The Importance of Succinylacetone: Tyrosinemia Type I Presenting with Hyperinsulinism and Multiorgan Failure Following Normal Newborn Screening

 , and
, and
Abstract
:1. Introduction
1.1. Tyrosinemia Type I
1.2. Screening for Tyrosinemia Type I
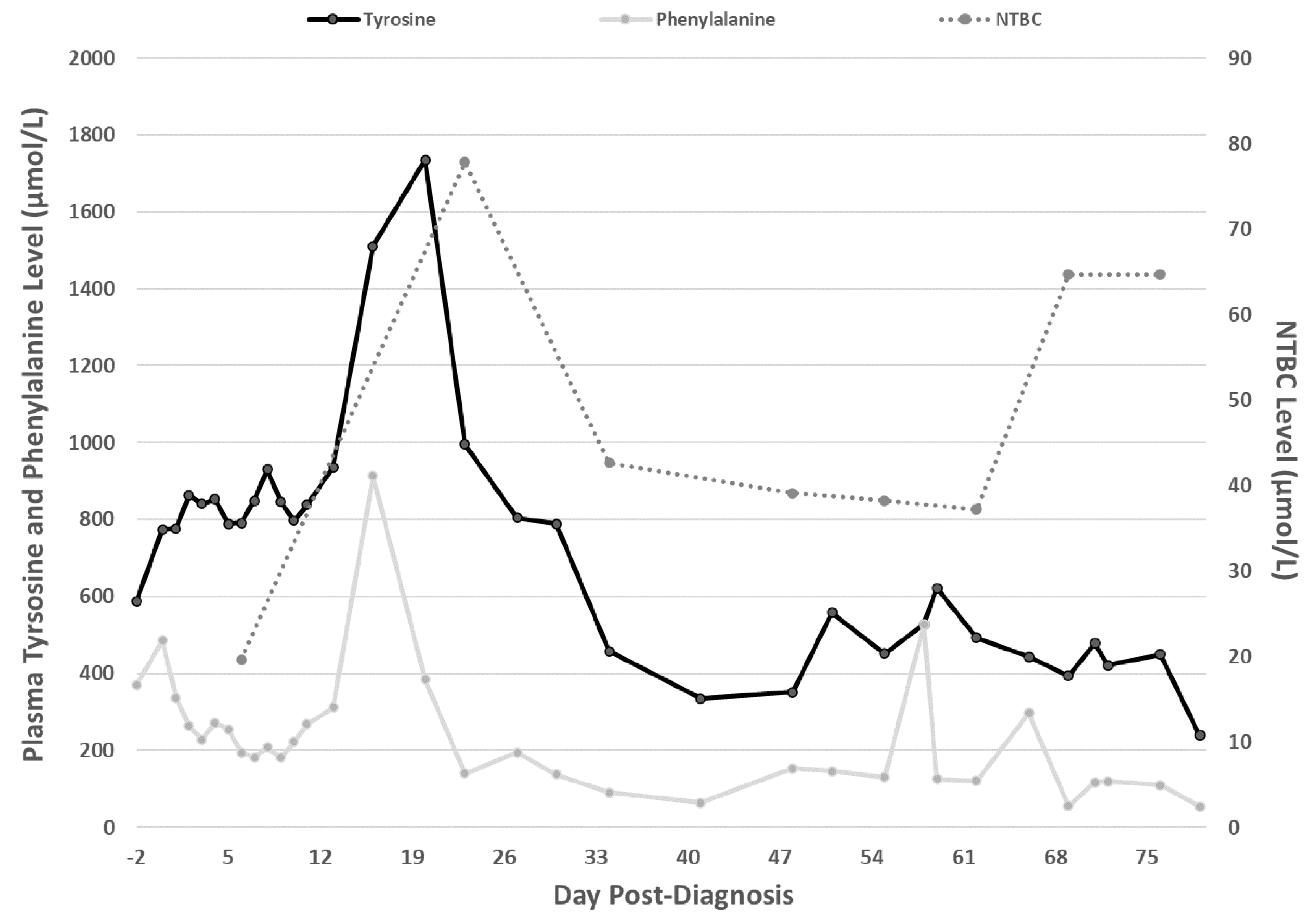
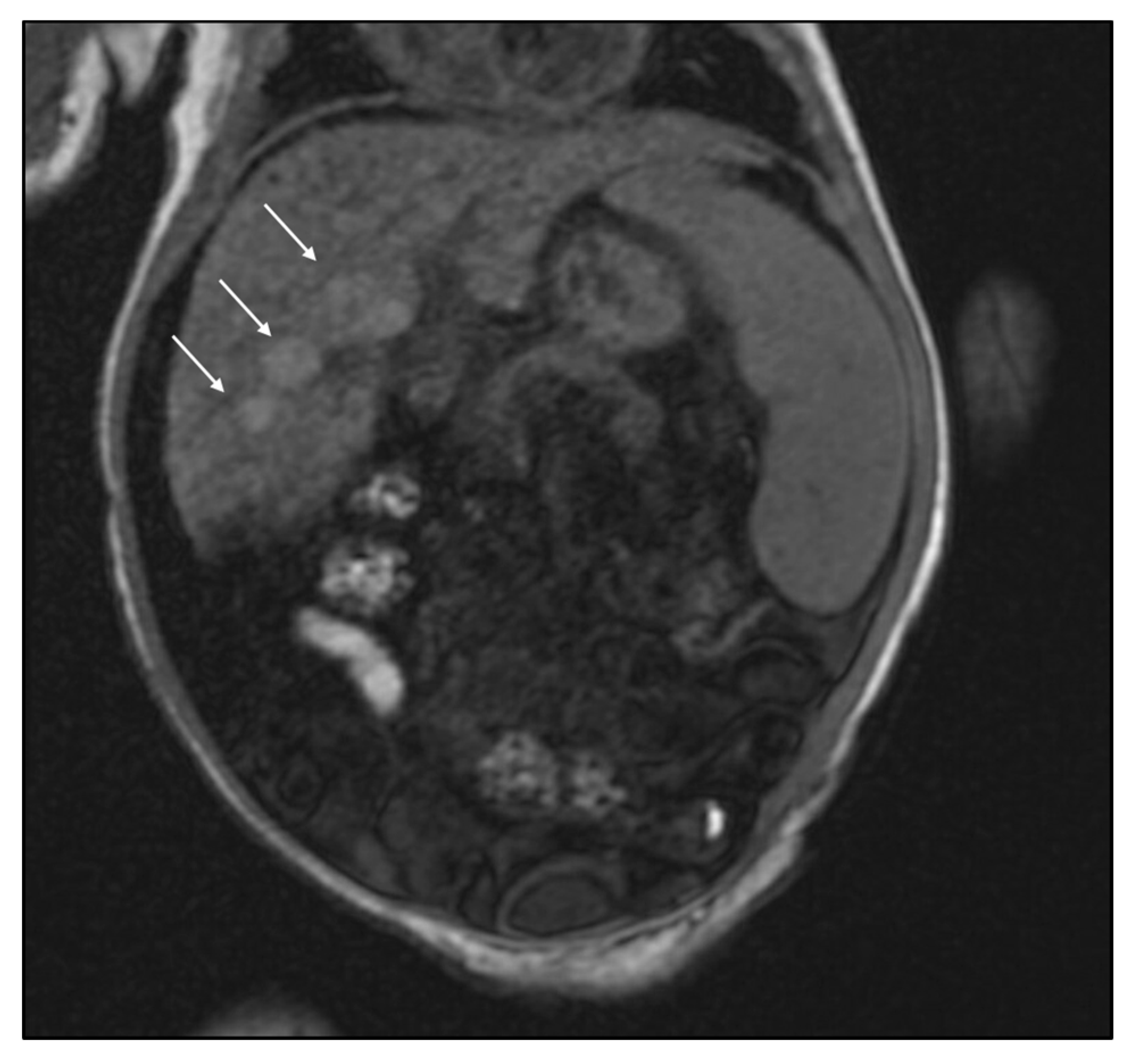
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gentz, J.; Jagenburg, R.; Zetterström, R. Tyrosinemia: An Inborn Error of Tyrosine Metabolism with Cirrhosis of the Liver and Multiple Renal Tubular Defects (de Toni-Debré-Fanconi Syndrome). J. Pediatr. 1965, 66, 670–696. [Google Scholar] [CrossRef]
- Lindblad, B.; Lindstedt, S.; Steen, G. On the Enzymic Defects in Hereditary Tyrosinemia. Proc. Natl. Acad. Sci. USA 1977, 74, 4641–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchesson, A.C.; Hall, S.K.; Preece, M.A.; Green, A. Screening for Tyrosinaemia Type I. Arch. Dis. Child. Fetal Neonatal Ed. 1996, 74, F191–F194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angileri, F.; Bergeron, A.; Morrow, G.; Lettre, F.; Gray, G.; Hutchin, T.; Ball, S.; Tanguay, R.M. Geographical and Ethnic Distribution of Mutations of the Fumarylacetoacetate Hydrolase Gene in Hereditary Tyrosinemia Type 1. JIMD Rep. 2015, 19, 43–58. [Google Scholar] [CrossRef] [Green Version]
- De Braekeleer, M.; Larochelle, J. Genetic Epidemiology of Hereditary Tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am. J. Hum. Genet. 1990, 47, 302–307. [Google Scholar]
- Mustonen, A.; Ploos van Amstel, H.K.; Berger, R.; Salo, M.K.; Viinikka, L.; Simola, K.O. Mutation Analysis for Prenatal Diagnosis of Hereditary Tyrosinaemia Type 1. Prenat. Diagn. 1997, 17, 964–966. [Google Scholar] [CrossRef]
- Bliksrud, Y.T.; Brodtkorb, E.; Backe, P.H.; Woldseth, B.; Rootwelt, H. Hereditary Tyrosinaemia Type I in Norway: Incidence and Three Novel Small Deletions in the Fumarylacetoacetase Gene. Scand. J. Clin. Lab. Investig. 2012, 72, 369–373. [Google Scholar] [CrossRef]
- Sniderman King, L.; Trahms, C.; Scott, C.R. Tyrosinemia Type I. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Spronsen, F.J.V.; Thomasse, Y.; Smit, G.P.A.; Leonard, J.V.; Clayton, P.T.; Fidler, V.; Berger, R.; Heymans, H.S.A. Hereditary Tyrosinemia Type I: A New Clinical Classification with Difference in Prognosis on Dietary Treatment. Hepatology 1994, 20, 1187–1191. [Google Scholar] [CrossRef]
