Microglial Expression of Hdac1 and Hdac2 is Dispensable for Experimental Autoimmune Encephalomyelitis (EAE) Progression


1
Institute of Neuropathology, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
2
Institute for Immunology, Faculty of Medicine, Ulm University, 89081 Ulm, Germany
3
Department of Pathology, Oslo University Hospital, 0424 Oslo, Norway
4
Berta-Ottenstein-Programme for Clinician Scientists, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
*
Author to whom correspondence should be addressed.
J 2020, 3(4), 358-365; https://0-doi-org.brum.beds.ac.uk/10.3390/j3040028
Submission received: 14 September 2020
/
Revised: 21 October 2020
/
Accepted: 29 October 2020
/
Published: 31 October 2020
(This article belongs to the Section Biology & Life Sciences)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Previously, we reported that microglial expression of histone deacetylases 1 and 2 (Hdac1 and Hdac2) is required for microglial maturation and modulates disease progression in a mouse model of Alzheimer’s disease. Here, we analyze the role of microglial expression of Hdac1 and Hdac2 in another disease paradigm, namely experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. The aim of this study was to ascertain whether microglial expression of these two epigenetic regulators modulates disease progression in the context of autoimmune disease. Hdac1 and Hdac2 were knocked out either individually or in combination using a microglia-specific, tamoxifen-inducible Cre-deleter line (Cx3cr1-CreERT2). The clinical course as well as histopathological changes during EAE were assessed in adult mice lacking microglial expression of these genes. Overall, no differences in disease onset, progression or severity could be detected in mice lacking microglial expression of either one or both of Hdac1 and Hdac2 genes. Similarly, the histopathology showed no differences in lymphocyte or macrophage infiltration or demyelination in either of the analyzed groups. As such, we conclude that unlike in neurodegenerative disease, microglial expression of Hdac1 and Hdac2 does not play a role in EAE.
1. Introduction
Microglia are the major brain resident type of myeloid cells and play major roles during maturation, homeostasis and various diseases of the central nervous system (CNS) [1,2]. During CNS homeostasis, they continuously survey their surroundings and react to neuronal damage as well as pathogen infiltration. Furthermore, they have been implicated in the regulation of adult neurogenesis as well as synaptic plasticity and myelination [3]. Microglia activation is an essential part of the innate immune response in the otherwise immune-privileged CNS and activated microglia can both serve to remove cellular debris or extracellular protein deposits, such as amyloid plaques, as well as participate in the inflammatory response during CNS disease [2,4]. In multiple sclerosis (MS) and animal models of the disease, such as experimental autoimmune encephalomyelitis (EAE), microglia function as part of the immune response. While activated, ramified microglia can be found predominantly in early active MS/EAE lesions, their morphology and activity is generally altered towards a phagocytic phenotype in older, chronic lesions. In early active disease, microglia are implicated as proinflammatory cells potentially driving disease initiation or progression [5]. Their phagocytic activity in later disease stages is important for clearance of cellular debris as well as initiation of remyelination [6,7]. As such, microglia function in MS/EAE can be both beneficial and detrimental to disease outcomes, depending on stage and regulation of their proinflammatory, phagocytic and neuroprotective functions.
Recently, we could show that both the homeostatic and neurodegeneration-related functions of microglia are regulated, at least in part, through epigenetic mechanisms and that the epigenetic regulators histone deacetylase (Hdac) 1 and 2 modulate the microglial response in a mouse model of Alzheimer’s disease [8]. Furthermore, microglial immune memory in response to repeated injection of lipopolysaccharide (LPS) is regulated through the function of Hdac1 and -2 [9].
However, in the context of neuroinflammatory disease, the role of microglial Hdac1 and -2 has not been extensively studied. Inhibition of class I and II Hdacs, of which Hdac1 and Hdac2 are ubiquitously expressed members, reduces disease severity and progression in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis [10,11,12]. Further, knockout of Hdac1 expression in T cells renders animals immune to EAE induction [13]. The objective of this study was to evaluate the contribution of microglial Hdac1 and -2 expression to the disease course and severity of EAE.
