Nanocomposite of Graphene Oxide Encapsulated in Polymethylmethacrylate (PMMA): Pre-Modification, Synthesis, and Latex Stability


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experiments
2.2.1. Preparation of Graphene Oxide (GO)
2.2.2. Pre-Modification of GO
2.2.3. Miniemulsion Polymerization of Methyl Methacrylate (MMA) in the Presence of Graphene Oxide (GO)
2.3. Characterization
3. Results
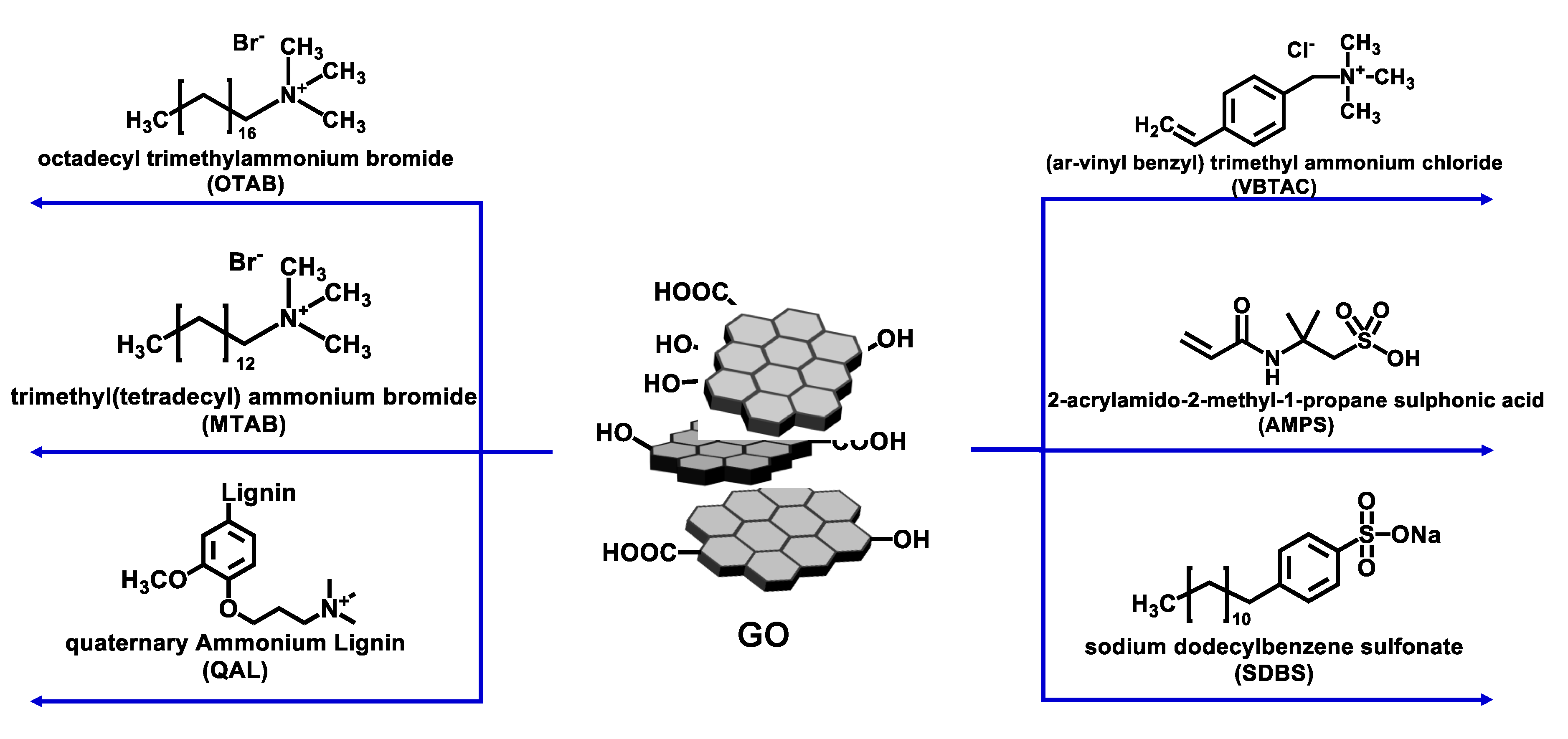
3.1. Modification of GO Using Different Modifiers
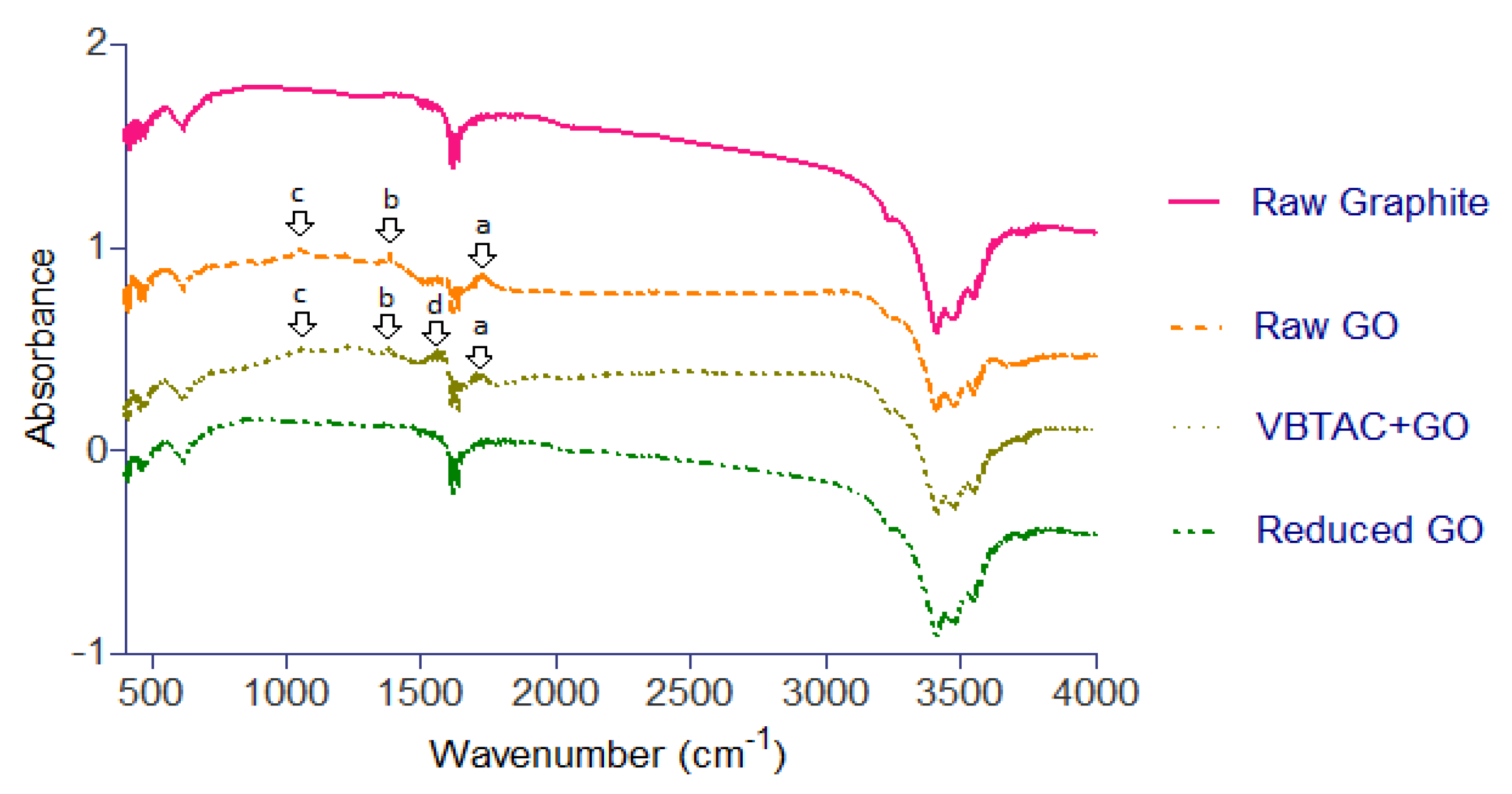
3.2. Fourier Transform Infrared Spectrometry (FTIR)
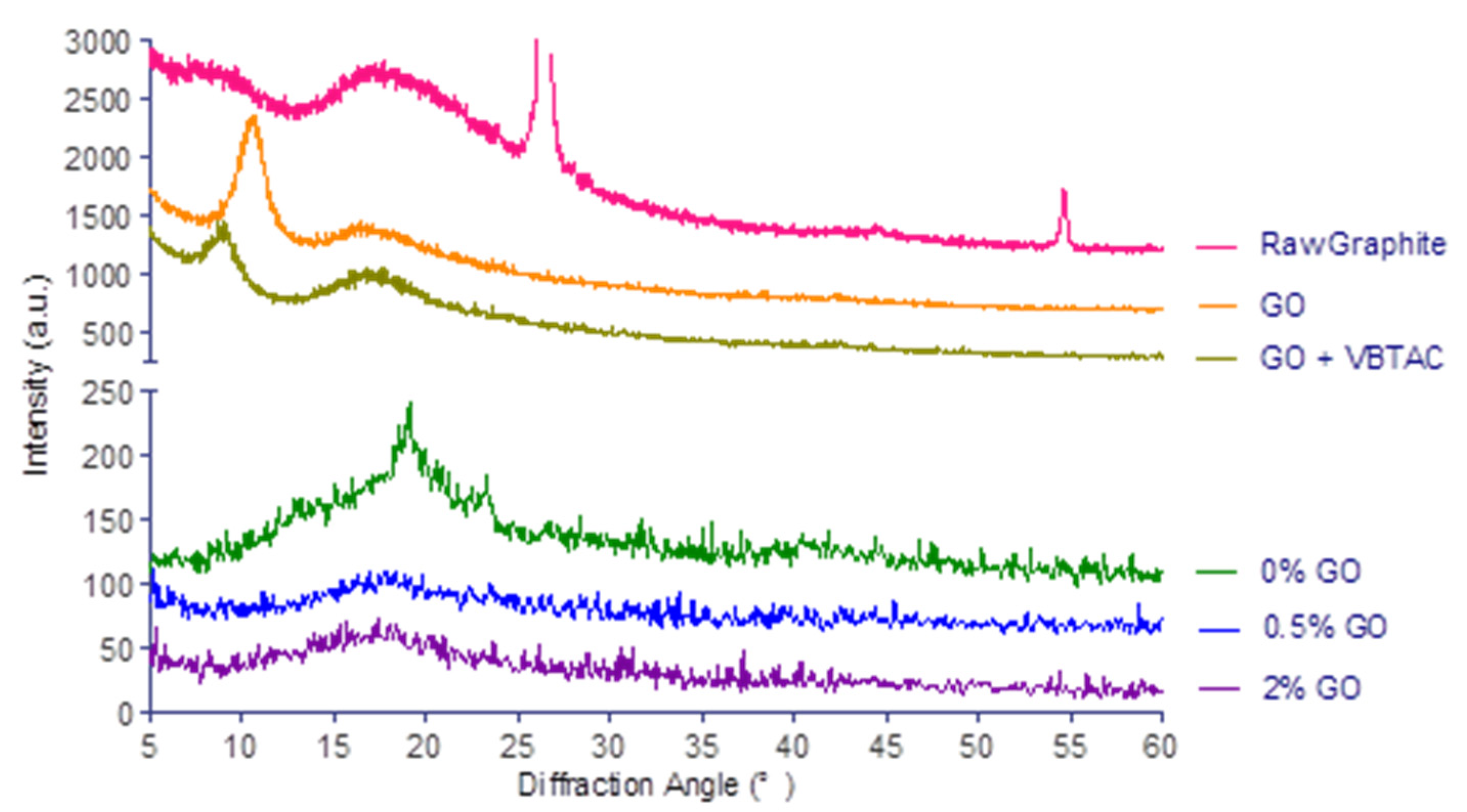
3.3. X-Ray Diffraction (XRD)
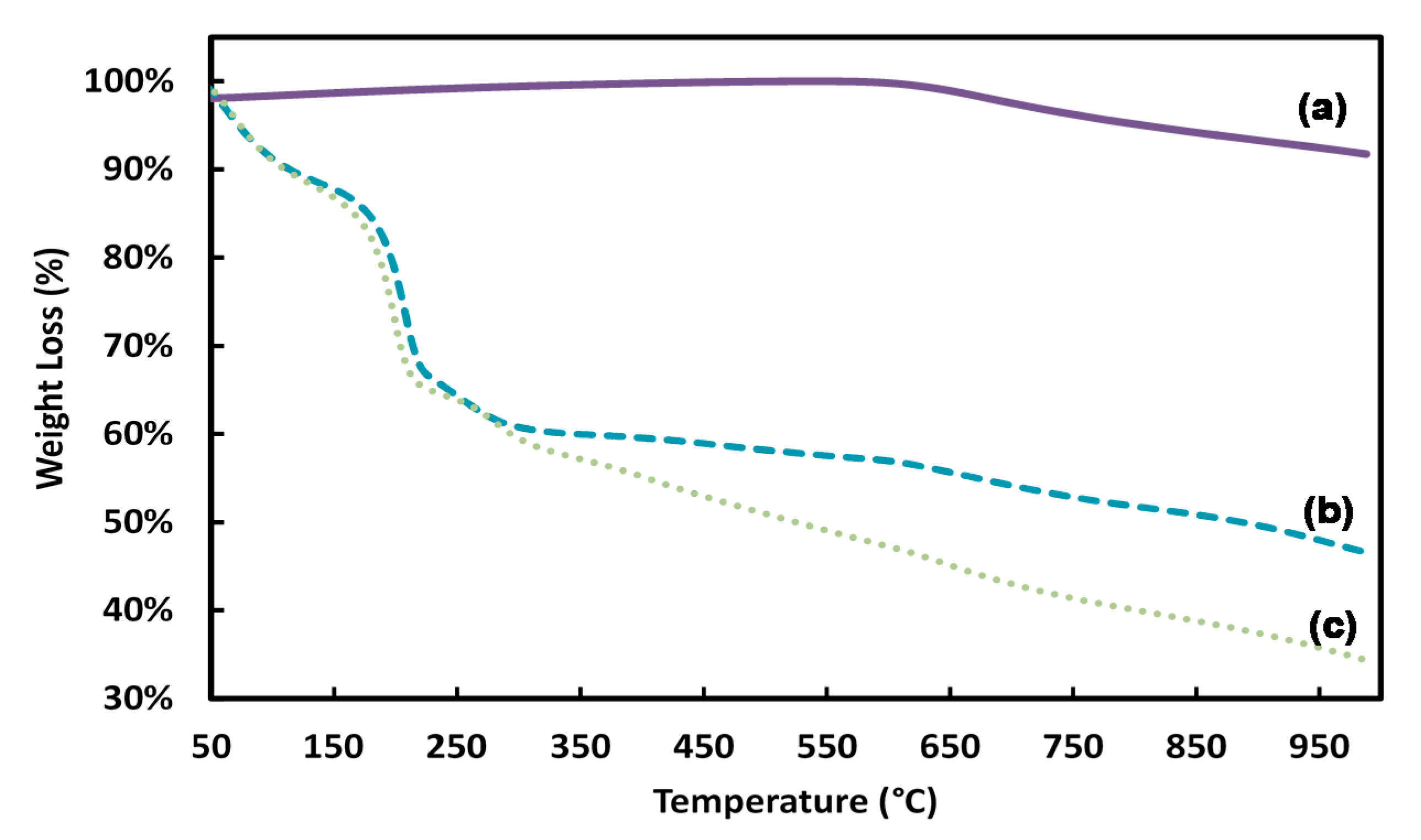
3.4. Thermal Stability Analysis
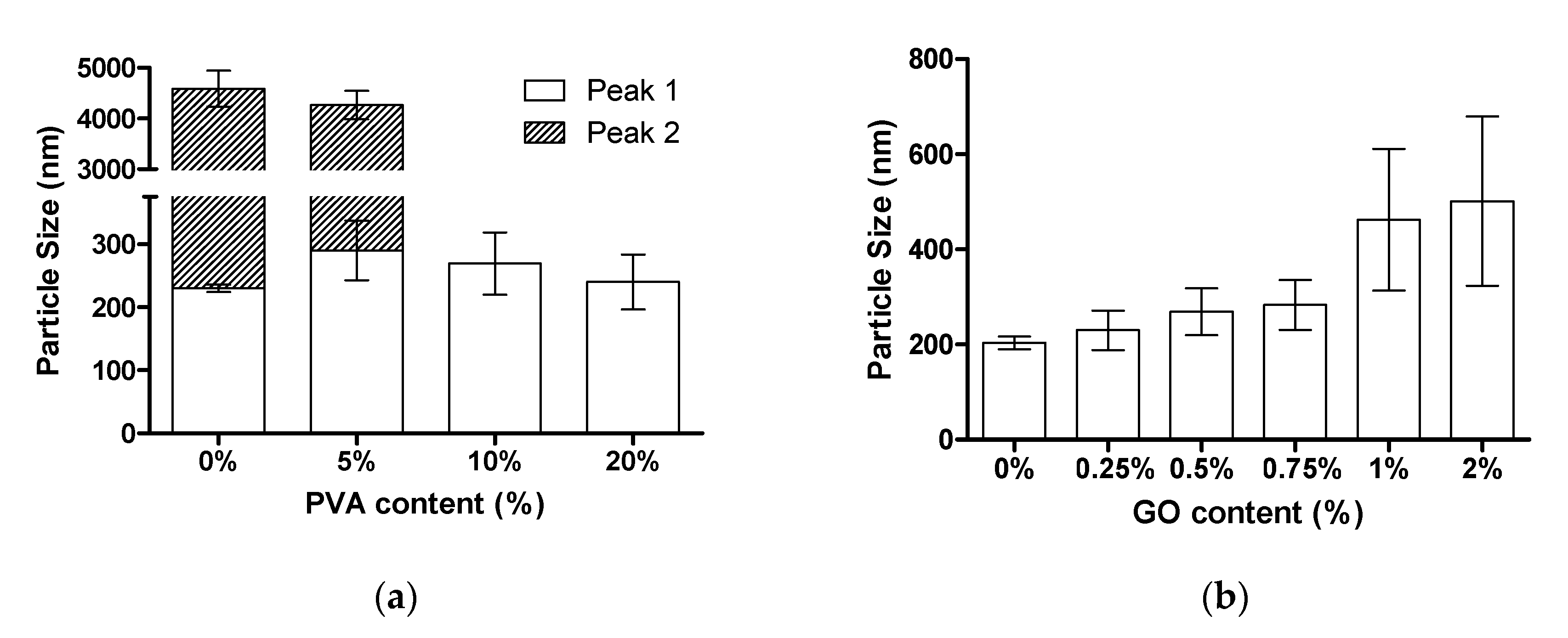
3.5. Particle Size Distribution of PMMA/GO Latex Droplets
3.6. Morphologies of GO, PMMA/GO Latex and Its Melt Films
3.7. Molecular Weight
3.8. Stability of Latex
4. Discussion & Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moniruzzaman, M.; Winey, K.I. Polymer nanocomposites containing carbon nanotubes. Macromolecules 2006, 39, 5194–5205. [Google Scholar] [CrossRef]
- Huang, J.C. Carbon black filled conducting polymers and polymer blends. Adv. Polym. Technol. J. Polym. Process. Inst. 2002, 21, 299–313. [Google Scholar]
- Tong, Z.; Deng, Y. Synthesis of polystyrene encapsulated nanosaponite composite latex via miniemulsion polymerization. Polymer 2007, 48, 4337–4343. [Google Scholar] [CrossRef]
- Jairam, S.; Tong, Z.; Wang, L.; Welt, B. Encapsulation of a biobased lignin–saponite nanohybrid into polystyrene co-butyl acrylate (PSBA) latex via miniemulsion polymerization. ACS Sustain. Chem. Eng. 2013, 1, 1630–1637. [Google Scholar] [CrossRef]
- Choi, S.; Han, S.I.; Kim, D.; Hyeon, T.; Kim, D.-H. High-performance stretchable conductive nanocomposites: Materials, processes, and device applications. Chem. Soc. Rev. 2019, 48, 1566–1595. [Google Scholar] [CrossRef]
- Liu, J.; Ma, Q.; Huang, Z.; Liu, G.; Zhang, H. Recent progress in graphene-based noble-metal nanocomposites for electrocatalytic applications. Adv. Mater. 2019, 31, 1800696. [Google Scholar] [CrossRef]