- Mieles, L.; Esquivel, C.; Van Thiel, D.; Koneru, B.; Makowka, L.; Tzakis, A.; Starzl, T. Liver Transplantation for Tyrosinemia. Dig. Dis. Sci. 1990, 35, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Arnon, R.; Annunziato, R.; Miloh, T.; Wasserstein, M.; Sogawa, H.; Wilson, M.; Suchy, F.; Kerkar, N. Liver Transplantation for Hereditary Tyrosinemia Type I: Analysis of the UNOS Database. Pediatr. Transplant. 2011, 15, 400–405. [Google Scholar] [CrossRef]
- Lindstedt, S.; Holme, E.; Lock, E.A.; Hjalmarson, O.; Strandvik, B. Treatment of Hereditary Tyrosinaemia Type I by Inhibition of 4-Hydroxyphenylpyruvate Dioxygenase. Lancet 1992, 340, 813–817. [Google Scholar] [CrossRef]
- Larochelle, J.; Alvarez, F.; Bussières, J.-F.; Chevalier, I.; Dallaire, L.; Dubois, J.; Faucher, F.; Fenyves, D.; Goodyer, P.; Grenier, A.; et al. Effect of Nitisinone (NTBC) Treatment on the Clinical Course of Hepatorenal Tyrosinemia in Québec. Mol. Genet. Metab. 2012, 107, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Masurel-Paulet, A.; Poggi-Bach, J.; Rolland, M.-O.; Bernard, O.; Guffon, N.; Dobbelaere, D.; Sarles, J.; de Baulny, H.O.; Touati, G. NTBC Treatment in Tyrosinaemia Type I: Long-Term Outcome in French Patients. J. Inherit. Metab. Dis. 2008, 31, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Holme, E.; Lindstedt, S. Tyrosinaemia Type I and NTBC (2-(2-Nitro-4-Trifluoromethylbenzoyl)-1,3-Cyclohexanedione). J. Inherit. Metab. Dis. 1998, 21, 507–517. [Google Scholar] [CrossRef]
- Chinsky, J.M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C.D.M.; Grompe, M.; Mitchell, G.; Waisbren, S.E.; Gucsavas-Calikoglu, M.; Wasserstein, M.P.; Coakley, K.; et al. Diagnosis and Treatment of Tyrosinemia Type I: A US and Canadian Consensus Group Review and Recommendations. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef] [Green Version]
- De Laet, C.; Dionisi-Vici, C.; Leonard, J.V.; McKiernan, P.; Mitchell, G.; Monti, L.; de Baulny, H.O.; Pintos-Morell, G.; Spiekerkötter, U. Recommendations for the Management of Tyrosinaemia Type 1. Orphanet J. Rare Dis. 2013, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R. Newborn Screening: Toward a Uniform Screening Panel and System. Genet. Med. 2006, 8, 1S–11S. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, F.; Atkinson, S.; Bouchard, M.; Brunel-Guitton, C.; Buhas, D.; Bussières, J.-F.; Dubois, J.; Fenyves, D.; Goodyer, P.; Gosselin, M.; et al. The Québec NTBC Study. In Hereditary Tyrosinemia: Pathogenesis, Screening and Management; Tanguay, R.M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 187–195. [Google Scholar] [CrossRef]
