity, as observed from SEM micrographs. Furthermore, it may be remarked that particles are homogeneous, separated, and lack impurities resulting from surfactants, crosslinkers, or CS-PEGA residues from faulty emulsion crosslinking.
The influence of the preparation parameters is well reflected in the size of CPH, especially the lowering of the concentration of CS-PEGA, which produced particulate gels with the lowest dimensions. A visual comparison between the A8 and the A9 samples (
Figure 5) clearly shows the dimensional decrease.
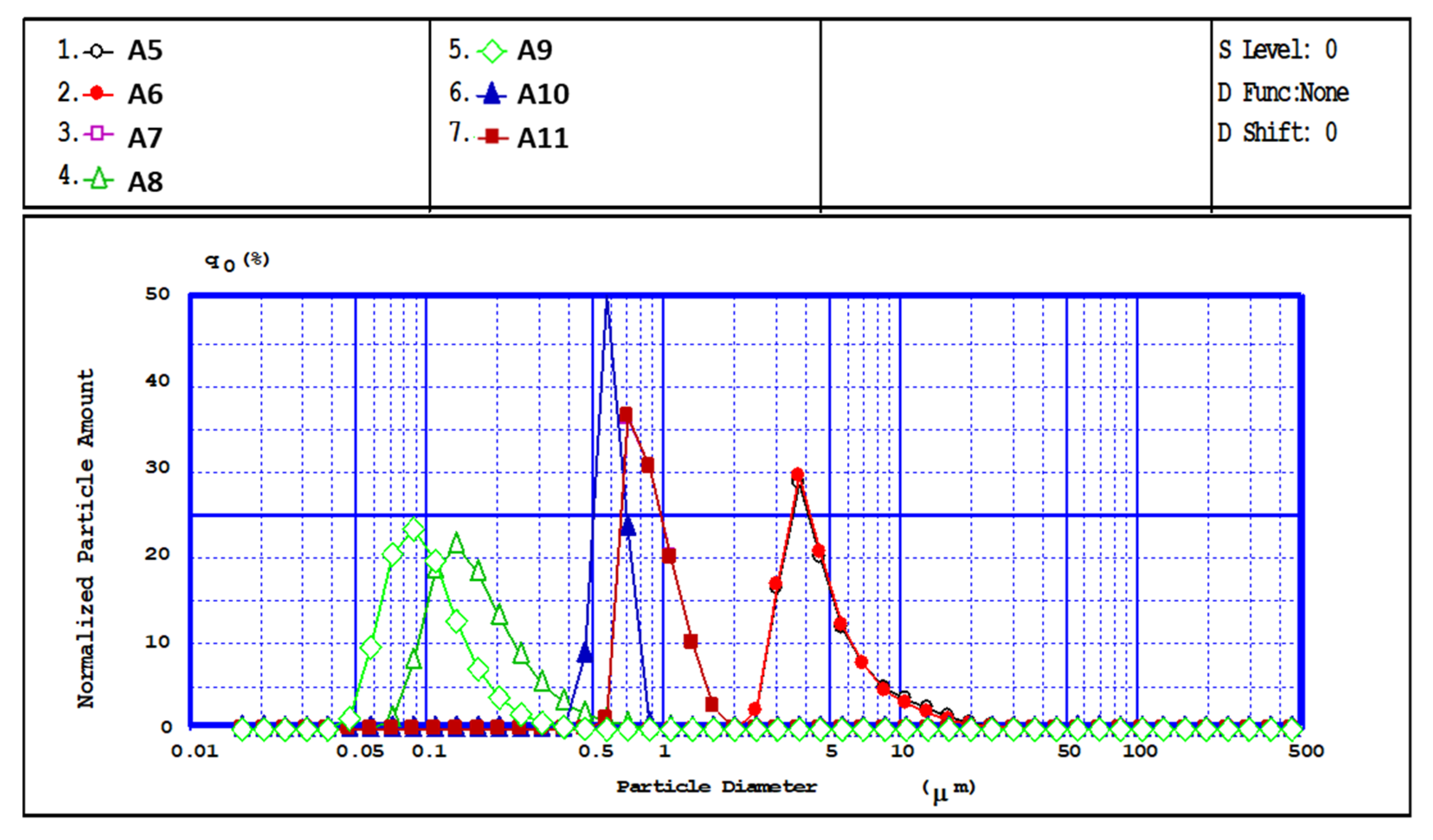
Furthermore, a more quantitative idea of the dimensional distribution of particle populations is obtained by dimensional analysis via laser diffraction (
Figure 6), which provides the diameter and dimensional polydispersity of CPH. The analysis was carried out in acetone, known as a poor solvent for CS, and the measured average diameters were in good agreement with SEM observations. The experimental data confirmed that the particle size is dependent on the polymer solution concentration and stirring speed used during preparation. Thus, a higher stirring speed implies higher energy dissipated in the stirred emulsion, i.e., a higher energy transfer, which causes the emulsion to break into smaller droplets, resulting in nanometric particles. The granulometric distribution curves of the CPH obtained (determined by laser beam diffraction (LD)) have a unimodal allure and a diameter falling in the submicronic range, depending on the preparation conditions (
Figure 6 and
Table 1). Thus, for samples A5–A8, after increasing the stirring rate from 5000 to 15,000 rpm/min, it was noticed that particle size decreases due to better dispersion and formation of small droplets in the emulsion phase.