- Farahani, R.D.; Dubé, M.; Therriault, D. Three-dimensional printing of multifunctional nanocomposites: Manufacturing techniques and applications. Adv. Mater. 2016, 28, 5794–5821. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Samanta, S.K. Soft-nanocomposites of nanoparticles and nanocarbons with supramolecular and polymer gels and their applications. Chem. Rev. 2016, 116, 11967–12028. [Google Scholar] [CrossRef]
- Boland, C.S.; Khan, U.; Ryan, G.; Barwich, S.; Charifou, R.; Harvey, A.; Backes, C.; Li, Z.; Ferreira, M.S.; Möbius, M.E. Sensitive electromechanical sensors using viscoelastic graphene-polymer nanocomposites. Science 2016, 354, 1257–1260. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Skachko, I.; Barker, A.; Andrei, E.Y. Approaching ballistic transport in suspended graphene. Nat. Nanotechnol. 2008, 3, 491–495. [Google Scholar] [PubMed] [Green Version]
- Bunch, J.S.; Verbridge, S.S.; Alden, J.S.; Van Der Zande, A.M.; Parpia, J.M.; Craighead, H.G.; McEuen, P.L. Impermeable atomic membranes from graphene sheets. Nano Lett. 2008, 8, 2458–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Wu, H. Graphene-based nanocomposites: Preparation, functionalization, and energy and environmental applications. Energy Environ. Sci. 2013, 6, 3483–3507. [Google Scholar] [CrossRef]
- Pei, S.; Wei, Q.; Huang, K.; Cheng, H.-M.; Ren, W. Green synthesis of graphene oxide by seconds timescale water electrolytic oxidation. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Y.; Zhang, M.; Yao, B.; Li, Y.; Huang, L.; Li, C.; Shi, G. Water-enhanced oxidation of graphite to graphene oxide with controlled species of oxygenated groups. Chem. Sci. 2016, 7, 1874–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Hu, Y.; Shi, M.; Lu, X.; Qin, C.; Li, C.; Ye, M. Fast and facile preparation of graphene oxide and reduced graphene oxide nanoplatelets. Chem. Mater. 2009, 21, 3514–3520. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Etmimi, H.M.; Sanderson, R.D. New approach to the synthesis of exfoliated polymer/graphite nanocomposites by miniemulsion polymerization using functionalized graphene. Macromolecules 2011, 44, 8504–8515. [Google Scholar] [CrossRef]
