Fingerprint-Based Detection of Non-Local Effects in the Electronic Structure of a Simple Single Component Covalent System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
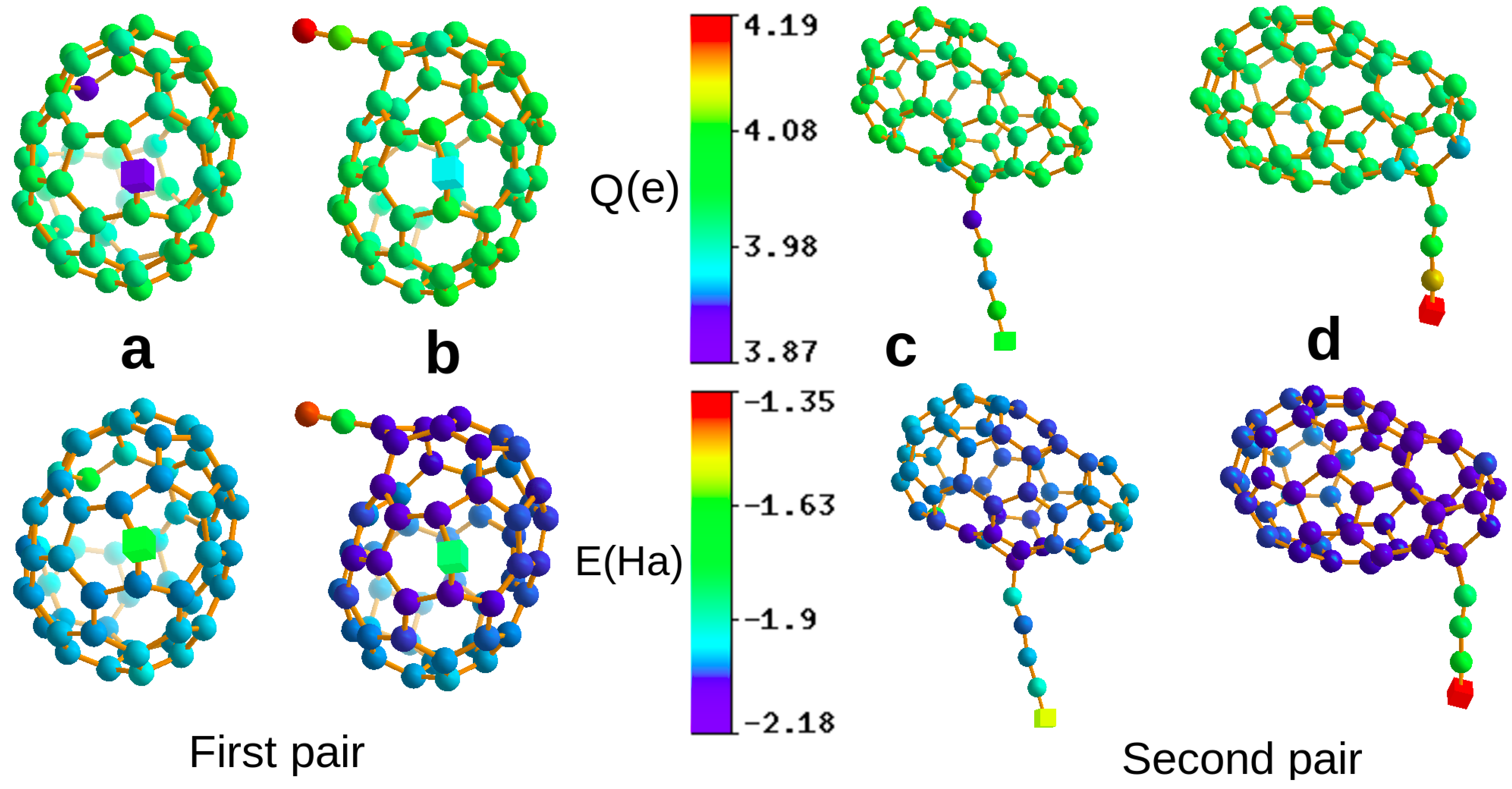
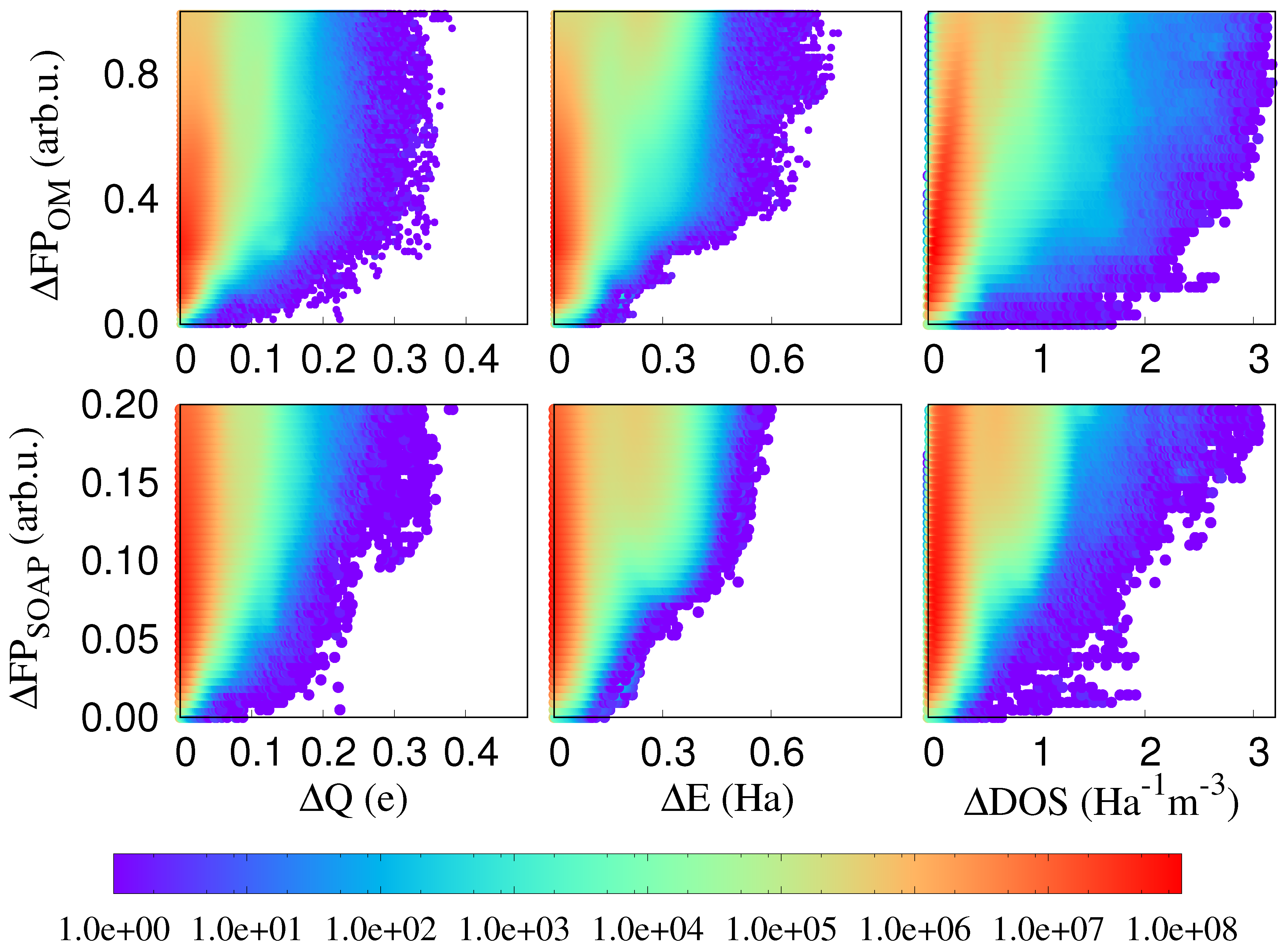
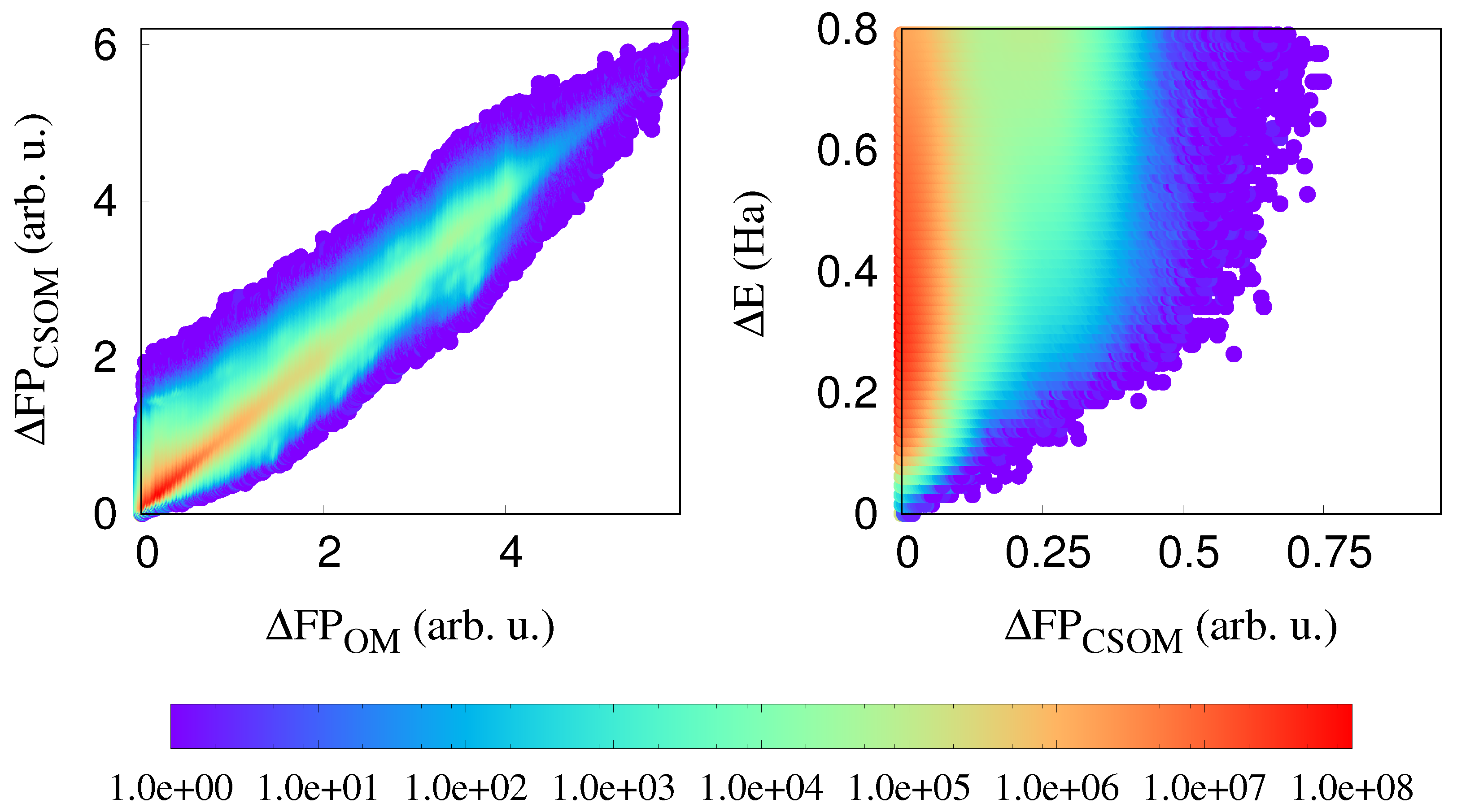
3. Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goedecker, S. Linear scaling electronic structure methods. Rev. Mod. Phys. 1999, 71, 1085. [Google Scholar] [CrossRef]
- Kohn, W. Density functional and density matrix method scaling linearly with the number of atoms. Phys. Rev. Lett. 1996, 76, 3168. [Google Scholar] [CrossRef]
- Prodan, E.; Kohn, W. Nearsightedness of electronic matter. Proc. Natl. Acad. Sci. USA 2005, 102, 11635–11638. [Google Scholar] [CrossRef] [Green Version]
- Prodan, E. Nearsightedness of electronic matter in one dimension. Phys. Rev. B 2006, 73, 085108. [Google Scholar] [CrossRef] [Green Version]
- Marks, N.A.; Cooper, N.; McKenzie, D.; McCulloch, D.; Bath, P.; Russo, S. Comparison of density-functional, tight-binding, and empirical methods for the simulation of amorphous carbon. Phys. Rev. B 2002, 65, 075411. [Google Scholar] [CrossRef] [Green Version]
- Marks, N.A. Generalizing the environment-dependent interaction potential for carbon. Phys. Rev. B 2000, 63, 035401. [Google Scholar] [CrossRef]
- Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 1988, 37, 6991. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458. [Google Scholar] [CrossRef] [PubMed]