The influence of the concentration of CS-PEGA solution on particle size was also analyzed for three different polymer concentrations, namely 0.35%, 0.5%, and 0.75%, while the other parameters were kept constant (samples A 8, A9, A11,
Table 1). The lowest polymer concentration led to particles with sizes in the nanometric range and an average diameter of 89 nm, and higher concentrations resulted in larger particles. Additionally, increasing the polymer solution concentration led to a considerable increase in the mean diameter and particle polydispersity, as well as a decrease in agglomeration tendency. The observed tendency is explained by the fact that higher concentrations lead to an increase in solution viscosity, and therefore to the formation of larger emulsion droplets, as presented by other studies [
35,
36].
Another studied parameter, the increased polymer/ionic crosslinker ratio (A8 vs. A10), caused a noticeable decrease in particle diameter, from 562 to 141 nm. Better availability of TPP may favor ionic gelation in smaller colloids in the early emulsification process. Furthermore, in the ionic crosslinking phase, due to the increase in the cohesion of the polymer matrix, the emulsion droplets should better maintain their colloidal stability. Covalent crosslinking improves and fixates the resulted particles which otherwise may suffer modifications in size.
Overall, considering the small size and dimensional uniformity of the prepared particulate hydrogels, the use of CS-PEGA in double crosslinking processes is an improvement on previous efforts. TPP ionotropic gelation alone leads to particle sizes of around 300 nm [
14] while previous double crosslinking attempts on native or modified CS led to a minimum particle size of 500 nm. The current work shows that PEG grafting up to 20% does not affect the capacity of CS to undergo crosslinking reactions because of sterical effects and amine moieties coverage.
2.2.3. Water Uptake
The swelling capacity was studied in two environments, which were closely related in terms of CPH drug loading and release potential. The acidic/basic standard buffer medium was chosen because, in general, the swelling behavior of CS hydrogels can be influenced/dependent on external triggers such as pH, ionic strength, the temperature of the environment, and the degree of cross-linking. Furthermore, in the case of CS hydrogels which are pH-dependent, the loading/release behavior of active principles is related to a higher swelling capacity in the acidic environment due to a significant increase in their volume which leads to an accelerated drug caption inside the network [
36].
To better understand the interaction between CPH and the aqueous environment, swelling studies were performed in acidic (pH = 3.3) and respectively alkaline (pH = 7.4) mediums, using the gravimetric method. The water swelling behavior gives insights into doubly crosslinked CS-PEGA interactions with selected environments and its adequacy for the drug loading and release process.