To this end, a microglia-specific knockout of Hdac1 and -2, either as single knockouts or in combination, was induced in adult mice using the microglia-specific Cx3CR1-CreERT2 line. These mice show a transient recombination of floxed targets in blood leukocytes within the first 14 days after tamoxifen induction; however, at later timepoints, the recombination persists only in long-living macrophage populations, most notably in microglia, while leukocytes, especially monocytes and T-lymphocytes, are no longer affected by the recombination [14]. EAE was induced in these mice 8 weeks after tamoxifen treatment to exclude effects on peripheral cells, most notably T cells. We then analyzed the effect of microglia-specific knockout of Hdac1 and -2 individually and in combination on EAE disease severity, as measured by maximal severity and total severity scores, as well as on the histological changes during EAE. Our data suggest that Hdac1 and Hdac2 function in microglia does not affect the severity or progression of EAE.
2. Materials and Methods
2.1. Mice
All animal experiments were approved by the Ministry for Nature, Environment and Consumers’ Protection of the state of Baden-Württemberg in accordance with EU directive 2010/63/EU and the German Animal Welfare Law under license G-12/103 and were performed in accordance with the respective EU, federal and institutional regulations. Experimental design and results reporting follow the ARRIVE guidelines [15].
Mice were housed in a specific pathogen-free (SPF) animal facility. They were group-housed with up to five animals per cage and kept on a 12-h light/dark cycle. Food and water were accessible ad libitum. Female mice only were used for experiments.
Cx3cr1-CreERT2 mice (B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung/J) [16], Hdac1loxP/loxP and Hdac2loxP/loxP [17] mice were kept on a C57BL/6 background and crossed to generate mouse lines heterozygous for Cx3cr1-CreERT2 and homozygous for floxed Hdac1 and Hdac2, either individually or in combination. The resulting lines are referred to as Hdac1-ko (Cx3cr1CreERT2/wtHdac1loxP/loxPHdac2wt/wt), Hdac2-ko (Cx3cr1CreERT2/wtHdac1wt/wtHdac2loxP/loxP) and DKO (Cx3cr1CreERT2/wtHdac1loxP/loxPHdac2loxP/loxP) throughout the manuscript.
2.2. Tamoxifen Treatment
Cre activity was induced by injection of tamoxifen at 6 weeks of age to induce gene knockout in adult microglia. To this end, 8 mg of tamoxifen (TAM, Sigma-Aldrich, St. Louis, MI, USA) dissolved in corn oil was applied subcutaneously twice with an interval of 48 h.
2.3. Experimental Autoimmune Encephalomyelitis (EAE)
EAE was induced in female mice 8 weeks after tamoxifen treatment in order to avoid residual effects of tamoxifen on EAE disease course [18,19]. Mice were immunized by subcutaneous injection with an emulsion of 200 μg of MOG35-55 peptide in Complete Freund’s Adjuvant (CFA) containing 0.1 mg of Mycobacterium tuberculosis (H37RA; Difco Laboratories, Detroit, MI, USA). Additionally, the animals were injected intraperitoneally with 200 ng of pertussis toxin (Sigma-Aldrich, Deisenhofen, Germany) 24 h and 48 h post-immunization. At the time of EAE induction, the mice weighed 20.8 g (± 2.4 g [SD]). Animals were evaluated for 30 days after induction of EAE. The experimental timeline is shown in Supplementary Figure S1.
The animals were housed in their respective home cages for the duration of the experiment and kept a litter cohort with multiple genotypes present in each cage and experimental batch. A total of 68 animals were analyzed in the four experimental groups (wildtype, Hdac1-ko, Hdac2-ko and DKO). The experimenters were blinded as to the genotype of the respective animals up until final data curation. As all animals received the same treatment, no randomization was performed.
EAE severity was scored according to a clinical assessment score daily following induction: Score 0—no disease; Score 1—decreased tail tone; Score 2—hindlimb weakness or partial paralysis; Score 3—complete hindlimb paralysis; Score 4—complete forelimb and hindlimb paralysis.
Animals reaching Score 4 were euthanized immediately and scores were analyzed only for the scores preceding the euthanasia. A total of 6 animals (3 in the wildtype and 3 in the DKO group) were euthanized as per these criteria. No additional adverse effects beyond the EAE signs as scored above were found.
