Diabetes Mellitus and Colon Carcinogenesis: Expectation for Inhibition of Colon Carcinogenesis by Oral Hypoglycemic Drugs

Abstract
:1. Introduction
2. Diabetes Mellitus and Colorectal Cancer: Basic Introduction and Epidemiological Relevance
2.1. Diabetes Mellitus
2.2. Colorectal Cancer
2.3. Epidemiological Relevance of Diabetes Mellitus and Colorectal Cancer Risk
2.4. Expectation for Reduction of Carcinogenic Risk by Diabetes Treatment, Especially Oral Hypoglycemic Drugs
3. Molecular Mechanisms of the Association between Diabetes Mellitus and Colorectal Cancer
3.1. The Insulin-like Growth Factor Signaling System
3.2. The Wnt Signaling System
3.3. Glucagon-like Peptide-1
3.4. Gut Microbiota
4. Suppressive Effect of Colonic Carcinogenesis by Oral Hypoglycemic Drugs
4.1. Types of Oral Hypoglycemic Drugs for Type 2 Diabetes Mellitus
4.2. Basic Pharmacological Action of Oral Hypoglycemic Drugs and Reports on Suppressive Effect of Colonic Carcinogenesis Risk
4.2.1. Sulfonylureas
4.2.2. Immediate-release Insulin Secretagogues (Glinides)
4.2.3. DPP-4 Inhibitors
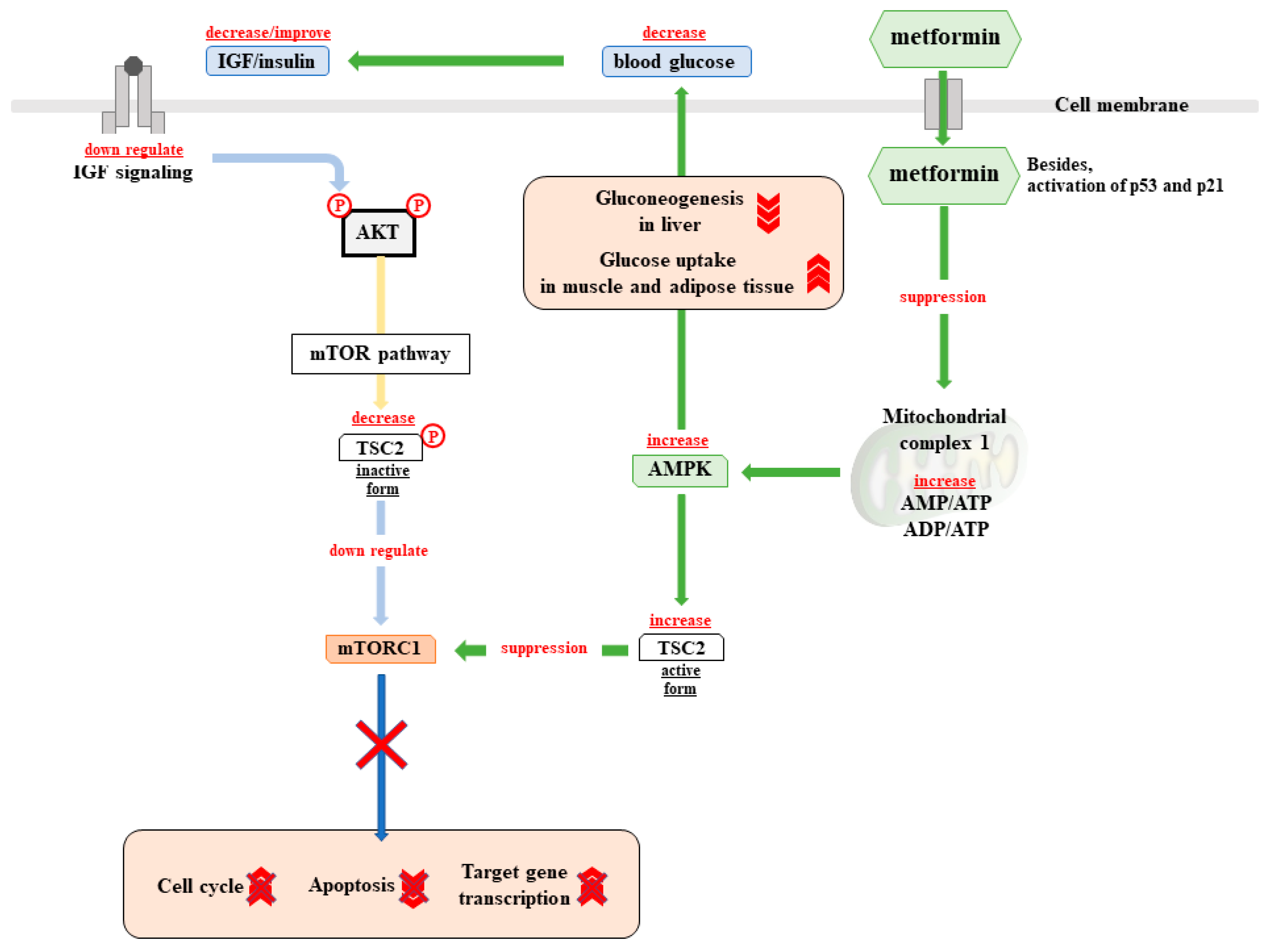
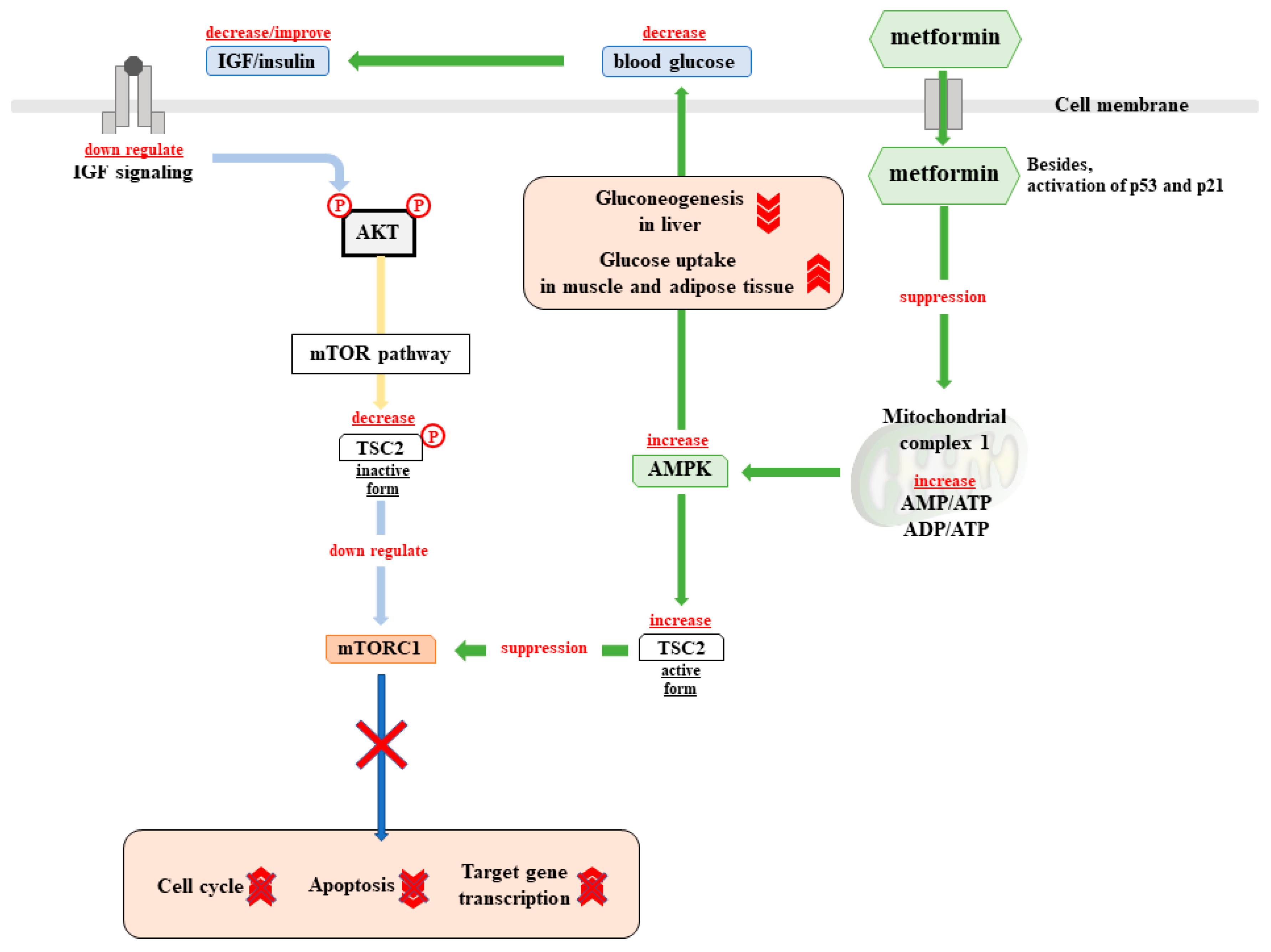
4.2.4. Biguanides
4.2.5. Thiazolidinedione
4.2.6. Alpha-glucosidase Inhibitors
4.2.7. SGLT2 Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonzalez, N.; Prieto, I.; Del Puerto-Nevado, L.; Portal-Nunez, S.; Ardura, J.A.; Corton, M.; Fernandez-Fernandez, B.; Aguilera, O.; Gomez-Guerrero, C.; Mas, S.; et al. 2017 update on the relationship between diabetes and colorectal cancer: Epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 2017, 8, 18456–18485. [Google Scholar] [CrossRef]
- Nilsen, T.I.; Vatten, L.J. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: Exploring the hyperinsulinaemia hypothesis. Br. J. Cancer 2001, 84, 417–422. [Google Scholar] [CrossRef]
- Jee, S.H.; Ohrr, H.; Sull, J.W.; Yun, J.E.; Ji, M.; Samet, J.M. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 2005, 293, 194–202. [Google Scholar] [CrossRef]
- Inoue, M.; Iwasaki, M.; Otani, T.; Sasazuki, S.; Noda, M.; Tsugane, S. Diabetes mellitus and the risk of cancer: Results from a large-scale population-based cohort study in Japan. Arch Intern. Med. 2006, 166, 1871–1877. [Google Scholar] [CrossRef]
- Noto, H.; Osame, K.; Sasazuki, T.; Noda, M. Substantially increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis of epidemiologic evidence in Japan. J. Diabetes Complicat. 2010, 24, 345–353. [Google Scholar] [CrossRef]
- Noto, H.; Tsujimoto, T.; Sasazuki, T.; Noda, M. Significantly increased risk of cancer in patients with diabetes mellitus: A systematic review and meta-analysis. Endocr. Pract. 2011, 17, 616–628. [Google Scholar] [CrossRef]
- Geraldine, N.; Marc, A.; Carla, T.; Chantal, M.; Stefaan, B.; Welcome, W.; Frank, B. Relation between diabetes, metformin treatment and the occurrence of malignancies in a Belgian primary care setting. Diabetes Res. Clin. Pract. 2012, 97, 331–336. [Google Scholar] [CrossRef]
- Lee, M.Y.; Lin, K.D.; Hsiao, P.J.; Shin, S.J. The association of diabetes mellitus with liver, colon, lung, and prostate cancer is independent of hypertension, hyperlipidemia, and gout in Taiwanese patients. Metabolism 2012, 61, 242–249. [Google Scholar] [CrossRef]
- Yeh, H.C.; Platz, E.A.; Wang, N.Y.; Visvanathan, K.; Helzlsouer, K.J.; Brancati, F.L. A prospective study of the associations between treated diabetes and cancer outcomes. Diabetes Care 2012, 35, 113–118. [Google Scholar] [CrossRef]
- Zhang, P.H.; Chen, Z.W.; Lv, D.; Xu, Y.Y.; Gu, W.L.; Zhang, X.H.; Le, Y.L.; Zhu, H.H.; Zhu, Y.M. Increased risk of cancer in patients with type 2 diabetes mellitus: A retrospective cohort study in China. BMC Public Health 2012, 12, 567. [Google Scholar] [CrossRef]
- Jarvandi, S.; Davidson, N.O.; Schootman, M. Increased risk of colorectal cancer in type 2 diabetes is independent of diet quality. PLoS ONE 2013, 8, e74616. [Google Scholar] [CrossRef]
- Lo, S.F.; Chang, S.N.; Muo, C.H.; Chen, S.Y.; Liao, F.Y.; Dee, S.W.; Chen, P.C.; Sung, F.C. Modest increase in risk of specific types of cancer types in type 2 diabetes mellitus patients. Int. J. Cancer 2013, 132, 182–188. [Google Scholar] [CrossRef]