- Los, J.H.; Ghiringhelli, L.M.; Meijer, E.J.; Fasolino, A. Improved long-range reactive bond-order potential for carbon. I. Construction. Phys. Rev. B 2005, 72, 214102. [Google Scholar] [CrossRef] [Green Version]
- Deringer, V.L.; Csányi, G. Machine learning based interatomic potential for amorphous carbon. Phys. Rev. B 2017, 95, 094203. [Google Scholar] [CrossRef] [Green Version]
- Rupp, M.; Tkatchenko, A.; Müller, K.R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef]
- Brockherde, F.; Vogt, L.; Li, L.; Tuckerman, M.E.; Burke, K.; Müller, K.R. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Behler, J. Constructing high-dimensional neural network potentials: A tutorial review. Int. J. Quantum Chem. 2015, 115, 1032–1050. [Google Scholar] [CrossRef]
- Grisafi, A.; Ceriotti, M. Incorporating long-range physics in atomic-scale machine learning. J. Chem. Phys. 2019, 151, 204105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bereau, T.; DiStasio, R.A., Jr.; Tkatchenko, A.; Von Lilienfeld, O.A. Non-covalent interactions across organic and biological subsets of chemical space: Physics-based potentials parametrized from machine learning. J. Chem. Phys. 2018, 148, 241706. [Google Scholar] [CrossRef]
- Yao, K.; Herr, J.E.; Toth, D.W.; Mckintyre, R.; Parkhill, J. The TensorMol-0.1 model chemistry: A neural network augmented with long-range physics. Chem. Sci. 2018, 9, 2261–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134, 074106. [Google Scholar] [CrossRef] [PubMed]
- Bartók, A.P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys. Rev. B 2013, 87, 184115. [Google Scholar] [CrossRef] [Green Version]