2.4. Histology and Immunohistochemistry
Histology was performed as previously described [14]. Mice were sacrificed by transcardial perfusion with phosphate-buffered saline (PBS). Spinal cord was isolated and fixed in 4% paraformaldehyde (PFA) in PBS overnight. The samples were subsequently embedded in paraffin and stained with hematoxylin/eosin (H&E), Luxol fast blue (LFB) to assess the degree of demyelination, rat anti-mouse MAC-3 (2.5 μg/mL, clone M3/84, BD Pharmingen) as a marker for macrophages and microglia, rat anti-human CD3 (3.5 μg/mL, clone CD3-12, Serotec, Düsseldorf, Germany) for T cells, rat anti-mouse B220 (2.5 μg/mL, clone RA3-6B2, BD Pharmingen, San Jose, CA, USA) for B cells and mouse anti-mouse APP (3 μg/mL, clone 22C11, Millipore, Burlington, MA, USA) to analyze axonal damage. For MAC-3, CD3, B220 and APP immunohistochemistry, tissue sections were incubated overnight with the primary antibody at 4 °C and then with biotin-labeled goat anti-rat, goat anti-mouse or goat anti-rabbit secondary antibodies (2.5 μg/mL, Southern Biotech, Birmingham, AL, USA) for 45 min at room temperature (RT). Streptavidin (Southern Biotech) incubation was performed for 45 min at RT, and finally, 3′ -Diaminobenzidine (DAB) brown chromogen (Dako) was used for resolution of specific antibody-binding.
Positively stained cells were counted in at least three tissue sections per animal analyzed and are reported as cells per mm2. Percentage of myelination was defined as the percentage of LFB (blue)-stained tissue compared to the total tissue area analyzed and determined using ImageJ/Fiji version 1.53c.
2.5. Statistical Analysis
For each animal, a severity score as described above was determined each day post-EAE induction. Total severity scores were determined as the sum of all severity scores. Area under the curve (AUC) was determined as previously published [20]. Day of disease onset was defined as the first day on which a severity score of 1 or greater was