- Putz, K.W.; Compton, O.C.; Palmeri, M.J.; Nguyen, S.T.; Brinson, L.C. High-nanofiller-content graphene oxide–polymer nanocomposites via vacuum-assisted self-assembly. Adv. Funct. Mater. 2010, 20, 3322–3329. [Google Scholar] [CrossRef]
- Xing, J.; Deng, B.; Liu, Q. Effect of graphene nanoplatelets on the performance of polyphenylene sulfide composites produced by melt intercalation. High Perform. Polym. 2018, 30, 519–526. [Google Scholar] [CrossRef]
- Bijarimi, M.; Amirul, M.; Norazmi, M.; Ramli, A.; Desa, M.S.Z.; Desa, M.D.A.; Samah, M.A.A. Preparation and characterization of poly (lactic acid)(PLA)/polyamide 6 (PA6)/graphene nanoplatelet (GNP) blends bio-based nanocomposites. Mater. Res. Express 2019, 6, 055044. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripathi, S.N.; Choudhary, V.; Gupta, B.D. Surface plasmon resonance-based fiber optic methane gas sensor utilizing graphene-carbon nanotubes-poly (methyl methacrylate) hybrid nanocomposite. Plasmonics 2015, 10, 1147–1157. [Google Scholar] [CrossRef]
- Rajabi, M.; Mahanpoor, K.; Moradi, O. Preparation of PMMA/GO and PMMA/GO-Fe3O4 nanocomposites for malachite green dye adsorption: Kinetic and thermodynamic studies. Compos. Part B Eng. 2019, 167, 544–555. [Google Scholar] [CrossRef]
- Thomassin, J.-M.; Trifkovic, M.; Alkarmo, W.; Detrembleur, C.; Jérôme, C.; Macosko, C. Poly (methyl methacrylate)/graphene oxide nanocomposites by a precipitation polymerization process and their dielectric and rheological characterization. Macromolecules 2014, 47, 2149–2155. [Google Scholar] [CrossRef]
- Wang, W.-P.; Pan, C.-Y. Preparation and characterization of polystyrene/graphite composite prepared by cationic grafting polymerization. Polymer 2004, 45, 3987–3995. [Google Scholar] [CrossRef]
- Hu, H.; Wang, X.; Wang, J.; Wan, L.; Liu, F.; Zheng, H.; Chen, R.; Xu, C. Preparation and properties of graphene nanosheets–polystyrene nanocomposites via in situ emulsion polymerization. Chem. Phys. Lett. 2010, 484, 247–253. [Google Scholar] [CrossRef]