- Oberaigner, W.; Ebenbichler, C.; Oberaigner, K.; Juchum, M.; Schonherr, H.R.; Lechleitner, M. Increased cancer incidence risk in type 2 diabetes mellitus: Results from a cohort study in Tyrol/Austria. BMC Public Health 2014, 14, 1058. [Google Scholar] [CrossRef]
- Harding, J.L.; Shaw, J.E.; Peeters, A.; Cartensen, B.; Magliano, D.J. Cancer risk among people with type 1 and type 2 diabetes: Disentangling true associations, detection bias, and reverse causation. Diabetes Care 2015, 38, 264–270. [Google Scholar] [CrossRef]
- Liu, X.; Hemminki, K.; Forsti, A.; Sundquist, K.; Sundquist, J.; Ji, J. Cancer risk in patients with type 2 diabetes mellitus and their relatives. Int. J. Cancer 2015, 137, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ 2015, 350, g7607. [Google Scholar] [CrossRef]
- Wang, M.; Hu, R.Y.; Wu, H.B.; Pan, J.; Gong, W.W.; Guo, L.H.; Zhong, J.M.; Fei, F.R.; Yu, M. Cancer risk among patients with type 2 diabetes mellitus: A population-based prospective study in China. Sci. Rep. 2015, 5, 11503. [Google Scholar] [CrossRef]
- Dankner, R.; Boffetta, P.; Balicer, R.D.; Boker, L.K.; Sadeh, M.; Berlin, A.; Olmer, L.; Goldfracht, M.; Freedman, L.S. Time-Dependent Risk of Cancer After a Diabetes Diagnosis in a Cohort of 2.3 Million Adults. Am. J. Epidemiol. 2016, 183, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- De Kort, S.; Simons, C.C.; van den Brandt, P.A.; Goldbohm, R.A.; Arts, I.C.; de Bruine, A.P.; Janssen-Heijnen, M.L.; Sanduleanu, S.; Masclee, A.A.; Weijenberg, M.P. Diabetes mellitus type 2 and subsite-specific colorectal cancer risk in men and women: Results from the Netherlands Cohort Study on diet and cancer. Eur. J. Gastroenterol. Hepatol. 2016, 28, 896–903. [Google Scholar] [CrossRef]
- Ballotari, P.; Vicentini, M.; Manicardi, V.; Gallo, M.; Chiatamone Ranieri, S.; Greci, M.; Giorgi Rossi, P. Diabetes and risk of cancer incidence: Results from a population-based cohort study in northern Italy. BMC Cancer 2017, 17, 703. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, F.; Saito, E.; Lin, Y.; Song, M.; Luu, H.N.; Gupta, P.C.; Sawada, N.; Tamakoshi, A.; Shu, X.O.; et al. Association between type 2 diabetes and risk of cancer mortality: A pooled analysis of over 771,000 individuals in the Asia Cohort Consortium. Diabetologia 2017, 60, 1022–1032. [Google Scholar] [CrossRef]
- De Jong, R.; Burden, A.M.; de Kort, S.; van Herk-Sukel, M.P.P.; Vissers, P.A.J.; Janssen, P.K.C.; Haak, H.R.; Masclee, A.A.M.; de Vries, F.; Janssen-Heijnen, M.L.G. Impact of detection bias on the risk of gastrointestinal cancer and its subsites in type 2 diabetes mellitus. Eur. J. Cancer 2017, 79, 61–71. [Google Scholar] [CrossRef]
- Saarela, K.; Tuomilehto, J.; Sund, R.; Keskimaki, I.; Hartikainen, S.; Pukkala, E. Cancer incidence among Finnish people with type 2 diabetes during 1989-2014. Eur. J. Epidemiol. 2018. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Habib, S.L.; Rojna, M. Diabetes and risk of cancer. ISRN Oncol. 2013, 2013, 583786. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakama, H.; Yamamoto, M.; Kamijo, N.; Li, T.; Wei, N.; Fattah, A.S.; Zhang, B. Colonoscopic evaluation of immunochemical fecal occult blood test for detection of colorectal neoplasia. Hepatogastroenterology 1999, 46, 228–231. [Google Scholar] [PubMed]
- Park, D.I.; Ryu, S.; Kim, Y.H.; Lee, S.H.; Lee, C.K.; Eun, C.S.; Han, D.S. Comparison of guaiac-based and quantitative immunochemical fecal occult blood testing in a population at average risk undergoing colorectal cancer screening. Am. J. Gastroenterol. 2010, 105, 2017–2025. [Google Scholar] [CrossRef]
- Rozen, P.; Shabtai, E.I.; Liphshitz, I.; Barchana, M. Risk for colorectal cancer in elderly persons and possible methodologies for their screening. Eur. J. Gastroenterol. Hepatol. 2011, 23, 431–437. [Google Scholar] [CrossRef]
- McCarthy, K.; Pearson, K.; Fulton, R.; Hewitt, J. Pre-operative chemoradiation for non-metastatic locally advanced rectal cancer. Cochrane Database Syst. Rev. 2012, 12, CD008368. [Google Scholar] [CrossRef]