- Faber, F.A.; Christensen, A.S.; Huang, B.; Von Lilienfeld, O.A. Alchemical and structural distribution based representation for universal quantum machine learning. J. Chem. Phys. 2018, 148, 241717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, A.; Ghasemi, S.A.; Schaefer, B.; Mohr, S.; Lill, M.A.; Goedecker, S. Metrics for measuring distances in configuration spaces. J. Chem. Phys. 2013, 139, 184118. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Amsler, M.; Fuhrer, T.; Schaefer, B.; Faraji, S.; Rostami, S.; Ghasemi, S.A.; Sadeghi, A.; Grauzinyte, M.; Wolverton, C.; et al. A fingerprint based metric for measuring similarities of crystalline structures. J. Chem. Phys. 2016, 144, 034203. [Google Scholar] [CrossRef] [Green Version]
- Parsaeifard, B.; De, D.S.; Christensen, A.S.; Faber, F.A.; Kocer, E.; De, S.; Behler, J.; von Lilienfeld, A.; Goedecker, S. An assessment of the structural resolution of various fingerprints commonly used in machine learning. Mach. Learn. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willand, A.; Kvashnin, Y.O.; Genovese, L.; Vázquez-Mayagoitia, Á.; Deb, A.K.; Sadeghi, A.; Deutsch, T.; Goedecker, S. Norm-conserving pseudopotentials with chemical accuracy compared to all-electron calculations. J. Chem. Phys. 2013, 138, 104109. [Google Scholar] [CrossRef] [Green Version]
- Genovese, L.; Neelov, A.; Goedecker, S.; Deutsch, T.; Ghasemi, S.A.; Willand, A.; Caliste, D.; Zilberberg, O.; Rayson, M.; Bergman, A.; et al. Daubechies wavelets as a basis set for density functional pseudopotential calculations. J. Chem. Phys. 2008, 129, 014109. [Google Scholar] [CrossRef]