- Poddar, M.K.; Pradhan, S.; Moholkar, V.S.; Arjmand, M.; Sundararaj, U. Ultrasound–assisted synthesis and characterization of polymethyl methacrylate/reduced graphene oxide nanocomposites. AIChE J. 2018, 64, 673–687. [Google Scholar] [CrossRef]
- Fadil, Y.; Agarwal, V.; Jasinski, F.; Thickett, S.C.; Minami, H.; Zetterlund, P.B. Electrically conductive polymer/rGO nanocomposite films at ambient temperature via miniemulsion polymerization using GO as surfactant. Nanoscale 2019, 11, 6566–6570. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, W.; Jiang, Z.; He, F.; Zhang, K.; Fan, J.; Wu, J. Graphene oxide-modified microencapsulated phase change materials with high encapsulation capacity and enhanced leakage-prevention performance. Appl. Energy 2017, 197, 354–363. [Google Scholar] [CrossRef]
- Thickett, S.C.; Wood, N.; Ng, Y.H.; Zetterlund, P.B. Hollow hybrid polymer–graphene oxide nanoparticles via Pickering miniemulsion polymerization. Nanoscale 2014, 6, 8590–8594. [Google Scholar] [CrossRef] [PubMed]
- Huynh, V.T.; Nguyen, D.; Such, C.H.; Hawkett, B.S. Polymer coating of graphene oxide via reversible addition–fragmentation chain transfer mediated emulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 1413–1421. [Google Scholar] [CrossRef]
- Pham, V.H.; Dang, T.T.; Hur, S.H.; Kim, E.J.; Chung, J.S. Highly conductive poly (methyl methacrylate)(PMMA)—Reduced graphene oxide composite prepared by self-assembly of PMMA latex and graphene oxide through electrostatic interaction. ACS Appl. Mater. Interfaces 2012, 4, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Sudol, E.D.; Dimonie, V.L.; El-Aasser, M.S. Nitroxide-mediated living free radical miniemulsion polymerization of styrene. Macromolecules 2001, 34, 481–488. [Google Scholar] [CrossRef]
- Taden, A.; Landfester, K. Crystallization of poly (ethylene oxide) confined in miniemulsion droplets. Macromolecules 2003, 36, 4037–4041. [Google Scholar] [CrossRef]
- Schork, F.J.; Luo, Y.; Smulders, W.; Russum, J.P.; Butté, A.; Fontenot, K. Miniemulsion polymerization. Adv. Polym. Sci. 2005, 175, 129–255. [Google Scholar]
- Asua, J.M. Miniemulsion polymerization. Prog. Polym. Sci. 2002, 27, 1283–1346. [Google Scholar] [CrossRef]