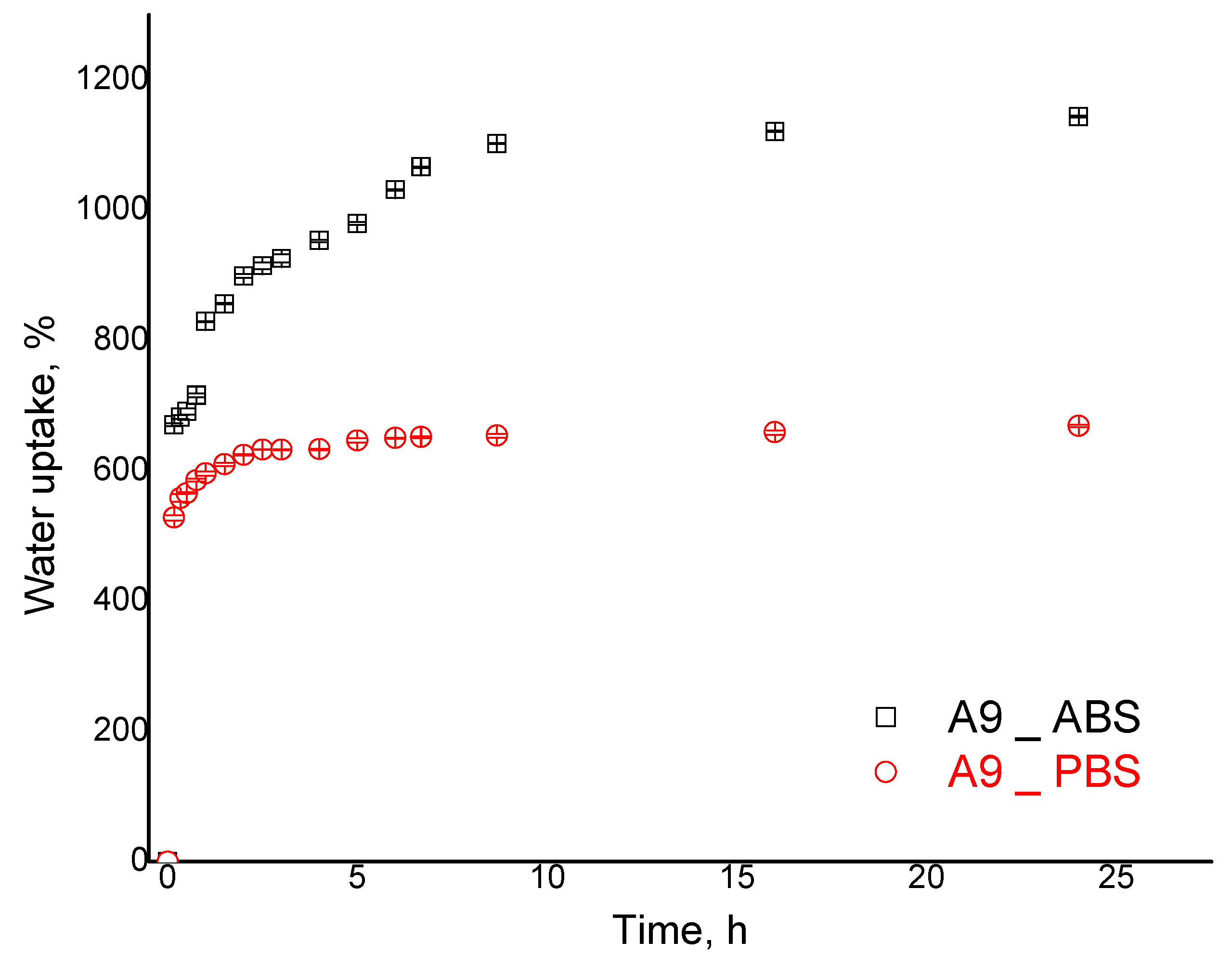
The kinetic data are presented as an example for sample A9 (
Figure 7). The results revealed two phases in the acidic environment: a fast increase up to 800% (sample A9) in the first 90 min, followed by a slower water uptake increase rate (90–480 min); the maximum value is reached after 24 h at 1144%. This behavior may reflect the double crosslinked nature of CPH. In the case of PBS, the maximum value of the swelling degree was smaller at 669%. Swelling kinetics presented in
Figure 7 reveal that an equilibrium phase is established in both acidic and alkaline environments, showing good stability of the particulate hydrogel’s formulations. Although in the alkaline environment such behavior would be expected because of diminished electrostatic repulsions among polysaccharide chains, the stability in the acidic environment confirms successful covalent crosslinking.
The water uptake in both environments proceeded quickly, demonstrating good water–CS interactions, most likely favored by hydrophilic PEG grafts. However, the remaining free amine groups dictate the swelling behavior as acidic ABS determined significant water swelling. Therefore, we can infer that CS-PEGA provides a good prospect for nanoparticulate hydrogels by diminishing the large difference in CS behaviors in acidic and alkaline environments.
Furthermore, the water interactions of CS-PEGA nanoparticulate hydrogels are discussed according to the maximal water uptake reached in the equilibrium swelling phase (
Table 2). Generally, water swelling of homogeneously crosslinked polymer networks is influenced by polymer–water interactions and the network’s capacity to expand its volume, influenced in turn by chain conformations, flexibility, and network crosslinking density. However, in the case of double crosslinked particulate hydrogels, the crosslinking is heterogeneous. GA covalent bridges are formed starting from the organic phase–water interface. In principle, because of its high reactivity with amine groups, GA diffusion into the water phase is limited and it can be considered that a layer on the surface of particles benefits from the covalent crosslinking while the inner polymer matrix remains mostly ionically crosslinked. The lack of crosslinking anisotropy further generates specific particle–water interactions. The outer layer of covalently crosslinked polymer is the key factor that determines the swelling behavior. By comparing the water uptake of samples A8 and A10 (
Table 2), with an increasing amount of ionic crosslinker, it was noticed that there were insignificant differences between the acidic and alkaline environments. Such behavior is in opposition to the increasing network crosslinking density and also to the possible network dissociation due to the neutralization of the ionic bridges in an alkaline environment.