- Andre, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef]
- Lin, C.M.; Huang, H.L.; Chu, F.Y.; Fan, H.C.; Chen, H.A.; Chu, D.M.; Wu, L.W.; Wang, C.C.; Chen, W.L.; Lin, S.H.; et al. Association between Gastroenterological Malignancy and Diabetes Mellitus and Anti-Diabetic Therapy: A Nationwide, Population-Based Cohort Study. PLoS ONE 2015, 10, e0125421. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Hense, H.W.; Kajuter, H.; Wellmann, J.; Batzler, W.U. Cancer incidence in type 2 diabetes patients - first results from a feasibility study of the D2C cohort. Diabetol. Metab. Syndr. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Will, J.C.; Galuska, D.A.; Vinicor, F.; Calle, E.E. Colorectal cancer: Another complication of diabetes mellitus? Am. J. Epidemiol. 1998, 147, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Giouleme, O.; Diamantidis, M.D.; Katsaros, M.G. Is diabetes a causal agent for colorectal cancer? Pathophysiological and molecular mechanisms. World J. Gastroenterol. 2011, 17, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R.; Hongo, A.; Rubini, M.; Prisco, M.; Valentinis, B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta 1997, 1332, F105–F126. [Google Scholar] [CrossRef]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Suh, Y. Regulation of IGF-1 signaling by microRNAs. Front. Genet. 2014, 5, 472. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Gupta, N.; Goh, T.; Naigamwalla, D.; Chia, M.C.; Koohestani, N.; Mehrotra, S.; McKeown-Eyssen, G.; Giacca, A.; Bruce, W.R. Direct measure of insulin sensitivity with the hyperinsulinemic-euglycemic clamp and surrogate measures of insulin sensitivity with the oral glucose tolerance test: Correlations with aberrant crypt foci promotion in rats. Cancer Epidemiol. Biomarkers Prev. 2003, 12, 47–56. [Google Scholar]
- Yakar, S.; Nunez, N.P.; Pennisi, P.; Brodt, P.; Sun, H.; Fallavollita, L.; Zhao, H.; Scavo, L.; Novosyadlyy, R.; Kurshan, N.; et al. Increased tumor growth in mice with diet-induced obesity: Impact of ovarian hormones. Endocrinology 2006, 147, 5826–5834. [Google Scholar] [CrossRef]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Manson, J.E.; Li, J.; Harris, T.G.; Rohan, T.E.; Xue, X.; Ho, G.Y.; et al. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 921–929. [Google Scholar] [CrossRef]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Howard, B.V.; Wylie-Rosett, J.; Anderson, G.L.; Ho, G.Y.; et al. Insulin, insulin-like growth factor-I, endogenous estradiol, and risk of colorectal cancer in postmenopausal women. Cancer Res. 2008, 68, 329–337. [Google Scholar] [CrossRef]
- Baxter, R.C.; Bryson, J.M.; Turtle, J.R. Somatogenic receptors of rat liver: Regulation by insulin. Endocrinology 1980, 107, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Palsgaard, J.; Emanuelli, B.; Winnay, J.N.; Sumara, G.; Karsenty, G.; Kahn, C.R. Cross-talk between insulin and Wnt signaling in preadipocytes: Role of Wnt co-receptor low density lipoprotein receptor-related protein-5 (LRP5). J. Biol. Chem. 2012, 287, 12016–12026. [Google Scholar] [CrossRef] [PubMed]
- Jin, T. Why diabetes patients are more prone to the development of colon cancer? Med. Hypotheses 2008, 71, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.I.; Dunn, W.B.; Griffin, J.L.; Allwood, J.W.; Goodacre, R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics 2007, 8, 1243–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tringe, S.G.; Hugenholtz, P. A renaissance for the pioneering 16S rRNA gene. Curr. Opin. Microbiol. 2008, 11, 442–446. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- DeFilippis, E.M.; Givertz, M.M. Treating Diabetes in Patients with Heart Failure: Moving from Risk to Benefit. Curr. Heart Fail Rep. 2016, 13, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Aw, W.; Fukuda, S. Understanding the role of the gut ecosystem in diabetes mellitus. J. Diabetes Investig. 2018, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Out, C.; Fuentes, S.; Jonker, L.; Reuling, I.; Kootte, R.S.; van Nood, E.; Holleman, F.; Knaapen, M.; Romijn, J.A.; et al. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J. Hepatol. 2014, 60, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Kanazawa, A.; Ikeda, F.; Yoshihara, T.; Goto, H.; Abe, H.; Komiya, K.; Kawaguchi, M.; Shimizu, T.; Ogihara, T.; et al. Gut dysbiosis and detection of "live gut bacteria" in blood of Japanese patients with type 2 diabetes. Diabetes Care 2014, 37, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Sola, D.; Rossi, L.; Schianca, G.P.; Maffioli, P.; Bigliocca, M.; Mella, R.; Corliano, F.; Fra, G.P.; Bartoli, E.; Derosa, G. Sulfonylureas and their use in clinical practice. Arch. Med. Sci. 2015, 11, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Becker, C.; Meier, C.; Jick, S.S.; Meier, C.R. Use of metformin is not associated with a decreased risk of colorectal cancer: A case-control analysis. Cancer Epidemiol. Biomark. Prev. 2012, 21, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Soranna, D.; Scotti, L.; Zambon, A.; Bosetti, C.; Grassi, G.; Catapano, A.; La Vecchia, C.; Mancia, G.; Corrao, G. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: A meta-analysis. Oncologist 2012, 17, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Rhoads, G.G.; Berlin, J.A.; Marcella, S.W.; Demissie, K. Initial metformin or sulphonylurea exposure and cancer occurrence among patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2013, 15, 349–357. [Google Scholar] [CrossRef]
- Chen, Y.C.; Kok, V.C.; Chien, C.H.; Horng, J.T.; Tsai, J.J. Cancer risk in patients aged 30 years and above with type 2 diabetes receiving antidiabetic monotherapy: A cohort study using metformin as the comparator. Ther. Clin. Risk Manag. 2015, 11, 1315–1323. [Google Scholar] [CrossRef]
- Mc Menamin, U.C.; Murray, L.J.; Hughes, C.M.; Cardwell, C.R. Metformin use and survival after colorectal cancer: A population-based cohort study. Int. J. Cancer 2016, 138, 369–379. [Google Scholar] [CrossRef]
- Baglia, M.L.; Cui, Y.; Zheng, T.; Yang, G.; Li, H.; You, M.; Xu, L.; Murff, H.; Gao, Y.T.; Zheng, W.; et al. Diabetes Medication Use in Association with Survival among Patients of Breast, Colorectal, Lung, or Gastric Cancer. Cancer Res. Treat. 2018. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Lin, J.W.; Wu, L.C.; Lai, M.S.; Chuang, L.M. Oral insulin secretagogues, insulin, and cancer risk in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, E1170–E1175. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, H.; Singh, P.P.; Murad, M.H.; Limburg, P.J. Antidiabetic medications and the risk of colorectal cancer in patients with diabetes mellitus: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hu, C.; Jia, W. Pharmacogenomics of glinides. Pharmacogenomics 2015, 16, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Plana-Ripoll, O.; Puente, D.; Morros, R.; Mundet, X.; Vilca, L.M.; Hernandez, C.; Fuentes, I.; Procupet, A.; Tabernero, J.M.; et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS ONE 2013, 8, e79968. [Google Scholar] [CrossRef]
- El Sharkawi, F.Z.; El Shemy, H.A.; Khaled, H.M. Possible anticancer activity of rosuvastatine, doxazosin, repaglinide and oxcarbazepin. Asian Pac. J. Cancer Prev. 2014, 15, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Omar, B.; Ahren, B. Pleiotropic mechanisms for the glucose-lowering action of DPP-4 inhibitors. Diabetes 2014, 63, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Abrahami, D.; Yin, H.; Yu, O.H.Y.; Pollak, M.N.; Azoulay, L. Incretin-based Drugs and the Incidence of Colorectal Cancer in Patients with Type 2 Diabetes. Epidemiology 2018, 29, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, I.; Sivaswamy, A.; Booth, C. Does the use of incretin-based medications increase the risk of cancer in patients with type-2 diabetes mellitus? Pharmacoepidemiol. Drug Saf. 2019, 28, 489–499. [Google Scholar] [CrossRef]
- Amritha, C.A.; Kumaravelu, P.; Chellathai, D.D. Evaluation of Anti Cancer Effects of DPP-4 Inhibitors in Colon Cancer- An Invitro Study. J. Clin. Diagn. Res. 2015, 9, FC14–FC16. [Google Scholar] [CrossRef]