- Che Man, S.; Thickett, S.C.; Whittaker, M.R.; Zetterlund, P.B. Synthesis of polystyrene nanoparticles “armoured” with nanodimensional graphene oxide sheets by miniemulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 47–58. [Google Scholar] [CrossRef]
- Che Man, S.; Mohd Yusof, N.; Whittaker, M.R.; Thickett, S.C.; Zetterlund, P.B. Influence of monomer type on miniemulsion polymerization systems stabilized by graphene oxide as sole surfactant. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 5153–5162. [Google Scholar] [CrossRef]
- Teo, G.H.; Ng, Y.H.; Zetterlund, P.B.; Thickett, S.C. Factors influencing the preparation of hollow polymer-graphene oxide microcapsules via Pickering miniemulsion polymerization. Polymer 2015, 63, 1–9. [Google Scholar] [CrossRef]
- Tokuda, M.; Yamane, M.; Thickett, S.C.; Minami, H.; Zetterlund, P.B. Synthesis of polymeric nanoparticles containing reduced graphene oxide nanosheets stabilized by poly (ionic liquid) using miniemulsion polymerization. Soft Matter 2016, 12, 3955–3962. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Fang, L.; Xiao, J.; Song, Y.; Zheng, Q. Grafting of copolymers onto graphene by miniemulsion polymerization for conductive polymer composites: Improved electrical conductivity and compatibility induced by interfacial distribution of graphene. Polym. Chem. 2013, 4, 2939–2944. [Google Scholar] [CrossRef]
- Fadil, Y.; Jasinski, F.; Guok, T.W.; Thickett, S.C.; Minami, H.; Zetterlund, P.B. Pickering miniemulsion polymerization using graphene oxide: Effect of addition of a conventional surfactant. Polym. Chem. 2018, 9, 3368–3378. [Google Scholar] [CrossRef]
- Man, S.C.; Ly, D.; Whittaker, M.R.; Thickett, S.C.; Zetterlund, P.B. Nano-sized graphene oxide as sole surfactant in miniemulsion polymerization for nanocomposite synthesis: Effect of pH and ionic strength. Polymer 2014, 55, 3490–3497. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Ten, E.; Vermerris, W. Recent developments in polymers derived from industrial lignin. J. Appl. Polym. Sci. 2015, 132, 42069. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Zhu, Z.; Wang, S.-Q. Synthesis and rheological properties of polystyrene/layered silicate nanocomposite. Polymer 2005, 46, 3006–3013. [Google Scholar] [CrossRef]