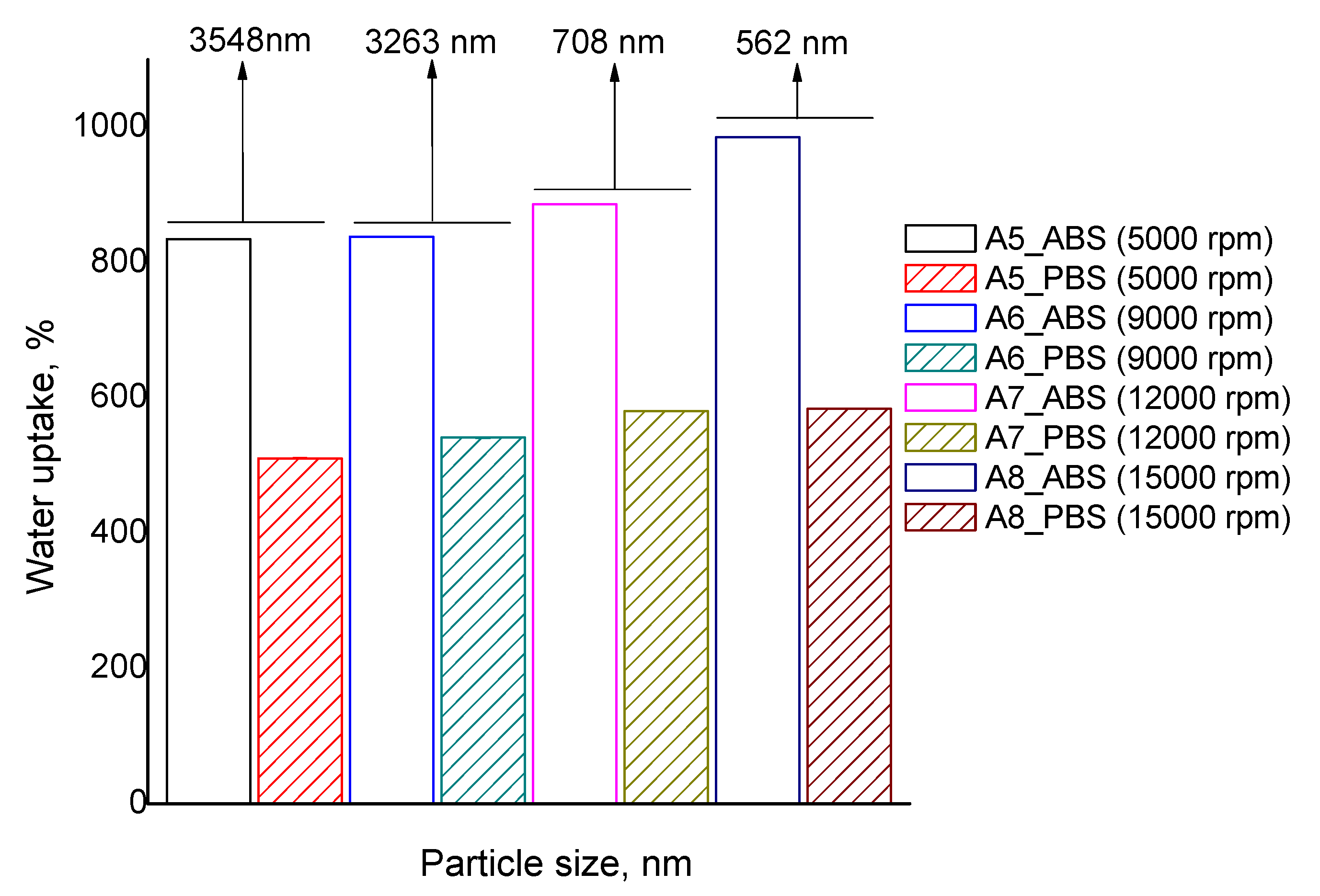
In principle, an increase in the surface of the particles is associated with a decrease in the particles’ average diameter. Taking this into consideration it can be inferred that particle size is influencing the swelling behavior. The evolution of water uptake according to particle size is presented in
Figure 8.
The decrease in particle size of the considered samples is only due to a more energetic initial emulsion stirring and the other parameters influencing crosslinking, such as ionic/covalent crosslinkers and polymer concentration, were kept constant. The acidic swelling for particles with an average diameter of 3540 nm is around 835% (sample A5), and a decrease in particle size to 562 nm leads to higher swelling, up to 985% (sample A8). Furthermore, in the alkaline environment, the swelling increases from 511 to 585%. Thus, water uptake shows a clear dependency on the average diameter of particles in both acidic and alkaline environments, but the effect is more pronounced in ABS swelling.