- Yorifuji, N.; Inoue, T.; Iguchi, M.; Fujiwara, K.; Kakimoto, K.; Nouda, S.; Okada, T.; Kawakami, K.; Abe, Y.; Takeuchi, T.; et al. The dipeptidyl peptidase-4 inhibitor sitagliptin suppresses mouse colon tumorigenesis in type 2 diabetic mice. Oncol. Rep. 2016, 35, 676–682. [Google Scholar] [CrossRef]
- Fujiwara, K.; Inoue, T.; Henmi, Y.; Hirata, Y.; Naka, Y.; Hara, A.; Kakimoto, K.; Nouda, S.; Okada, T.; Kawakami, K.; et al. Sitagliptin, a dipeptidyl peptidase-4 inhibitor, suppresses CXCL5 and SDF-1 and does not accelerate intestinal neoplasia formation in Apc(Min/+) mice fed a high-fat diet. Oncol. Lett. 2017, 14, 4355–4360. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Ferrannini, E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: A systematic review. Diabetologia 2006, 49, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Mulherin, A.J.; Oh, A.H.; Kim, H.; Grieco, A.; Lauffer, L.M.; Brubaker, P.L. Mechanisms underlying metformin-induced secretion of glucagon-like peptide-1 from the intestinal L cell. Endocrinology 2011, 152, 4610–4619. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Hsu, C.C.; Wahlqvist, M.L.; Tsai, H.N.; Chang, Y.H.; Huang, Y.C. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: A representative population prospective cohort study of 800,000 individuals. BMC Cancer 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeon, S.M.; Hong, S.P.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Metformin use is associated with a decreased incidence of colorectal adenomas in diabetic patients with previous colorectal cancer. Dig. Liver Dis. 2012, 44, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef]
- Tseng, C.H. Diabetes, metformin use, and colon cancer: A population-based cohort study in Taiwan. Eur. J. Endocrinol. 2012, 167, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef] [PubMed]
- Kanadiya, M.K.; Gohel, T.D.; Sanaka, M.R.; Thota, P.N.; Shubrook, J.H., Jr. Relationship between type-2 diabetes and use of metformin with risk of colorectal adenoma in an American population receiving colonoscopy. J. Diabetes Complicat. 2013, 27, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, H.; Tan, X.; Chen, L.; Wang, S. Association of metformin use with cancer incidence and mortality: A meta-analysis. Cancer Epidemiol. 2013, 37, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Ko, B.M.; Kim, S.H.; Myung, Y.S.; Choi, J.H.; Han, J.P.; Hong, S.J.; Jeon, S.R.; Kim, H.G.; Kim, J.O.; et al. Does metformin affect the incidence of colonic polyps and adenomas in patients with type 2 diabetes mellitus? Intest. Res. 2014, 12, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Noh, R.; Cho, S.Y.; Park, S.J.; Jeon, S.M.; Shin, H.D.; Kim, S.B.; Shin, J.E. Inhibitory effect of metformin therapy on the incidence of colorectal advanced adenomas in patients with diabetes. Intest. Res. 2015, 13, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Shih, Y.C.; Vekhter, B.; Bissonnette, M.B.; Olopade, O.I.; Polite, B.N. Metformin for primary colorectal cancer prevention in patients with diabetes: A case-control study in a US population. Cancer 2015, 121, 1071–1078. [Google Scholar] [CrossRef]
- He, X.K.; Su, T.T.; Si, J.M.; Sun, L.M. Metformin Is Associated With Slightly Reduced Risk of Colorectal Cancer and Moderate Survival Benefits in Diabetes Mellitus: A Meta-Analysis. Medicine (Baltimore) 2016, 95, e2749. [Google Scholar] [CrossRef]
- Nie, Z.; Zhu, H.; Gu, M. Reduced colorectal cancer incidence in type 2 diabetic patients treated with metformin: A meta-analysis. Pharm. Biol. 2016, 54, 2636–2642. [Google Scholar] [CrossRef]
- Rokkas, T.; Portincasa, P. Colon neoplasia in patients with type 2 diabetes on metformin: A meta-analysis. Eur. J. Intern. Med. 2016, 33, 60–66. [Google Scholar] [CrossRef]
- Rosato, V.; Tavani, A.; Gracia-Lavedan, E.; Guino, E.; Castano-Vinyals, G.; Villanueva, C.M.; Kogevinas, M.; Polesel, J.; Serraino, D.; Pisa, F.E.; et al. Type 2 Diabetes, Antidiabetic Medications, and Colorectal Cancer Risk: Two Case-Control Studies from Italy and Spain. Front. Oncol. 2016, 6, 210. [Google Scholar] [CrossRef]