- McKiernan, P.J.; Preece, M.A.; Chakrapani, A. Outcome of Children with Hereditary Tyrosinaemia Following Newborn Screening. Arch. Dis. Child. 2015, 100, 738–741. [Google Scholar] [CrossRef]
- Couce, M.L.; Sánchez-Pintos, P.; Aldámiz-Echevarría, L.; Vitoria, I.; Navas, V.; Martín-Hernández, E.; García-Volpe, C.; Pintos, G.; Peña-Quintana, L.; Hernández, T.; et al. Evolution of Tyrosinemia Type 1 Disease in Patients Treated with Nitisinone in Spain. Medicine 2019, 98, e17303. [Google Scholar] [CrossRef]
- Gagné, R.; Lescault, A.; Grenier, A.; Laberge, C.; Mélançon, S.B.; Dallaire, L. Prenatal Diagnosis of Hereditary Tyrosinaemia: Measurement of Succinylacetone in Amniotic Fluid. Prenat. Diagn. 1982, 2, 185–188. [Google Scholar] [CrossRef]
- Grenier, A.; Lescault, A.; Laberge, C.; Gagné, R.; Marner, O. Detection of Succinylacetone and the Use of Its Measurement in Mass Screening for Hereditary Tyrosinemia. Clin. Chim. Acta 1982, 123, 93–99. [Google Scholar] [CrossRef]
- Schulze, A.; Frommhold, D.; Hoffmann, G.F.; Mayatepek, E. Spectrophotometric Microassay for Delta-Aminolevulinate Dehydratase in Dried-Blood Spots as Confirmation for Hereditary Tyrosinemia Type I. Clin. Chem. 2001, 47, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giguère, Y.; Berthier, M.-T. Newborn Screening for Hereditary Tyrosinemia Type I in Québec: Update. In Hereditary Tyrosinemia: Pathogenesis, Screening and Management; Tanguay, R.M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 139–146. [Google Scholar] [CrossRef]
- Allard, P.; Grenier, A.; Korson, M.S.; Zytkovicz, T.H. Newborn Screening for Hepatorenal Tyrosinemia by Tandem Mass Spectrometry: Analysis of Succinylacetone Extracted from Dried Blood Spots. Clin. Biochem. 2004, 37, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, C.; Magera, M.J.; Allard, P.; Tortorelli, S.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Matern, D. Combined Newborn Screening for Succinylacetone, Amino Acids, and Acylcarnitines in Dried Blood Spots. Clin. Chem. 2008, 54, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jesús, V.R.; Adam, B.W.; Mandel, D.; Cuthbert, C.D.; Matern, D. Succinylacetone as Primary Marker to Detect Tyrosinemia Type I in Newborns and Its Measurement by Newborn Screening Programs. Mol. Genet. Metab. 2014, 113, 67–75. [Google Scholar] [CrossRef] [Green Version]