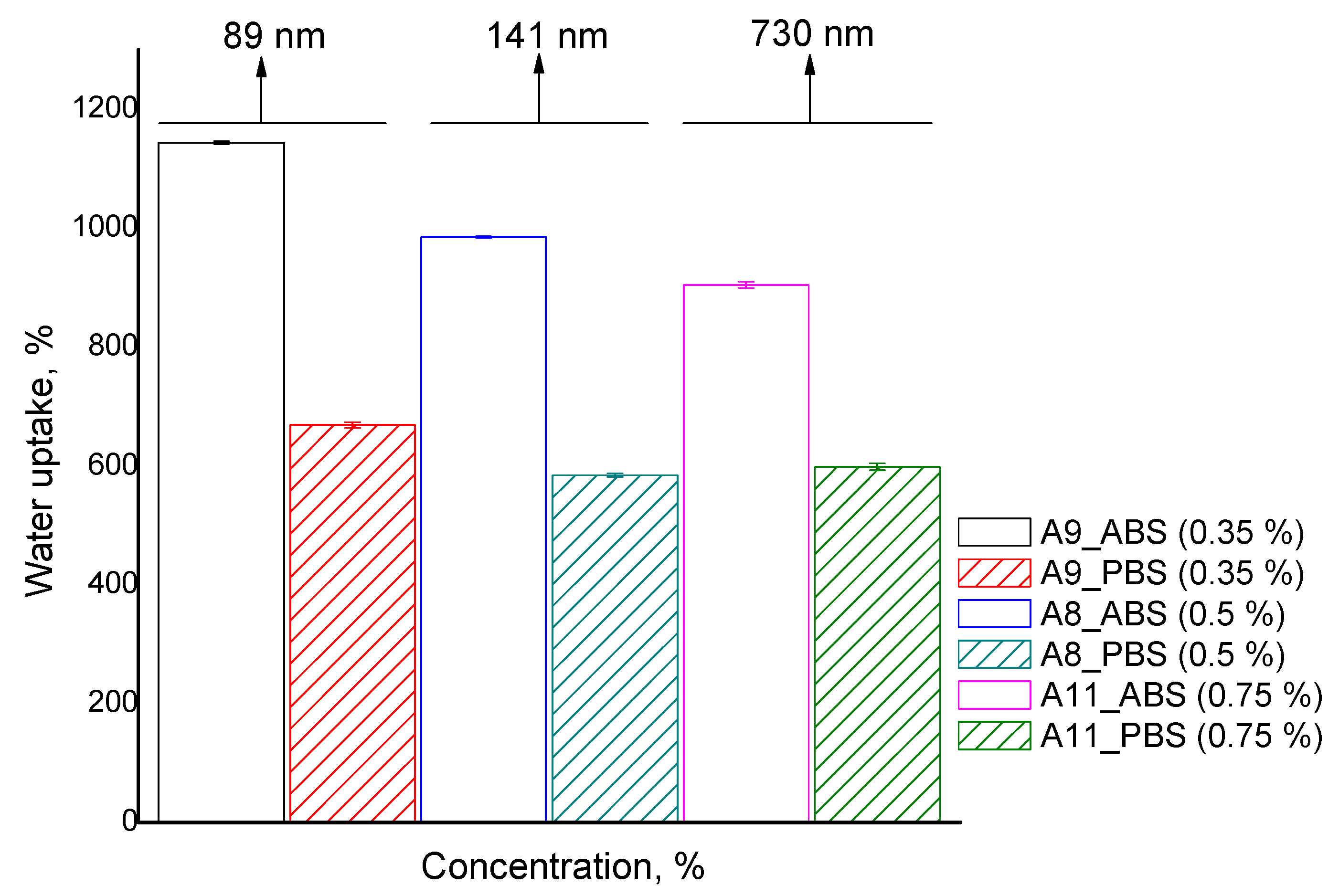
The water uptake experiments were performed to investigate the dependence between the swelling degree and polymer concentration (
Table 2 and
Figure 9). The data presented in
Figure 9 show the evolution of the swelling degree for three different concentrations and it is observed that increasing the CS-PEGA concentration in the emulsion feed diminishes the particle water uptake in both ABS and PBS. However, the swelling decrease is more obvious in an acidic environment, from 1175% (sample A9) to 900% (sample A11). This behavior is to be expected due to more dense polymer networks with a concentration increase.
Taking into consideration the significant differences observed in water uptake behavior in ABS and PBS, it can be affirmed that CS-PEGA particulate hydrogels have a pH-sensitive character. The effect is determined by the amine groups of the CS derivative which didn’t participate in the crosslinking process (both ionic and covalent). In an acidic environment, the amine groups are quaternized by passing into ammonium cations, and the electrostatic rejections between them increase the distance between network chains, and their meshes, leading to higher water uptake. The maximum swelling values are lower in a basic pH compared to an acidic pH, because the amine groups of the CS derivative are deprotonated, therefore, hydrogen bonds are formed in the newly formed polymer network.
The best swelling ratio was achieved by sample A9, obtained at a low polymer solution concentration (0.35%) and with the smallest particle size (89 nm). The higher water uptake for CPH obtained is probably due to the increased agglomeration tendency, which is mainly evident in sample A9, and a low cross-linking density of the polymer matrix.
