
Harnessing Intronic microRNA Structures to Improve Tolerance and Expression of shRNAs in Animal Cells

 , , and
, , and 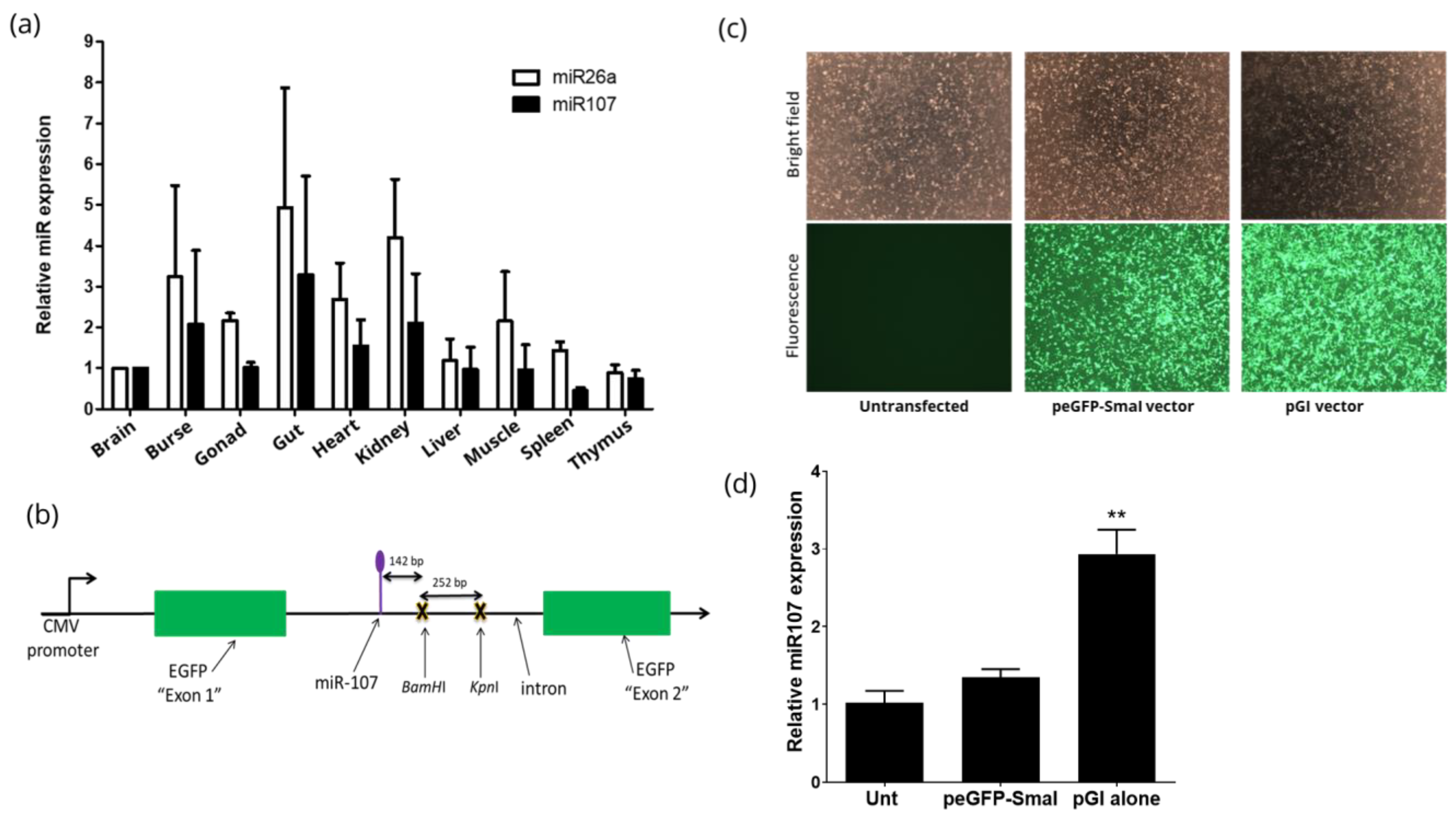{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primers
2.2. Construction of pGFP-Intron (pGI) Vector
2.3. Cloning of shRNAs into pGI Vector
2.4. Cell Culture and Transfections
2.5. RNA Extraction
2.6. Polyadenylation of RNA, cDNA Synthesis and qRT-PCR
2.7. miniTol2 Plasmids
2.8. Assessment of Toxicity in Chicken Embryos
2.9. Statistical Analysis
3. Results
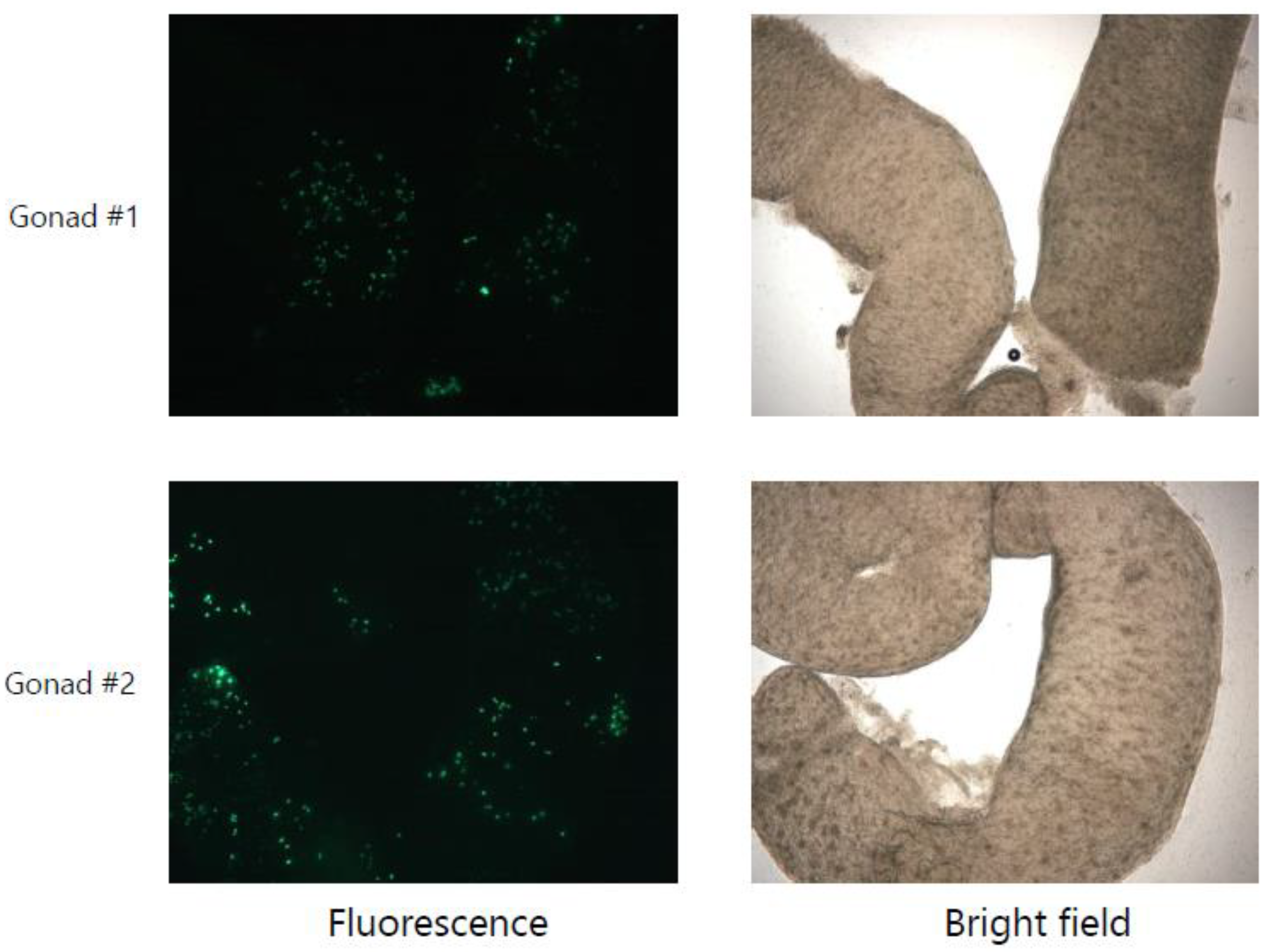
3.1. Identification of a Universally Expressed Intronic miRNA
3.2. Construction and Characterisation of peGFP-Intron
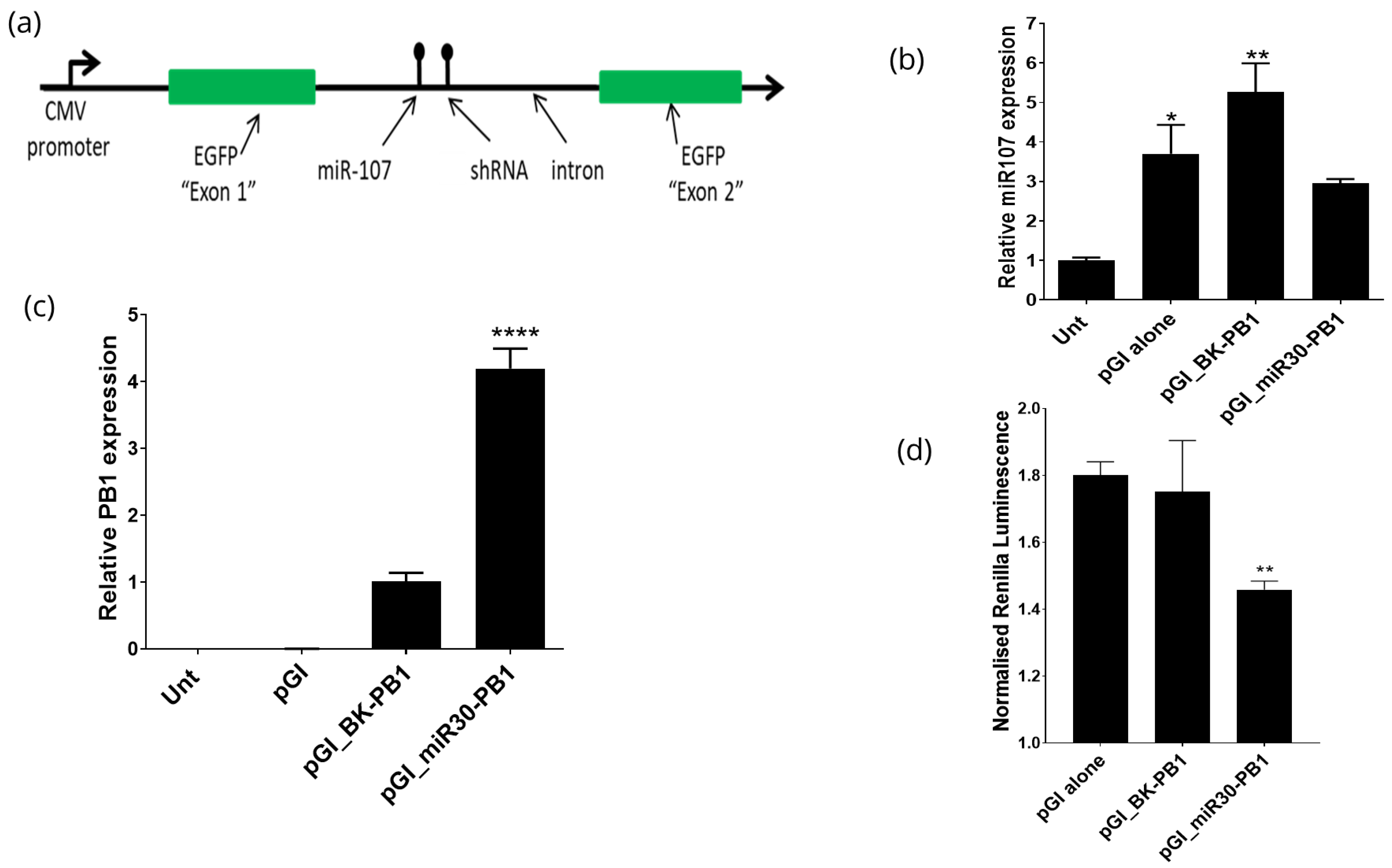
3.3. Expression of shRNAs from the pGI Vector
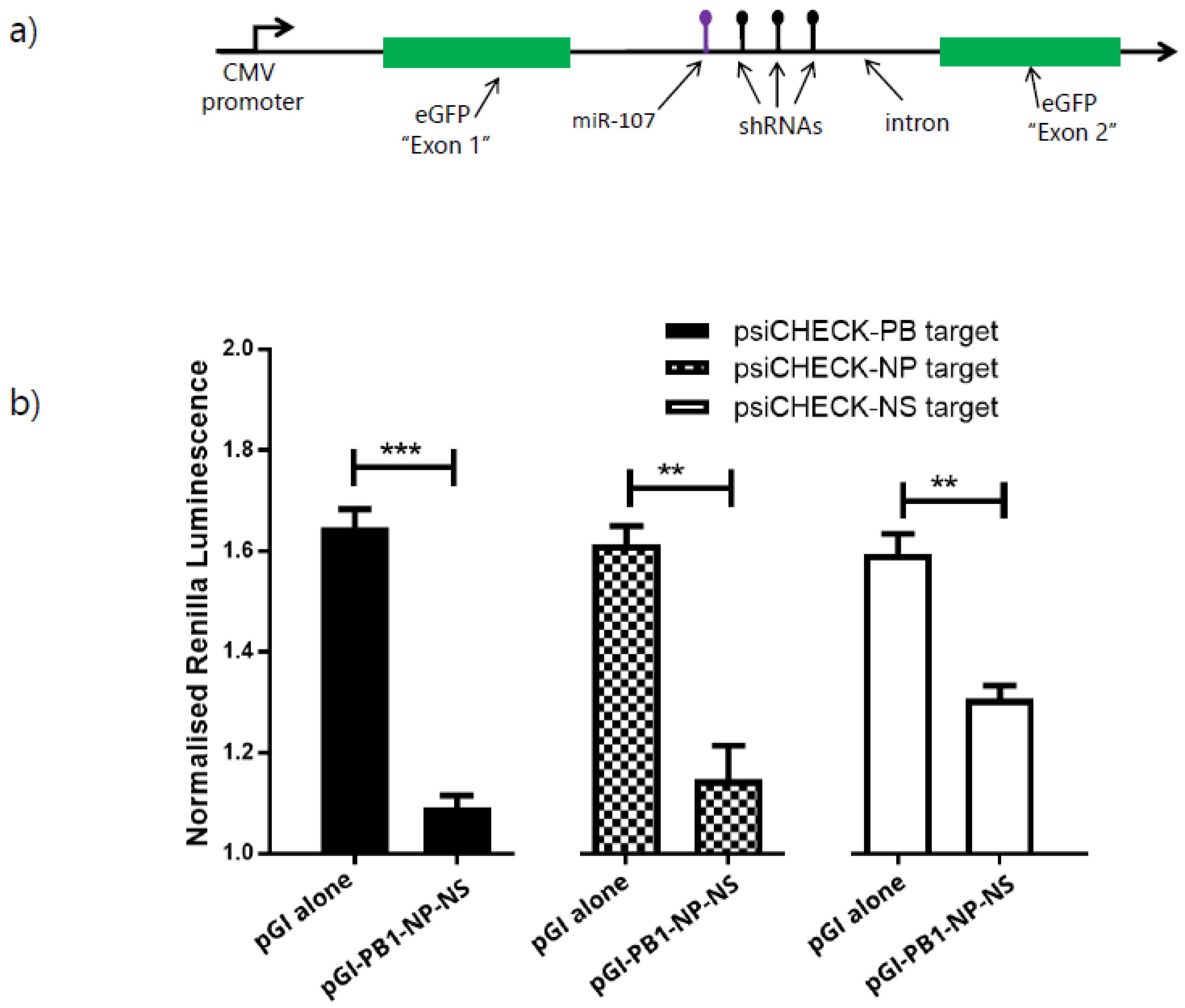
3.4. Expression of Multiple shRNAs from the pGI Vector
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Hinton, T.M.; Challagulla, A.; Stewart, C.R.; Guerrero-Sanchez, C.; Grusche, F.A.; Shi, S.; Bean, A.G.; Monaghan, P.; Gunatillake, P.A.; Thang, S.H.; et al. Inhibition of influenza virus in vivo by siRNA delivered using ABA triblock copolymer synthesized by reversible addition-fragmentation chain-transfer polymerization. Nanomedicine 2014, 9, 1141–1154. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.D.; Cummins, D.M.; McColl, K.A.; Ward, A.C.; Doran, T.J. Characterization of zebrafish polymerase III promoters for the expression of short-hairpin RNA interference molecules. Zebrafish 2013, 10, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, L.S.; Moore, R.J.; Muralitharan, M.; Dalrymple, B.P.; McWilliam, S.; Doran, T.J. Characterisation and application of a bovine U6 promoter for expression of short hairpin RNAs. BMC Biotechnol. 2005, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, T.G.; Schafer, D.J.; Lambeth, L.S.; Tyack, S.G.; Bruce, M.P.; Moore, R.J.; Doran, T.J. Characterization and comparison of chicken U6 promoters for the expression of short hairpin RNAs. Anim. Biotechnol. 2007, 18, 153–162. [Google Scholar] [CrossRef]
- Bannister, S.C.; Wise, T.G.; Cahill, D.M.; Doran, T.J. Comparison of chicken 7SK and U6 RNA polymerase III promoters for short hairpin RNA expression. BMC Biotechnol. 2007, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Cummins, D.M.; Tyack, S.G.; Doran, T.J. Characterisation and comparison of the chicken H1 RNA polymerase III promoter for short hairpin RNA expression. Biochem. Biophys. Res. Commun. 2011, 416, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ni, W.; Hazi, W.; Zhang, H.; Zhang, N.; Meng, R.; Chen, C. Cloning and functional analysis of sheep U6 promoters. Anim. Biotechnol. 2011, 22, 170–174. [Google Scholar] [CrossRef]
- Bish, L.T.; Sleeper, M.M.; Reynolds, C.; Gazzara, J.; Withnall, E.; Singletary, G.E.; Buchlis, G.; Hui, D.; High, K.A.; Gao, G.; et al. Cardiac gene transfer of short hairpin RNA directed against phospholamban effectively knocks down