- La Marca, G.; Malvagia, S.; Pasquini, E.; Cavicchi, C.; Morrone, A.; Ciani, F.; Funghini, S.; Villanelli, F.; Zammarchi, E.; Guerrini, R. Newborn Screening for Tyrosinemia Type I: Further Evidence That Succinylacetone Determination on Blood Spot Is Essential. JIMD Rep. 2011, 1, 107–109. [Google Scholar] [CrossRef] [Green Version]
- La Marca, G.; Malvagia, S.; Funghini, S.; Pasquini, E.; Moneti, G.; Guerrini, R.; Zammarchi, E. The Successful Inclusion of Succinylacetone as a Marker of Tyrosinemia Type I in Tuscany Newborn Screening Program. Rapid Commun. Mass Spectrom. 2009, 23, 3891–3893. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, K.S.; Bhandal, A.S.; Aznar, C.P.; Lorey, F.W.; Neogi, P. Improved Tandem Mass Spectrometry (MS/MS) Derivatized Method for the Detection of Tyrosinemia Type I, Amino Acids and Acylcarnitine Disorders Using a Single Extraction Process. Clin. Chim. Acta 2011, 412, 873–879. [Google Scholar] [CrossRef]
- Metz, T.F.; Mechtler, T.P.; Merk, M.; Gottschalk, A.; Lukačin, R.; Herkner, K.R.; Kasper, D.C. Evaluation of a Novel, Commercially Available Mass Spectrometry Kit for Newborn Screening Including Succinylacetone without Hydrazine. Clin. Chim. Acta 2012, 413, 1259–1264. [Google Scholar] [CrossRef]
- Stinton, C.; Geppert, J.; Freeman, K.; Clarke, A.; Johnson, S.; Fraser, H.; Sutcliffe, P.; Taylor-Phillips, S. Newborn Screening for Tyrosinemia Type 1 Using Succinylacetone—A Systematic Review of Test Accuracy. Orphanet J. Rare Dis. 2017, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Tanguay, R.M. Hereditary Tyrosinemia: Pathogenesis, Screening and Management; Springer: New York, NY, USA, 2017. [Google Scholar]
- Blackburn, P.R.; Hickey, R.D.; Nace, R.A.; Giama, N.H.; Kraft, D.L.; Bordner, A.J.; Chaiteerakij, R.; McCormick, J.B.; Radulovic, M.; Graham, R.P.; et al. Silent Tyrosinemia Type I Without Elevated Tyrosine or Succinylacetone Associated with Liver Cirrhosis and Hepatocellular Carcinoma. Hum. Mutat. 2016, 37, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L. Tyrosinemia Associated with Hypermethioninemia and Islet Cell Hyperplasia. Can. Med. Assoc. J. 1967, 97, 1067–1075. [Google Scholar] [PubMed]