2.2.4. Drug Loading and Release
The ability of CPH to encapsulate active principles was analyzed using levofloxacin (LEV) as a model drug. LEV is a synthetic antibacterial drug of a broad-spectrum antibiotic belonging to the class of fluoroquinolones, generally used to treat antibacterial infections. LEV is a hydrophilic drug and therefore a suitable candidate for hydrogel particulate systems designed for the treatment of bacterial infections [
35,
37]. Taking into account the swelling results, which demonstrate that in acid pH the swelling ability of CPH is much higher than in weak alkaline conditions, it was decided to load the LEV in acid conditions ensuring a higher amount of loaded LEV. The LEV content in the supernatant was determined by UV-Vis spectrophotometry (at 287 nm wavelength) based on an established calibration curve. CPH were able to encapsulate between 0.48–0.75 mg drug/mg particles (Equation (6)). Therefore, the LEV loading efficiency values calculated with Equation (7) were between 47.0–76.0%, showing a high capacity of the CPH to support the biologically active compound. The amounts of encapsulated LEV in CPH and loading efficiencies are outlined in
Table 3.
The values obtained at different time intervals showed that after 72 h the amount of encapsulated LEV ranged from 0.48–0.75 mg per mg of CPH. The kinetic data presented as an example for samples A8 and A9 (
Figure S2), showed that LEV is loaded in two phases, a fast process during the first 10h, followed by a slower drug loading until 72 h. When comparing the drug loading profile with the swelling results, a similarity could be observed.
For the LEV release study, pH = 7.4 was considered appropriate for a future systemic administration, acidic release conditions being used only for oral formulations. The LEV release capacity of CPH was analyzed by the diffusion method in phosphate buffer pH = 7.4 at 37 °C. The released LEV for all samples is presented in
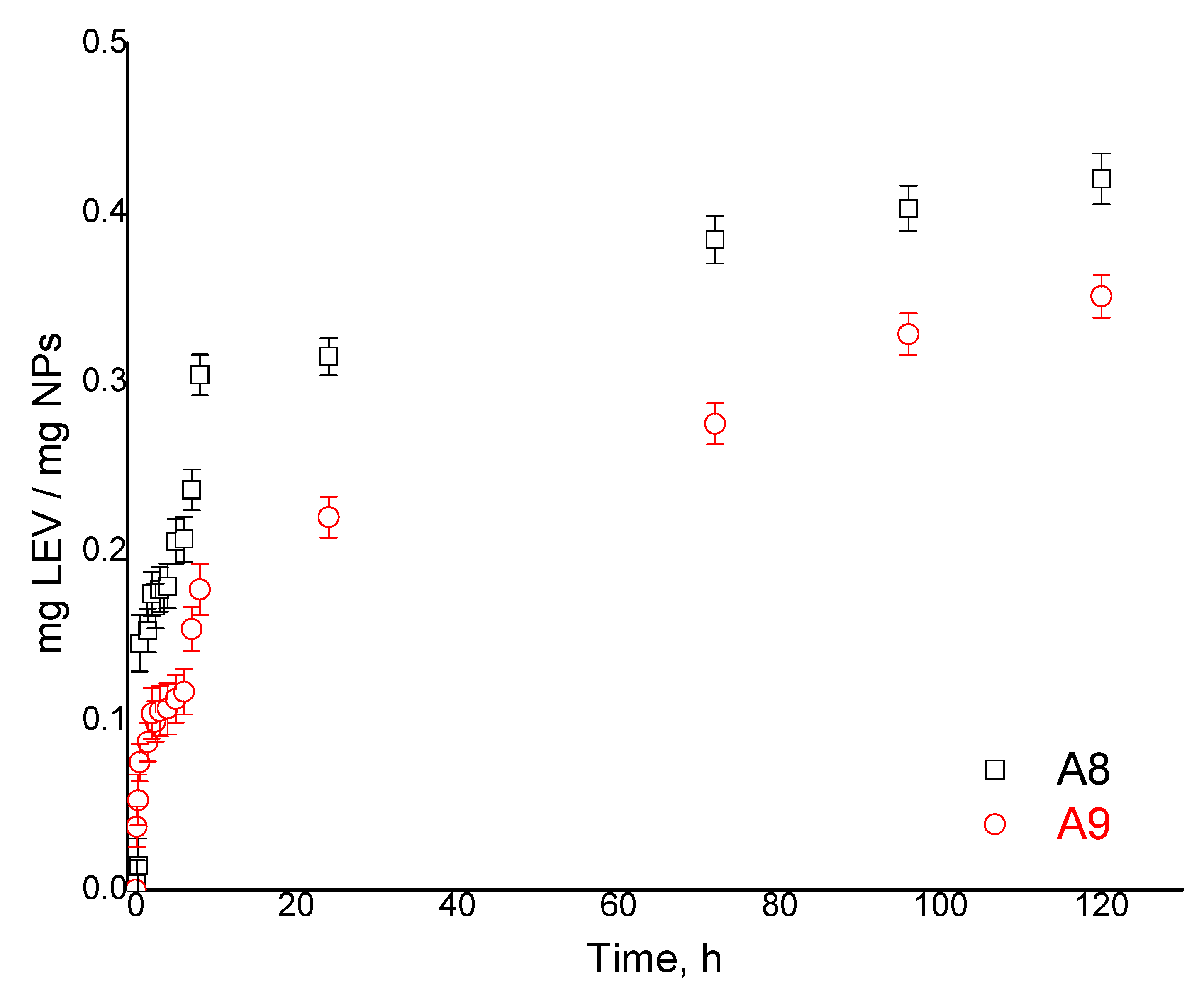
Table 4 and the kinetic data for two representative samples, A8 and A9, are displayed in
Figure 10. One can observe a fast release phase, which is reached within the initial 10 h, followed by a slower phase (characterized by a slow release) until 120 h. The sustained release of LEV can be explained by the fact that the released drug was adsorbed in the CPH network due to the excellent hydrophilicity of the polymer network, which it seems is retained after the crosslinking procedures.