gene expression but causes cellular toxicity in canines. Hum. Gene Ther. 2011, 22, 969–977. [Google Scholar] [CrossRef] [Green Version]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial microRNAs as siRNA shuttles: Improved safety as compared to shRNAs in vitro and in vivo. Mol. Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- An, D.S.; Qin, F.X.; Auyeung, V.C.; Mao, S.H.; Kung, S.K.; Baltimore, D.; Chen, I.S. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol. Ther. 2006, 14, 494–504. [Google Scholar] [CrossRef]
- Giering, J.C.; Grimm, D.; Storm, T.A.; Kay, M.A. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol. Ther. 2008, 16, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Challagulla, A.; Jenkins, K.A.; O’Neil, T.E.; Morris, K.R.; Wise, T.G.; Tizard, M.L.; Bean, A.G.D.; Schat, K.A.; Doran, T.J. Germline engineering of the chicken genome using CRISPR/Cas9 by in vivo transfection of PGCs. Anim. Biotechnol. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challagulla, A.; Jenkins, K.A.; O’Neil, T.E.; Shi, S.; Morris, K.R.; Wise, T.G.; Paradkar, P.N.; Tizard, M.L.; Doran, T.J.; Schat, K.A. In Vivo Inhibition of Marek’s Disease Virus in Transgenic Chickens Expressing Cas9 and gRNA against ICP4. Microorganisms 2021, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 2005, 39, 519–525. [Google Scholar] [CrossRef]