present. For AUC and maximal as well as the total severity scores, the Kruskal–Wallis test was performed using GraphPad Prism 5 software (GraphPad Software Inc., Auckland, New Zealand). For all other analyses, one-way analysis of variance (ANOVA) was used for significance testing. Detailed results of the statistical analysis are shown in Tables S1 to S8.
3. Results
In order to investigate the role of Hdac1 and Hdac2 expression in microglia during neuroinflammatory disease, we analyzed the disease course of experimental autoimmune encephalomyelitis (EAE) in mice with a microglia-specific knockout of these genes, as well as mice with a combined knockout of Hdac1 and -2 in microglia. This stemmed from our previously published findings that in steady state as well as neurodegenerative disease, single knockout of Hdac1 or -2 in microglia did not exhibit detectable changes from the wildtype condition, while combined gene knockout did show differences in microglia response to disease [8].
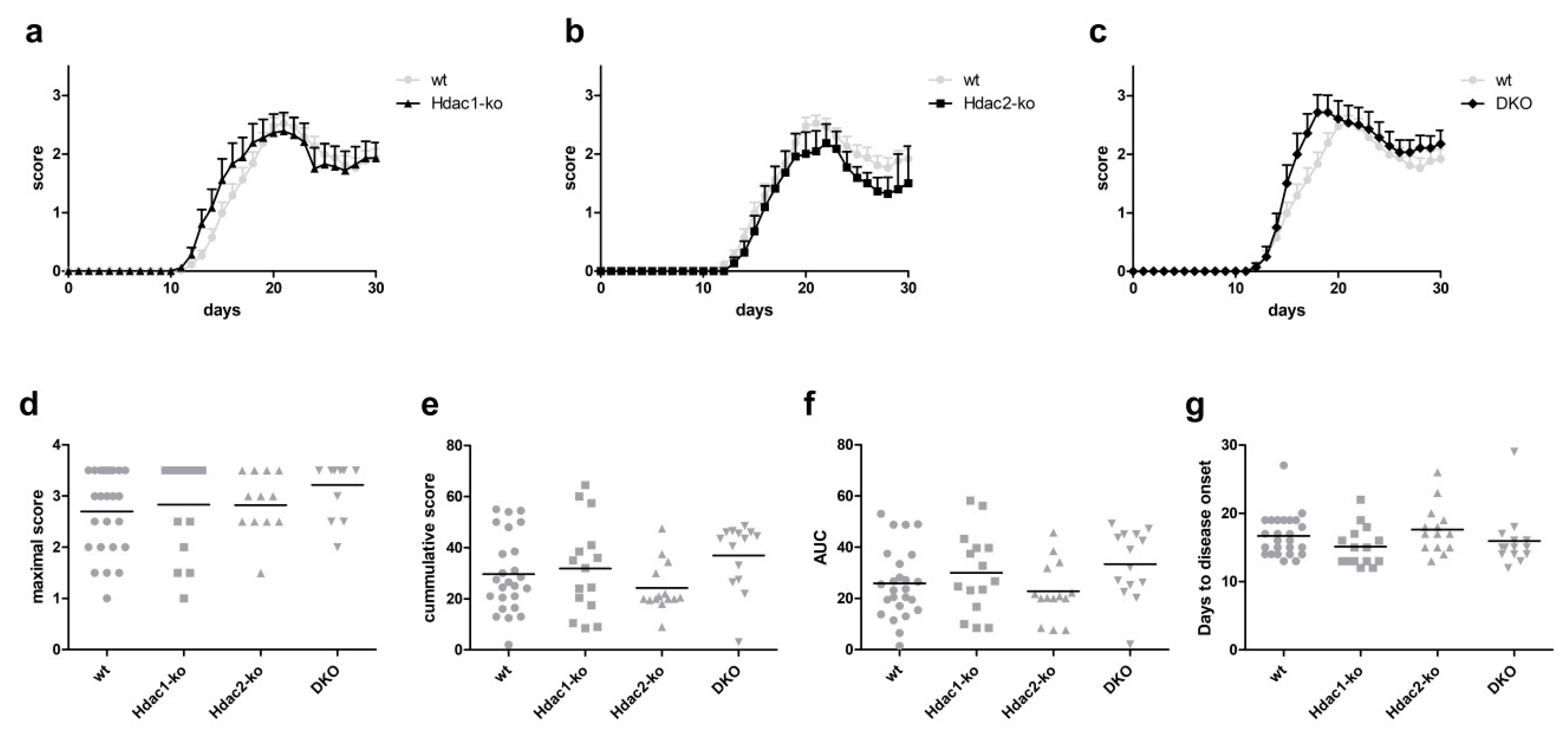
EAE disease course in mice with Hdac1 knockout alone was not altered when compared to wildtype mice (Figure 1a). Similarly, the disease course in Hdac2-ko mice appeared unaltered compared to wildtype mice (Figure 1b). As we found previously that double knockout animals showed alterations in microglia functions not apparent in single knockout animals, we also performed EAE in mice harboring a combined knockout of Hdac1 and Hdac2 (DKO) (Figure 1c). While there appeared to be a trend towards a slightly earlier peak of disease, neither the time course nor severity scores of the disease differed significantly from wildtype animals. To investigate disease course and severity in more depth, we also analyzed the maximal scores as well as the cumulative scores and area under the curve (AUC) in all animals (Figure 1d–f). Neither of the strains analyzed showed significant differences in these measurements when compared to wildtype mice. Days to disease onset were also not significantly altered in any of the strains when compared to wildtype controls (Figure 1g).