By analyzing the influence of preparation parameters on the release ability of the CPH, similar behavior as for drug loading was observed. The maximum released amount of LEV varied between 0.27 and 0.42 mg/mg CPH (
Table 4). Furthermore, when calculating the efficiency of LEV release for all samples, values between 41 and 60% were obtained (
Figure S3), the highest efficiency being in sample A8, which has also shown a high water uptake. The analysis of the release kinetics was achieved based on the Korsmeyer–Peppas mathematical model (
Figure S4).
According to the literature [
38,
39,
40,
41], the drug release process is dependent on the method of obtaining the particulate systems, their dimensions, and the physical–chemical properties of the polymer support used. After analyzing the experimental kinetic data for the interval 0–480 min, it was possible to determine the diffusion exponent based on the equations listed in
Table S3. As an example,
Figure S4 shows the results obtained for samples A8 and A9. One can observe that for sample A8, the value of the exponent
n, which characterized the release mechanism, is quite close to 0.5 (0.5 <
n < 1.0), indicating a transport/release mechanism dominated by diffusion. Furthermore, it can be stated that sample A11 exhibits an abnormal, non-Fickian diffusion, the drug transport process through the polymer matrix being governed by both diffusion and swelling.
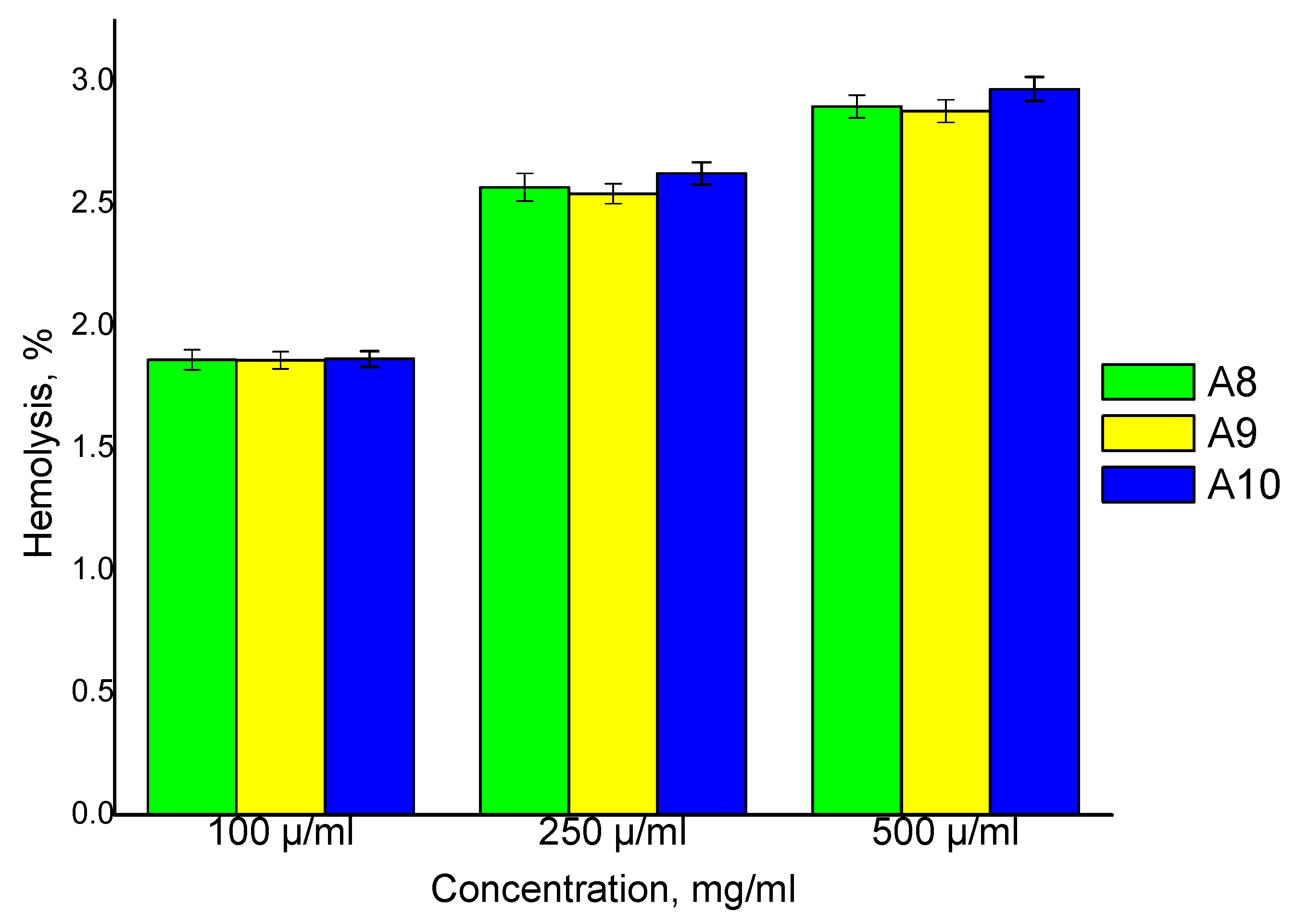
The CPH hemolytic potential test is a mandatory requirement for blood-borne materials, since their interaction with blood components may lead to erythrocytes lysis. For this reason, the effects of the prepared CPH on blood were evaluated using a hemolysis assay. The results obtained from this study revealed that the degree of hemolysis increases with an increase in the CPH concentration (
Figure 11). The degree of hemolysis for three different samples (A8, A9, A10) was analyzed and the results showed that there were no significant differences between them at the same concentration. It has been found that the prepared CPH shows very good compatibility with normal blood (<10% compared to the positive control) [
41].
The viability of HDMVEC cells (Primary Dermal Microvascular Endothelial Cells) in 2D culture treated with nanoparticulated formulations (A9) was not affected by the presence of glutaraldehyde in the CPH structure (
Figure S5).
2.2.5. Cell Proliferation
For cell viability, a CellTiter-Blue
® Cell Viability Assay (Promega, Madison, WI, USA) was used. Cells were seeded into a 96-well flat-bottomed tissue culture plate at a density of 3000 cells/well and allowed to adhere to the plate by incubating at 37 °C under 5% CO
2 overnight. Following cell attachment, the cells were incubated with 0.5 and 1% CPH for 72 h. After 72 h treatment periods, 50 μL of cell viability solution was added to each well and the plate was reincubated for 4 h before fluorescence recording using a multiplate microplate reader (FilterMax F5, Sunnyvale, CA, USA). As one can see from