- Hou, Y.C.; Hu, Q.; Huang, J.; Fang, J.Y.; Xiong, H. Metformin therapy and the risk of colorectal adenoma in patients with type 2 diabetes: A meta-analysis. Oncotarget 2017, 8, 8843–8853. [Google Scholar] [CrossRef]
- Su, T.; Liao, B.; Dong, Y.; Peng, Z.; Zhou, Q.; Li, B.; Peng, S.; Zhang, N. [Effect of metformin on colorectal carcinoma in type 2 diabetes mellitus patients: A Markov model analysis]. Zhonghua Wei Chang Wai Ke Za Zhi 2017, 20, 689–693. [Google Scholar] [CrossRef]
- Tian, S.; Lei, H.B.; Liu, Y.L.; Chen, Y.; Dong, W.G. The association between metformin use and colorectal cancer survival among patients with diabetes mellitus: An updated meta-analysis. Chronic Dis. Transl. Med. 2017, 3, 169–175. [Google Scholar] [CrossRef]
- Tseng, C.H. Metformin is associated with a lower risk of colorectal cancer in Taiwanese patients with type 2 diabetes: A retrospective cohort analysis. Diabetes Metab. 2017, 43, 438–445. [Google Scholar] [CrossRef]
- Zhu, R.C.; Rattanakorn, K.; Pham, S.; Mallam, D.; McIntyre, T.; Salifu, M.O.; Youssef, I.; McFarlane, S.I.; Vignesh, S. Survival benefits in colorectal adenocarcinoma with the use of metformin among a black diabetic inner city population. Colorectal Cancer 2017, 6, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Al Omari, A.; Abdelkhaleq, H.; Al-Hussaini, M.; Turfa, R.; Awad, N.; Hassan, M.M.; Alfaqih, M.A.; Garrett, C.R. Validation of the Survival Benefits of Metformin in Middle Eastern Patients With Type II Diabetes Mellitus and Colorectal Cancer. J. Glob. Oncol. 2018, 4, 1–10. [Google Scholar] [CrossRef]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P., Jr.; Habel, L.A. A Cohort Study of Metformin and Colorectal Cancer Risk among Patients with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2018, 27, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Tsai, H.L.; Kung, Y.T.; Yeh, Y.S.; Huang, C.W.; Ma, C.J.; Chiu, H.C.; Wang, J.Y. Dose-Dependent Relationship Between Metformin and Colorectal Cancer Occurrence Among Patients with Type 2 Diabetes-A Nationwide Cohort Study. Transl. Oncol. 2018, 11, 535–541. [Google Scholar] [CrossRef]
- Smiechowski, B.; Azoulay, L.; Yin, H.; Pollak, M.N.; Suissa, S. The use of metformin and colorectal cancer incidence in patients with type II diabetes mellitus. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1877–1883. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Capothanassi, D.; Allen, N.E.; Rizos, E.C.; Lopez, D.S.; van Veldhoven, K.; Sacerdote, C.; Ashby, D.; Vineis, P.; Tzoulaki, I.; et al. Metformin does not affect cancer risk: A cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care 2014, 37, 2522–2532. [Google Scholar] [CrossRef]
- Kowall, B.; Stang, A.; Rathmann, W.; Kostev, K. No reduced risk of overall, colorectal, lung, breast, and prostate cancer with metformin therapy in diabetic patients: Database analyses from Germany and the UK. Pharmacoepidemiol. Drug Saf. 2015, 24, 865–874. [Google Scholar] [CrossRef]
- Mansourian, M.; Karimi, R.; Vaseghi, G. Different effects of metformin and insulin on primary and secondary chemoprevention of colorectal adenoma in diabetes type 2: Traditional and Bayesian meta-analysis. EXCLI J. 2018, 17, 45–56. [Google Scholar] [CrossRef]
- Vicentini, M.; Ballotari, P.; Giorgi Rossi, P.; Venturelli, F.; Sacchettini, C.; Greci, M.; Mangone, L.; Pezzarossi, A.; Manicardi, V. Effect of different glucose-lowering therapies on cancer incidence in type 2 diabetes: An observational population-based study. Diabetes Res. Clin. Pract. 2018, 143, 398–408. [Google Scholar] [CrossRef]
- Farmer, R.E.; Ford, D.; Mathur, R.; Chaturvedi, N.; Kaplan, R.; Smeeth, L.; Bhaskaran, K. Metformin use and risk of cancer in patients with type 2 diabetes: A cohort study of primary care records using inverse probability weighting of marginal structural models. Int. J. Epidemiol. 2019. [Google Scholar] [CrossRef]