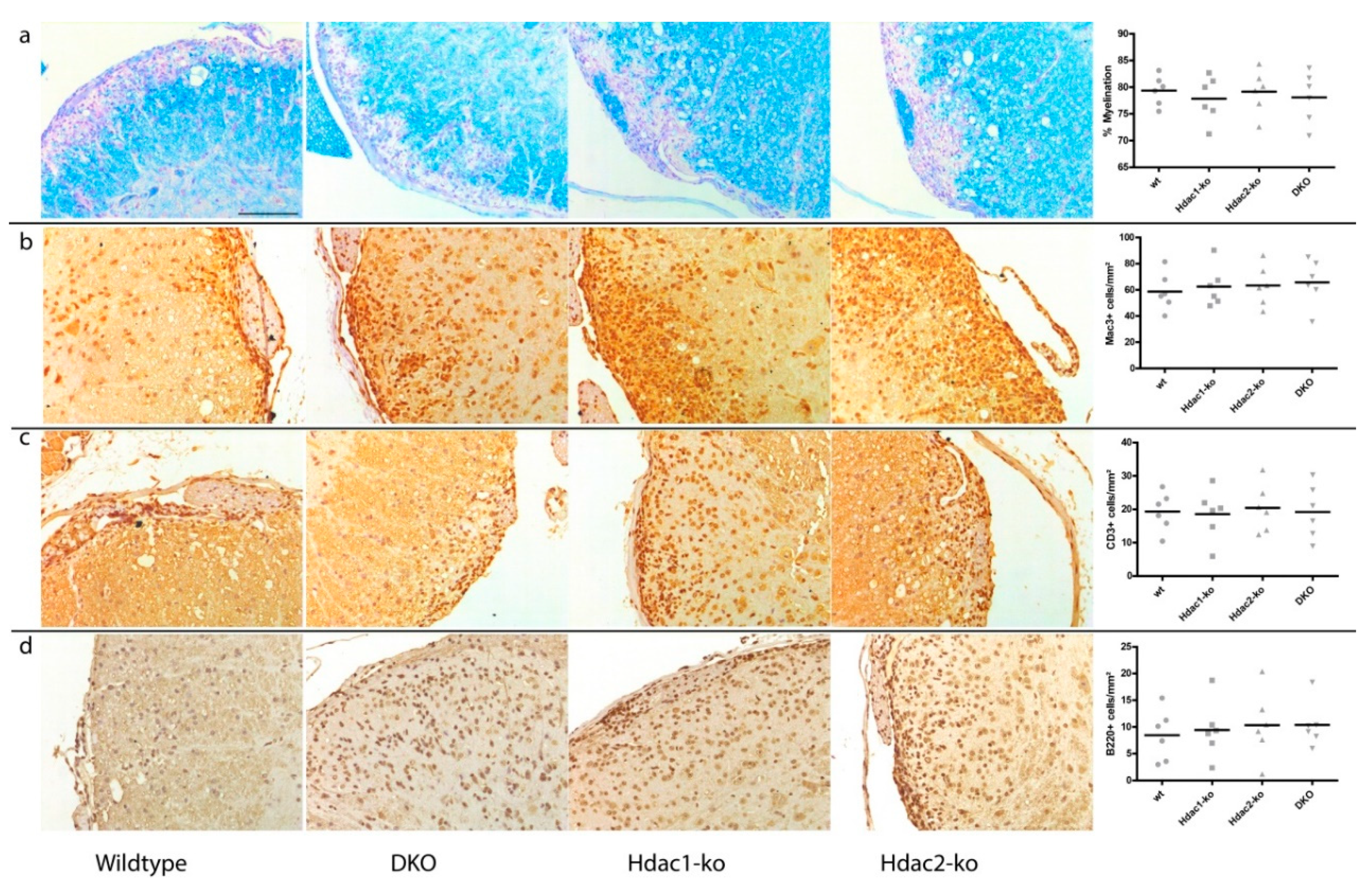
Mice were sacrificed on day 30 after EAE induction and brains and spinal cords were analyzed for histological changes. Spinal cord lesions showed no histological differences between genotypes (Figure 2a). Spinal cord demyelination as measured by Luxol fast blue (LFB) staining was also not altered in Hdac1-ko, Hdac2-ko or DKO mice when compared to wildtype mice (Figure 2b and Figure 3a). Similarly, no overt changes in axonal damage could be found in APP immunohistochemistry (Figure 2f). Macrophage infiltration as well as the number of microglia in EAE lesions were similarly not significantly altered in any of the mouse lines investigated, as evidenced by Mac-3 immunohistochemistry (Figure 2c and Figure 3b). Finally, T-cell infiltration, as visualized by CD3 immunohistochemistry (Figure 2d and Figure 3c), and B-cell infiltrates, as shown by B220 immunohistochemistry (Figure 2e and Figure 3d), exhibited no alteration in the analyzed mouse lines.
4. Conclusions
Here, we report on the effect of microglia-specific knockout of histone deacetylases Hdac1 and Hdac2 on the disease progression in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis (MS). While Hdac1/2 knockout alters microglia response in a murine model for Alzheimer’s disease (AD) as well as immune memory [8,9], its effect on neuroinflammatory disease had not been previously studied.
We find that neither individual knockout of Hdac1 or Hdac2, nor a combined knockout of these two genes in microglia leads to statistically significant alterations in disease course or disease severity in EAE. Furthermore, immune cell infiltration into the CNS lesions did not appear to be altered in either the individual or combined knockout mice. As such, microglial Hdac1 and Hdac2 expression appears to be dispensable for microglial function in EAE.