- Deraguin, B.V.; Landau, L. Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solution of electrolytes. Acta Physicochim USSR 1941, 14, 633–662. [Google Scholar]
- Verwey, E.J.W. Theory of the stability of lyophobic colloids. J. Phys. Chem. 1947, 51, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, L.; Guo, Y.; Cheng, C.; Yang, L.; Jiang, L.; Yu, G.; Hu, W.; Liu, Y.; Zhu, D. Reduction of graphene oxide to highly conductive graphene by Lawesson’s reagent and its electrical applications. J. Mater. Chem. C 2013, 1, 3104–3109. [Google Scholar] [CrossRef]
- Zangmeister, C.D. Preparation and evaluation of graphite oxide reduced at 220 C. Chem. Mater. 2010, 22, 5625–5629. [Google Scholar] [CrossRef]
- Nethravathi, C.; Rajamathi, M. Chemically modified graphene sheets produced by the solvothermal reduction of colloidal dispersions of graphite oxide. Carbon 2008, 46, 1994–1998. [Google Scholar] [CrossRef]
- Titelman, G.I.; Gelman, V.; Bron, S.; Khalfin, R.L.; Cohen, Y.; Bianco-Peled, H. Characteristics and microstructure of aqueous colloidal dispersions of graphite oxide. Carbon 2005, 43, 641–649. [Google Scholar] [CrossRef]
- Uhl, F.M.; Yao, Q.; Nakajima, H.; Manias, E.; Wilkie, C.A. Expandable graphite/polyamide-6 nanocomposites. Polym. Degrad. Stab. 2005, 89, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; An, J.; Potts, J.R.; Velamakanni, A.; Murali, S.; Ruoff, R.S. Hydrazine-reduction of graphite-and graphene oxide. Carbon 2011, 49, 3019–3023. [Google Scholar] [CrossRef]
- Khezri, K.; Haddadi-Asl, V.; Roghani-Mamaqani, H.; Salami-Kalajahi, M. Encapsulation of organomodified montmorillonite with PMMA via in situ SR&NI ATRP in miniemulsion. J. Polym. Res. 2012, 19, 9868. [Google Scholar]
- Lan, F.; Liu, K.-X.; Jiang, W.; Zeng, X.-B.; Wu, Y.; Gu, Z.-W. Facile synthesis of monodisperse superparamagnetic Fe3O4/PMMA composite nanospheres with high magnetization. Nanotechnology 2011, 22, 225604. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Liu, Y.; Wang, W.; Wynn, J.; Gao, J. Using glucosamine as a reductant to prepare reduced graphene oxide and its nanocomposites with metal nanoparticles. J. Nanopart. Res. 2012, 14, 875. [Google Scholar] [CrossRef]