Figure S5, analyzed CPH does not affect cell viability according to statistical analysis. The statistical analysis of significance in differences was performed using a GraphPad Prism 7 Anova
t-test.
CPH formulation-based CS-PEGA does not affect cell viability, thus confirming that they are cytocompatible and can be used as a drug delivery system.
3. Conclusions
This work demonstrates the suitability of CS-PEGA for the preparation of particulate hydrogel systems through double emulsion crosslinking. The relatively low substitution degree of chitosan with PEG-acrylate, about 20% as determined by 1H NMR, ensures sufficient availability of the remaining amino functions for further ionotropic gelation with TPP and covalent crosslinking with glutaraldehyde in water in oil emulsions. The outcome of the particle preparation procedure may be finely tuned by playing with process parameters such as water phase concentration, emulsification stirring speed, and amount of ionic crosslinker. According to SEM analysis, the CPH have a spherical shape, are homogeneous, separated, and lack impurities. The particle average diameter, measured by laser diffractometry, depends on specific preparation conditions and ranges from 3500 to 90 nm, in agreement with SEM estimations. Particularly, emulsification speed and water phase concentration significantly influence the CPH size and allow fine-tuning, according to the envisaged applications.
CPH behavior in two different aqueous media, acid (pH = 3.3) and basic (pH = 7.4,) was studied, demonstrating a higher water retention capacity for the acidic media, compared to basic media. Furthermore, the water uptake properties are significantly influenced by polymer concentration and ionic crosslinker amount employed in the preparation stage, and they are correlated with the particle size. The drug loading and release capacity of CPH, tested using levofloxacin, showed a good diffusional loading capacity, up to 0.75 mg of LEV per mg of CPH in an acidic environment. Drug rapid release in physiological pH was observed within the first 10 h, followed by a slower phase, characterized by a constant release up to 120 h. Drug release kinetics analysis demonstrated that the drug transport/release mechanism is predominantly diffusional.
CPH have also been tested by hemocompatibility analysis, the results showing a hemolysis percentage of less than 3%, indicating that they are hemocompatible. Cytocompatiblity studies demonstrated that CPH formulations based on CS-PEGA do not affect cell viability, thus confirming their low cytotoxicity and recommending them as pH-dependent drug delivery systems. Further experiments will consider the antibacterial potential of the levofloxacin-loaded chitosan particulate hydrogels.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}