- Balciunas, D.; Wangensteen, K.J.; Wilber, A.; Bell, J.; Geurts, A.; Sivasubbu, S.; Wang, X.; Hackett, P.B.; Largaespada, D.A.; McIvor, R.S.; et al. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006, 2, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyack, S.G.; Jenkins, K.A.; O’Neil, T.E.; Wise, T.G.; Morris, K.R.; Bruce, M.P.; McLeod, S.; Wade, A.J.; McKay, J.; Moore, R.J.; et al. A new method for producing transgenic birds via direct in vivo transfection of primordial germ cells. Transgenic Res. 2013, 22, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Glazov, E.A.; Cottee, P.A.; Barris, W.C.; Moore, R.J.; Dalrymple, B.P.; Tizard, M.L. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008, 18, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, G.W.; Yatskievych, T.A.; Antin, P.B. GEISHA, a whole-mount in situ hybridization gene expression screen in chicken embryos. Dev. Dyn. 2004, 229, 677–687. [Google Scholar] [CrossRef]
- Rock, C.O.; Calder, R.B.; Karim, M.A.; Jackowski, S. Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J. Biol. Chem. 2000, 275, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [Green Version]
- Chandra, P.K.; Kundu, A.K.; Hazari, S.; Chandra, S.; Bao, L.; Ooms, T.; Morris, G.F.; Wu, T.; Mandal, T.K.; Dash, S. Inhibition of hepatitis C virus replication by intracellular delivery of multiple siRNAs by nanosomes. Mol. Ther. 2012, 20, 1724–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebbink, R.J.; Lowe, M.; Chan, T.; Khine, H.; Wang, X.; McManus, M.T. Polymerase II promoter strength determines efficacy of microRNA adapted shRNAs. PLoS ONE 2011, 6, e26213. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Jin, L.; Zhang, Y.; Huang, Y.; Zhang, F.; Valdmanis, P.N.; Kay, M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell 2012, 151, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, D.; Pusch, O.; Silbermann, R.; Lee, F.; Tucker, L.; Ramratnam, B. Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins. Nucleic Acids Res. 2004, 32, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Hoffmann, T.; Sridhar, V.; Hopfgartner, B.; Muhar, M.; Roth, M.; Lai, D.Y.; Barbosa, I.A.; Kwon, J.S.; Guan, Y.; et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013, 5, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Hinton, T.M.; Wise, T.G.; Cottee, P.A.; Doran, T.J. Native microRNA loop sequences can improve short hairpin RNA processing for virus gene silencing in animal cells. J. RNAi Gene Silencing 2008, 4, 295–301. [Google Scholar]
- Zhang, T.; Cheng, T.; Wei, L.; Cai, Y.; Yeo, A.E.; Han, J.; Yuan, Y.A.; Zhang, J.; Xia, N. Efficient inhibition of HIV-1 replication by an artificial polycistronic miRNA construct. Virol. J. 2012, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challagulla, A.; Schat, K.A.; Doran, T.J. In Vitro Inhibition of Influenza Virus Using CRISPR/Cas13a in Chicken Cells. Methods Protoc. 2021, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Tizard, M.L.; Jenkins, K.A.; Cooper, C.A.; Woodcock, M.E.; Challagulla, A.; Doran, T.J. Potential benefits of gene editing for the future of poultry farming. Transgenic Res. 2019, 28 (Suppl. 2), 87–92. [Google Scholar] [CrossRef]