- Wu, T.; Wang, X.; Qiu, H.; Gao, J.; Wang, W.; Liu, Y. Graphene oxide reduced and modified by soft nanoparticles and its catalysis of the Knoevenagel condensation. J. Mater. Chem. 2012, 22, 4772–4779. [Google Scholar] [CrossRef]
- Wang, X.; Wen, X.; Liu, Z.; Tan, Y.; Yuan, Y.; Zhang, P. Rapid and efficient synthesis of soluble graphene nanosheets using N-methyl-p-aminophenol sulfate as a reducing agent. Nanotechnology 2012, 23, 485604. [Google Scholar] [CrossRef]
- Wang, P.; Tang, Y.; Dong, Z.; Chen, Z.; Lim, T.-T. Ag–AgBr/TiO 2/RGO nanocomposite for visible-light photocatalytic degradation of penicillin G. J. Mater. Chem. A 2013, 1, 4718–4727. [Google Scholar] [CrossRef]
- Kim, N.; Sudol, E.D.; Dimonie, V.L.; El-Aasser, M.S. Poly (vinyl alcohol) stabilization of acrylic emulsion polymers using the miniemulsion approach. Macromolecules 2003, 36, 5573–5579. [Google Scholar] [CrossRef]
- Kim, N.; Sudol, E.D.; Dimonie, V.L.; El-Aasser, M.S. Comparison of conventional and miniemulsion copolymerizations of acrylic monomers using poly (vinyl alcohol) as the sole stabilizer. Macromolecules 2004, 37, 2427–2433. [Google Scholar] [CrossRef]
- Yuki, K.; Nakamae, M.; Sato, T.; Maruyama, H.; Okaya, T. Physical properties of acrylic copolymer emulsions using poly (vinyl alcohol) as a protective colloid in comparison with those using surfactants. Polym. Int. 2000, 49, 1629–1635. [Google Scholar] [CrossRef]
- Robeson, L.M.; Vratsanos, M.S. Mechanical characterization of vinyl acetate based emulsion polymer blends. In Macromolecular Symposia; WILEY-VCH Verlag: Weinheim, Germany, 2000; Volume 1, pp. 117–138. [Google Scholar]
- Choi, K.S.; Liu, F.; Choi, J.S.; Seo, T.S. Fabrication of free-standing multilayered graphene and poly (3, 4-ethylenedioxythiophene) composite films with enhanced conductive and mechanical properties. Langmuir 2010, 26, 12902–12908. [Google Scholar] [CrossRef] [PubMed]