As Hdac inhibition has been discussed as a potential therapeutic target for MS [12,21,22], understanding the cellular targets of such inhibitor treatment is valuable for understanding the biological effects leading to the amelioration of MS disease severity due to the treatment. While previous reports show that T cell-specific Hdac1 knockout does ameliorate the disease in EAE [13], the effects on CNS cells had not previously been reported. With the results presented here showing that Hdac1/2 function in microglia, the predominant CNS-resident immune cell type, does not affect EAE disease course, it appears likely that broad Hdac inhibitor treatment in EAE/MS does, indeed, act predominantly on the infiltrating lymphocytic immune cell compartment during disease and/or that other Hdacs are involved in regulation of cellular activity in this setting. Specific inhibition of the class IIb Hdac6, for example, has been reported as ameliorating EAE progression, potentially through alterations in B and T cell activity [23]. Another recent report implicated Hdac11 in regulation of monocyte function during EAE [24]. Whether and how these Hdacs are involved in regulation of microglia function during MS/EAE warrants further investigation.
Taken together with our previously reported evidence that Hdac1 and 2 function is required for microglial development, maturation and immune memory and is involved in the regulation of microglial response to amyloid deposits in an AD model [8,9], these results indicate that Hdac1 and Hdac2 function in microglia is stimulus-dependent and not required in all types of microglial activation. As microglia both in steady state and in disease conditions are heterogeneous cell populations [25,26], it appears possible that only some subpopulations of microglia are regulated directly through the activity of Hdac1 and/or -2 and that these subpopulations predominate only in specific contexts, such as the response to amyloid deposition in AD. Further study of the stimuli required for Hdac1/2 activity in microglia could further improve the understanding of microglia biology.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2571-8800/3/4/28/s1, Figure S1: Timeline of experiments, Tables S1 to S8: Detailed results of statistical analysis for data shown in Figure 1 and Figure 3.
Author Contributions
Conceptualization, M.D. and O.S.; formal analysis, M.D. and O.S.; investigation, M.D., S.M.H. and O.S.; data curation, M.D.; writing—original draft preparation, O.S.; writing—review and editing, M.D. and S.M.H.; visualization, M.D. and O.S. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for this project was provided in part by the German Research Foundation (DFG) CRC/SFB 992. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding programme Open Access Publishing. Funding agencies were not involved in the design of the study nor in data collection, analysis or interpretation.
Acknowledgments
We thank Marco Prinz, Institute of Neuropathology Freiburg, for his support of this study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Schafer, D.P.; Stevens, B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 7, a020545. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.; Barak, B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia 2019, 67, 2125–2141. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell. Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Datta, M.; Staszewski, O.; Raschi, E.; Frosch, M.; Hagemeyer, N.; Tay, T.L.; Blank, T.; Kreutzfeldt, M.; Merkler, D.; et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity 2018, 48, 514–529.e6. [Google Scholar] [CrossRef]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Faraco, G.; Cavone, L.; Chiarugi, A. The therapeutic potential of HDAC inhibitors in the treatment of multiple sclerosis. Mol. Med. 2011, 17, 442–447. [Google Scholar] [CrossRef]
- Dasgupta, S.; Zhou, Y.; Jana, M.; Banik, N.L.; Pahan, K. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. J. Immunol. 2003, 170, 3874–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.G.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef]
- Goschl, L.; Preglej, T.; Hamminger, P.; Bonelli, M.; Andersen, L.; Boucheron, N.; Gülich, A.F.; Müller, L.; Saferding, V.; Mufazalov, I.A.; et al. A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis. J. Autoimmun. 2018, 86, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Elloso, M.M.; Phiel, K.; Henderson, R.A.; Harris, H.A.; Adelman, S.J. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J. Endocrinol. 2005, 185, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Bebo, B.F., Jr.; Dehghani, B.; Foster, S.; Kurniawan, A.; Lopez, F.J.; Sherman, L.S. Treatment with selective estrogen receptor modulators regulates myelin specific T-cells and suppresses experimental autoimmune encephalomyelitis. Glia 2009, 57, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Fleming, K.K.; Bovaird, J.A.; Mosier, M.C.; Emerson, M.R.; LeVine, S.M.; Marquis, J.G. Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 170, 71–84. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.Y.; Wu, Y.; Schluesener, H.J. Valproic acid ameliorates inflammation in experimental autoimmune encephalomyelitis rats. Neuroscience 2012, 221, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P. The Selective HDAC6 Inhibitor ACY-738 Impacts Memory and Disease Regulation in an Animal Model of Multiple Sclerosis. Front. Neurol. 2019, 10, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1, e201800039. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; Groß, O.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef]
Figure 1.