- Challagulla, A.; Shi, S.; Nair, K.; O’Neil, T.E.; Morris, K.R.; Wise, T.G.; Cahill, D.M.; Tizard, M.L.; Doran, T.J.; Jenkins, K.A. Marker counter-selection via CRISPR/Cas9 co-targeting for efficient generation of genome edited avian cell lines and germ cells. Anim. Biotechnol. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.A.; Doran, T.J.; Challagulla, A.; Tizard, M.L.V.; Jenkins, K.A. Innovative approaches to genome editing in avian species. J. Anim. Sci. Biotechnol. 2018, 9, 15. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Challagulla, A.; Tizard, M.L.; Doran, T.J.; Cahill, D.M.; Jenkins, K.A. Harnessing Intronic microRNA Structures to Improve Tolerance and Expression of shRNAs in Animal Cells. Methods Protoc. 2022, 5, 18. https://0-doi-org.brum.beds.ac.uk/10.3390/mps5010018
Challagulla A, Tizard ML, Doran TJ, Cahill DM, Jenkins KA. Harnessing Intronic microRNA Structures to Improve Tolerance and Expression of shRNAs in Animal Cells. Methods and Protocols. 2022; 5(1):18. https://0-doi-org.brum.beds.ac.uk/10.3390/mps5010018
Chicago/Turabian StyleChallagulla, Arjun, Mark L. Tizard, Timothy J. Doran, David M. Cahill, and Kristie A. Jenkins. 2022. "Harnessing Intronic microRNA Structures to Improve Tolerance and Expression of shRNAs in Animal Cells" Methods and Protocols 5, no. 1: 18. https://0-doi-org.brum.beds.ac.uk/10.3390/mps5010018