- Fransgaard, T.; Thygesen, L.C.; Gogenur, I. Metformin Increases Overall Survival in Patients with Diabetes Undergoing Surgery for Colorectal Cancer. Ann. Surg. Oncol. 2016, 23, 1569–1575. [Google Scholar] [CrossRef]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res 2013, 19, 6741–6750. [Google Scholar] [CrossRef]
- Jeong, Y.K.; Kim, M.S.; Lee, J.Y.; Kim, E.H.; Ha, H. Metformin Radiosensitizes p53-Deficient Colorectal Cancer Cells through Induction of G2/M Arrest and Inhibition of DNA Repair Proteins. PLoS ONE 2015, 10, e0143596. [Google Scholar] [CrossRef]
- Miranda, V.C.; Braghiroli, M.I.; Faria, L.D.; Bariani, G.; Alex, A.; Bezerra Neto, J.E.; Capareli, F.C.; Sabbaga, J.; Lobo Dos Santos, J.F.; Hoff, P.M.; et al. Phase 2 Trial of Metformin Combined With 5-Fluorouracil in Patients With Refractory Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2016, 15, 321–328.e1. [Google Scholar] [CrossRef]
- Mussin, N.; Oh, S.C.; Lee, K.W.; Park, M.Y.; Seo, S.; Yi, N.J.; Kim, H.; Yoon, K.C.; Ahn, S.W.; Kim, H.S.; et al. Sirolimus and Metformin Synergistically Inhibits Colon Cancer In Vitro and In Vivo. J. Korean Med. Sci. 2017, 32, 1385–1395. [Google Scholar] [CrossRef]
- Higurashi, T.; Hosono, K.; Takahashi, H.; Komiya, Y.; Umezawa, S.; Sakai, E.; Uchiyama, T.; Taniguchi, L.; Hata, Y.; Uchiyama, S.; et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: A multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016, 17, 475–483. [Google Scholar] [CrossRef]
- Thent, Z.C.; Zaidun, N.H.; Azmi, M.F.; Senin, M.I.; Haslan, H.; Salehuddin, R. Is Metformin a Therapeutic Paradigm for Colorectal Cancer: Insight into the Molecular Pathway? Curr. Drug Targets 2017, 18, 734–750. [Google Scholar] [CrossRef]
- Barros, F.W.; Bezerra, D.P.; Ferreira, P.M.; Cavalcanti, B.C.; Silva, T.G.; Pitta, M.G.; de Lima Mdo, C.; Galdino, S.L.; Pitta Ida, R.; Costa-Lotufo, L.V.; et al. Inhibition of DNA topoisomerase I activity and induction of apoptosis by thiazacridine derivatives. Toxicol. Appl. Pharmacol. 2013, 268, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Ban, J.O.; Kwak, D.H.; Oh, J.H.; Park, E.J.; Cho, M.C.; Song, H.S.; Song, M.J.; Han, S.B.; Moon, D.C.; Kang, K.W.; et al. Suppression of NF-kappaB and GSK-3beta is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chem. Biol. Interact. 2010, 188, 75–85. [Google Scholar] [CrossRef]
- Bischoff, H. The mechanism of alpha-glucosidase inhibition in the management of diabetes. Clin. Investig. Med. 1995, 18, 303–311. [Google Scholar]
- Tseng, Y.H.; Tsan, Y.T.; Chan, W.C.; Sheu, W.H.; Chen, P.C. Use of an alpha-Glucosidase Inhibitor and the Risk of Colorectal Cancer in Patients With Diabetes: A Nationwide, Population-Based Cohort Study. Diabetes Care 2015, 38, 2068–2074. [Google Scholar] [CrossRef]
- Ron, Y.; Wainstein, J.; Leibovitz, A.; Monastirsky, N.; Habot, B.; Avni, Y.; Segal, R. The effect of acarbose on the colonic transit time of elderly long-term care patients with type 2 diabetes mellitus. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, M111–M114. [Google Scholar] [CrossRef]
- Citronberg, J.; Kantor, E.D.; Potter, J.D.; White, E. A prospective study of the effect of bowel movement frequency, constipation, and laxative use on colorectal cancer risk. Am. J. Gastroenterol. 2014, 109, 1640–1649. [Google Scholar] [CrossRef]
- Weaver, G.A.; Tangel, C.T.; Krause, J.A.; Parfitt, M.M.; Stragand, J.J.; Jenkins, P.L.; Erb, T.A.; Davidson, R.H.; Alpern, H.D.; Guiney, W.B., Jr.; et al. Biomarkers of human colonic cell growth are influenced differently by a history of colonic neoplasia and the consumption of acarbose. J. Nutr. 2000, 130, 2718–2725. [Google Scholar] [CrossRef]
- Pili, R.; Chang, J.; Partis, R.A.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The alpha-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 1995, 55, 2920–2926. [Google Scholar]
- Horibe, Y.; Adachi, S.; Ohno, T.; Goto, N.; Okuno, M.; Iwama, M.; Yamauchi, O.; Kojima, T.; Saito, K.; Ibuka, T.; et al. Alpha-glucosidase inhibitor use is associated with decreased colorectal neoplasia risk in patients with type 2 diabetes mellitus receiving colonoscopy: A retrospective study. Oncotarget 2017, 8, 97862–97870. [Google Scholar] [CrossRef]