- Bader, R.F. The quantum mechanical basis of conceptual chemistry. Monatshefte Chem. Chem. Mon. 2005, 136, 819–854. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H. Energy density analysis with Kohn–Sham orbitals. Chem. Phys. Lett. 2002, 363, 73–79. [Google Scholar] [CrossRef]
- Harris, J. Simplified method for calculating the energy of weakly interacting fragments. Phys. Rev. B 1985, 31, 1770. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S. Minima hopping: An efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 2004, 120, 9911–9917. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Goedecker, S.; Hellmann, V. Bell-Evans-Polanyi principle for molecular dynamics trajectories and its implications for global optimization. Phys. Rev. E 2008, 77, 056707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicher, M.; Mohr, S.; Goedecker, S. Efficient moves for global geometry optimization methods and their application to binary systems. J. Chem. Phys. 2011, 134, 044106. [Google Scholar] [CrossRef] [Green Version]
- Aradi, B.; Hourahine, B.; Frauenheim, T. DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A 2007, 111, 5678–5684. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.; Sun, H.; Govind, N.; Kowalski, K.; Autschbach, J. Charge-Transfer Versus Charge-Transfer-Like Excitations Revisited. J. Chem. Theory Comput. 2015, 11, 3305–3320. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ratcliff, L.E.; Degomme, A.; Flores-Livas, J.A.; Goedecker, S.; Genovese, L. Affordable and accurate large-scale hybrid-functional calculations on GPU-accelerated supercomputers. J. Phys. Condens. Matter 2018, 30, 095901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirn, M.; Mallat, S.; Poilvert, N. Wavelet scattering regression of quantum chemical energies. Multiscale Model. Simul. 2017, 15, 827–863. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, S.A.; Hofstetter, A.; Saha, S.; Goedecker, S. Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network. Phys. Rev. B 2015, 92, 045131. [Google Scholar] [CrossRef] [Green Version]
- Amsler, M.; Rostami, S.; Tahmasbi, H.; Rahmatizad, E.; Faraji, S.; Rasoulkhani, R.; Ghasemi, S.A. FLAME: A library of atomistic modeling environments. Comput. Phys. Commun. 2020, 256, 107415. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsaeifard, B.; De, D.S.; Finkler, J.A.; Goedecker, S. Fingerprint-Based Detection of Non-Local Effects in the Electronic Structure of a Simple Single Component Covalent System. Condens. Matter 2021, 6, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/condmat6010009
Parsaeifard B, De DS, Finkler JA, Goedecker S. Fingerprint-Based Detection of Non-Local Effects in the Electronic Structure of a Simple Single Component Covalent System. Condensed Matter. 2021; 6(1):9. https://0-doi-org.brum.beds.ac.uk/10.3390/condmat6010009
Chicago/Turabian StyleParsaeifard, Behnam, Deb Sankar De, Jonas A. Finkler, and Stefan Goedecker. 2021. "Fingerprint-Based Detection of Non-Local Effects in the Electronic Structure of a Simple Single Component Covalent System" Condensed Matter 6, no. 1: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/condmat6010009