(a–c) Experimental autoimmune encephalomyelitis (EAE) disease course in Hdac1-ko (a), Hdac2-ko (b) and DKO (c) mice compared to wildtype (wt). Error bars depict S.D. (d) Maximal disease scores in Hdac1-ko, Hdac2-ko, DKO and wt mice. (e) Cumulative disease scores in Hdac1-ko, Hdac2-ko, DKO and wt mice. (f) Area under the curve for disease course for each animal by genotype. (g) Days to disease onset for each animal. No statistical difference at any time point was found. The numbers of animals analyzed were 25 (wt), 15 (Hdac1-ko), 14 (Hdac2-ko) and 14 (DKO).
Figure 1.
(a–c) Experimental autoimmune encephalomyelitis (EAE) disease course in Hdac1-ko (a), Hdac2-ko (b) and DKO (c) mice compared to wildtype (wt). Error bars depict S.D. (d) Maximal disease scores in Hdac1-ko, Hdac2-ko, DKO and wt mice. (e) Cumulative disease scores in Hdac1-ko, Hdac2-ko, DKO and wt mice. (f) Area under the curve for disease course for each animal by genotype. (g) Days to disease onset for each animal. No statistical difference at any time point was found. The numbers of animals analyzed were 25 (wt), 15 (Hdac1-ko), 14 (Hdac2-ko) and 14 (DKO).

Figure 2.
Representative histological and immunohistochemical stainings of (a) spinal cord lesions (H&E), (b) myelination (Luxol fast blue (LFB)), (c) microglia/macrophage infiltrates (Mac-3), (d) T-cell infiltrates (CD3), (e) B-cell infiltrates (B220) and (f) axonal damage (APP). Scale bar = 500 µm; all images have the same scaling.
Figure 2.
Representative histological and immunohistochemical stainings of (a) spinal cord lesions (H&E), (b) myelination (Luxol fast blue (LFB)), (c) microglia/macrophage infiltrates (Mac-3), (d) T-cell infiltrates (CD3), (e) B-cell infiltrates (B220) and (f) axonal damage (APP). Scale bar = 500 µm; all images have the same scaling.

Figure 3.
Higher-magnification images of histological and immunohistochemical stains and quantification of six samples for each genotype. (a) Myelination as identified by LFB staining; blue-stained tissue is myelinated. (b) Mac-3-positive macrophages/microglia in spinal cord lesions. (c) CD3-positive T cell and (d) B220-positive B cell counts. No statistically significant changes were found in either analysis. Scale bar = 100 µm; all images have the same scaling.
Figure 3.
Higher-magnification images of histological and immunohistochemical stains and quantification of six samples for each genotype. (a) Myelination as identified by LFB staining; blue-stained tissue is myelinated. (b) Mac-3-positive macrophages/microglia in spinal cord lesions. (c) CD3-positive T cell and (d) B220-positive B cell counts. No statistically significant changes were found in either analysis. Scale bar = 100 µm; all images have the same scaling.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Datta, M.; Hansen, S.M.; Staszewski, O. Microglial Expression of Hdac1 and Hdac2 is Dispensable for Experimental Autoimmune Encephalomyelitis (EAE) Progression. J 2020, 3, 358-365. https://0-doi-org.brum.beds.ac.uk/10.3390/j3040028
AMA Style
Datta M, Hansen SM, Staszewski O. Microglial Expression of Hdac1 and Hdac2 is Dispensable for Experimental Autoimmune Encephalomyelitis (EAE) Progression. J. 2020; 3(4):358-365. https://0-doi-org.brum.beds.ac.uk/10.3390/j3040028
Chicago/Turabian StyleDatta, Moumita, Stefanie M. Hansen, and Ori Staszewski. 2020. "Microglial Expression of Hdac1 and Hdac2 is Dispensable for Experimental Autoimmune Encephalomyelitis (EAE) Progression" J 3, no. 4: 358-365. https://0-doi-org.brum.beds.ac.uk/10.3390/j3040028