- Imamura, M.; Nakanishi, K.; Suzuki, T.; Ikegai, K.; Shiraki, R.; Ogiyama, T.; Murakami, T.; Kurosaki, E.; Noda, A.; Kobayashi, Y.; et al. Discovery of Ipragliflozin (ASP1941): A novel C-glucoside with benzothiophene structure as a potent and selective sodium glucose co-transporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. 2012, 20, 3263–3279. [Google Scholar] [CrossRef]
- Tahrani, A.A.; Barnett, A.H.; Bailey, C.J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013, 1, 140–151. [Google Scholar] [CrossRef]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Hediger, M.A.; Rhoads, D.B. Molecular physiology of sodium-glucose cotransporters. Physiol. Rev. 1994, 74, 993–1026. [Google Scholar] [CrossRef]
- Clar, C.; Gill, J.A.; Court, R.; Waugh, N. Systematic review of SGLT2 receptor inhibitors in dual or triple therapy in type 2 diabetes. BMJ Open 2012, 2. [Google Scholar] [CrossRef]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13, 11–26. [Google Scholar] [CrossRef]
- Briasoulis, A.; Al Dhaybi, O.; Bakris, G.L. SGLT2 Inhibitors and Mechanisms of Hypertension. Curr. Cardiol. Rep. 2018, 20, 1. [Google Scholar] [CrossRef]

{kind=link}
| References | Study | Country | Sample | Period (Years) | Risk of CRC Among DM Participants (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Males | Females | Overall | |||||
| Nilsen et al., 2001 [2] | Cohort | Norway | 751,922 | 12 | RR 0.66 (0.35–1.34) | RR 1.55 (1.04–2.31) | N.A. |
| Inoue et al., 2006 [4] | Cohort | Japan | 97,771 | 12 | HR 1.36 (1.00–1.85) | HR 0.83 (0.42–1.68) | N.A. |
| Jarvandi et al., 2013 [11] | Cohort | USA | 484,020 | 12 | C: HR 1.24 (1.12–1.38) R: HR 1.34 (1.14–1.57) | C: HR 1.37 (1.16–1.60) R: HR 1.43 (1.08–1.88) | C: HR 1.27 (1.17–1.39) R: HR 1.36 (1.18–1.56) |
| Oberaigner et al., 2014 [13] | Cohort | Austria | 5,709 | 22 | SIR 1.11 (0.81–1.49) | SIR 0.94 (0.62–1.36) | N.A. |
| Wang et al., 2015 [17] | Cohort | China | 327,000 | 7 | C: SIR 1.47 (1.29–1.67) R: SIR 1.25 (1.09–1.43) | C: SIR 1.33 (1.15–1.54) R: SIR 1.29 (1.10–1.51) | C: SIR 1.40 (1.27–1.55) R: SIR 1.26 (1.14–1.40) |
| Harding et al. 2015 [14] | Cohort | Australia | 953,382 | 12 | SIR 1.18 (1.15–1.21) | SIR 1.16 (1.13–1.20) | N.A. |
| Liu et al., 2015 [15] | Cohort | Sweden | 380,196 | 47 | N.A. | N.A. | C: SIR 1.33 (1.28–1.38) R: SIR 1.19 (1.13–1.25) |
| Dankner et al., 2016 [18] | Cohort | Israel | 218,6196 | 11 | SIR 1.45 (1.37–1.55) | SIR 1.48 (1.39–1.57) | N.A. |
| de Kort et al., 2016 [19] | Cohort | Netherlands | 120,852 | 21 | HR 0.95 (0.75–1.20) | HR 1.08 (0.85–1.37) | N.A. |
| Ballotari et al., 2017 [20] | Cohort | Italy | 383,799 | 4 | IRR 1.44 (1.25–1.55) | IRR 1.44 (1.25–2.60) | IRR 1.32 (1.12–1.55) |
| de Jong et al., 2017 [22] | Cohort | Netherlands | 34,038 | 14 | N.A. | N.A. | C: HR 1.40 (1.10–1.70) R: HR 0.88 (0.63–1.20) |
| Chen et al., 2017 [21] | Cohort | Asia | 771,297 | 23 | C: HR 1.57 (1.27–1.93) R: HR 1.26 (0.99–1.61) | C: HR 1.48 (1.20–1.81) R: HR 1.55 (1.12–2.16) | HR 1.41 (1.26–1.57) |
| Saarela et al., 2018 [23] | Cohort | Finland | 428,326 | 27 | C: SIR 1.29 (1.24–1.33) R: SIR 1.16 (1.10–1.21) | C: SIR 1.16 (1.12–1.20) R: SIR 1.13 (1.06–1.19) | C: SIR 1.22 (1.19–1.25) R: SIR 1.14 (1.10–1.18) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, J.; Shirakami, Y.; Shimizu, M. Diabetes Mellitus and Colon Carcinogenesis: Expectation for Inhibition of Colon Carcinogenesis by Oral Hypoglycemic Drugs. Gastrointest. Disord. 2019, 1, 273-289. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1020023
Kato J, Shirakami Y, Shimizu M. Diabetes Mellitus and Colon Carcinogenesis: Expectation for Inhibition of Colon Carcinogenesis by Oral Hypoglycemic Drugs. Gastrointestinal Disorders. 2019; 1(2):273-289. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1020023
Chicago/Turabian StyleKato, Junichi, Yohei Shirakami, and Masahito Shimizu. 2019. "Diabetes Mellitus and Colon Carcinogenesis: Expectation for Inhibition of Colon Carcinogenesis by Oral Hypoglycemic Drugs" Gastrointestinal Disorders 1, no. 2: 273-289. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1020023