- Carson, N.A.; Biggart, J.D.; Bittles, A.H.; Donovan, D. Hereditary Tyrosinaemia. Clinical, Enzymatic, and Pathological Study of an Infant with the Acute Form of the Disease. Arch. Dis. Child. 1976, 51, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, U.; Preece, M.A.; Green, A.; Kelly, D.A.; McKiernan, P.J. Hyperinsulinism in Tyrosinaemia Type I. J. Inherit. Metab. Dis. 2005, 28, 131–135. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Age at Collection | Tyrosine (µmol/L) | Succinylacetone (µmol/L) | Reported Result | Comments |
|---|---|---|---|---|
| 1 day 9 h | 151 | 5.23 | Normal | Full screen |
| 6 days 2 h | 437 | 4.65 | Normal | Only screened for congenital hypothyroidism and congenital adrenal hyperplasia |
| 13 days 2 h | 312 | 5.57 | Normal | Only screened for congenital hypothyroidism and congenital adrenal hyperplasia |
| 18 days 20 h | 409 | 10.34 | Abnormal | Low T4, TSH within acceptable limits |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priestley, J.R.C.; Alharbi, H.; Callahan, K.P.; Guzman, H.; Payan-Walters, I.; Smith, L.; Ficicioglu, C.; Ganetzky, R.D.; Ahrens-Nicklas, R.C. The Importance of Succinylacetone: Tyrosinemia Type I Presenting with Hyperinsulinism and Multiorgan Failure Following Normal Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 39. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020039
Priestley JRC, Alharbi H, Callahan KP, Guzman H, Payan-Walters I, Smith L, Ficicioglu C, Ganetzky RD, Ahrens-Nicklas RC. The Importance of Succinylacetone: Tyrosinemia Type I Presenting with Hyperinsulinism and Multiorgan Failure Following Normal Newborn Screening. International Journal of Neonatal Screening. 2020; 6(2):39. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020039
Chicago/Turabian StylePriestley, Jessica R. C., Hana Alharbi, Katharine Press Callahan, Herodes Guzman, Irma Payan-Walters, Ligia Smith, Can Ficicioglu, Rebecca D. Ganetzky, and Rebecca C. Ahrens-Nicklas. 2020. "The Importance of Succinylacetone: Tyrosinemia Type I Presenting with Hyperinsulinism and Multiorgan Failure Following Normal Newborn Screening" International Journal of Neonatal Screening 6, no. 2: 39. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020039