- Samakande, A.; Sanderson, R.D.; Hartmann, P.C. Encapsulated clay particles in polystyrene by RAFT mediated miniemulsion polymerization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7114–7126. [Google Scholar] [CrossRef]
- Tong, Z.; Deng, Y. Kinetics of miniemulsion polymerization of styrene in the presence of organoclays. Macromol. Mater. Eng. 2008, 293, 529–537. [Google Scholar] [CrossRef]
- Roghani-Mamaqani, H.; Haddadi-Asl, V.; Najafi, M.; Salami-Kalajahi, M. Preparation of tailor-made polystyrene nanocomposite with mixed clay-anchored and free chains via atom transfer radical polymerization. AIChE J. 2011, 57, 1873–1881. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixtures | Component | Amount Added (g) | Percentage/Total (wt.%) | Percentage/Monomer (wt.%) |
|---|---|---|---|---|
| Oil Phase | Methyl Methacrylate | 6 | 20.00% | 100.00% |
| Hexadecane | 0.5 | 1.67% | 8.33% | |
| AIBN | 0.1 | 0.33% | 1.67% | |
| Water Phase | Poly Vinyl Alcohol (10% solution) | 0.6 | 2.00% | 10.00% |
| Triton-405 | 0.17 | 0.57% | 2.83% | |
| Deionized Water | 24 | 80.00% | 400.00% |
| GO Loading% | Mw | Mn | PDI |
|---|---|---|---|
| 0% GO | 1.28E + 06 | 4.14E + 05 | 3.11 |
| 0.5% GO | 2.15E + 05 | 4.49E + 04 | 4.80 |
| 1% GO | 2.99E + 05 | 6.55E + 04 | 4.57 |
| 2% GO | 3.93E + 05 | 6.14E + 04 | 6.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wang, L.; Meng, S.; Lin, H.; Correll, M.; Tong, Z. Nanocomposite of Graphene Oxide Encapsulated in Polymethylmethacrylate (PMMA): Pre-Modification, Synthesis, and Latex Stability. J. Compos. Sci. 2020, 4, 118. https://0-doi-org.brum.beds.ac.uk/10.3390/jcs4030118
Wang H, Wang L, Meng S, Lin H, Correll M, Tong Z. Nanocomposite of Graphene Oxide Encapsulated in Polymethylmethacrylate (PMMA): Pre-Modification, Synthesis, and Latex Stability. Journal of Composites Science. 2020; 4(3):118. https://0-doi-org.brum.beds.ac.uk/10.3390/jcs4030118
Chicago/Turabian StyleWang, Hui, Letian Wang, Shanyu Meng, Hanxue Lin, Melanie Correll, and Zhaohui Tong. 2020. "Nanocomposite of Graphene Oxide Encapsulated in Polymethylmethacrylate (PMMA): Pre-Modification, Synthesis, and Latex Stability" Journal of Composites Science 4, no. 3: 118. https://0-doi-org.brum.beds.ac.uk/10.3390/jcs4030118