
High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts


1
Department of Chemistry and Biochemistry, Fort Lewis College, 1000 Rim Dr., Durango, CO 81301, USA
2
Materials and Molecular Analysis Center, Analytical Resource Core, Colorado State University, 200 W. Lake St., Fort Collins, CO 80523, USA
*
Author to whom correspondence should be addressed.
Reactions 2022, 3(1), 59-69; https://0-doi-org.brum.beds.ac.uk/10.3390/reactions3010005
Submission received: 8 December 2021
/
Revised: 27 December 2021
/
Accepted: 30 December 2021
/
Published: 4 January 2022
(This article belongs to the Special Issue Traditional and Innovative Catalysts for Reactions of Industrial Interest)
Abstract
:Synthetic access to poly(indazolyl)methanes has limited their study despite their structural similarity to the highly investigated chelating poly(pyrazolyl)methanes and their potentially important indazole moiety. Herein is presented a high yielding, one-pot synthesis for the 3d-metal catalyzed formation of bis(1H-indazol-1-yl)methane from 1H-indazole utilizing dimethylsulfoxide as the methylene source. Complete characterization of bis(1H-indazol-1-yl)methane is given with 1H and 13C NMR, UV/Vis, FTIR, high resolution mass spectrometry and for the first time, single crystal X-ray diffraction. This simple, inexpensive pathway to yield exclusively bis(1H-indazol-1-yl)methane provides synthetic access to further investigate the coordination and potential applications of the family of bis(indazolyl)methanes.
1. Introduction
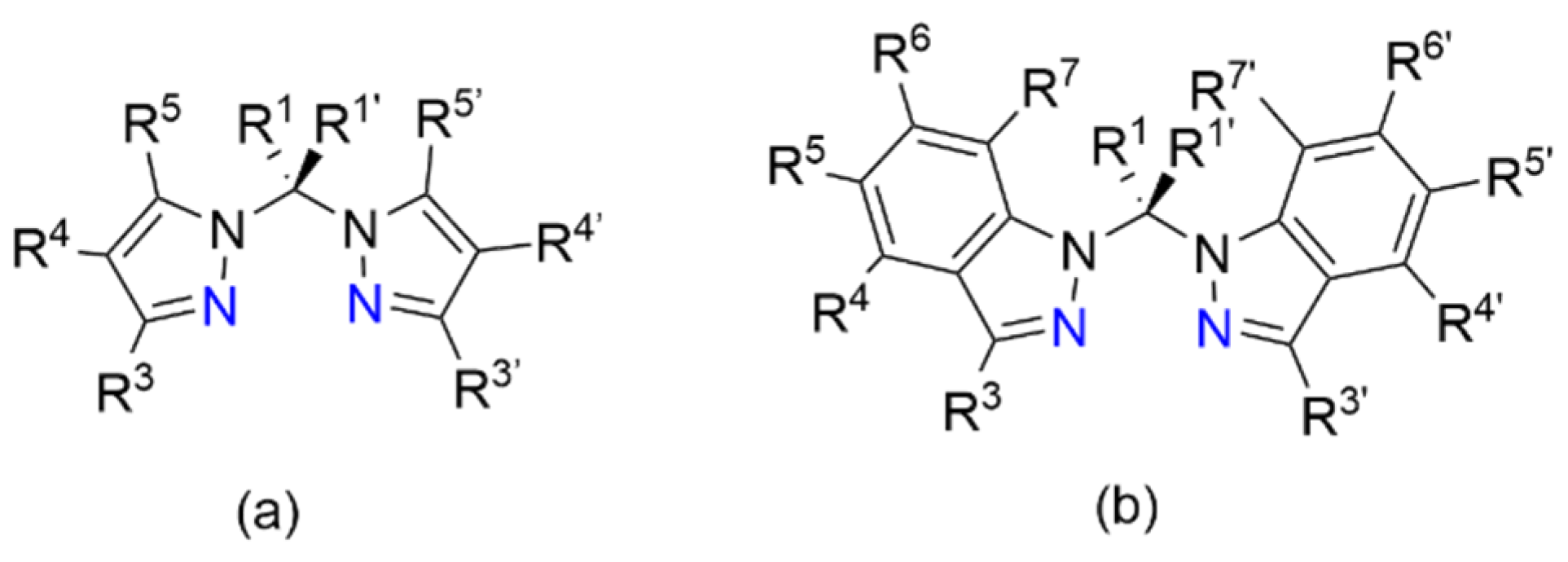
Poly(1H-pyrazol-1-yl)alkanes are a class of ligands that have been investigated due to their overall neutral charge and unique bidentate chelating capabilities through the formation of a six- or seven-membered ring in a boat conformation upon binding to a metal center [1]. The first synthesis of a poly(1H-pyrazol-1-yl)methane was reported by Hückel and Chneider in 1937 [2]. Using the potassium salt of pyrazole and chloroform as the carbon source, tri(1H-pyrazol-1-yl)methane was prepared [2,3]. In 1970, Trofimenko reported on the synthesis and coordination chemistry of a family of methane and/or pyrazole substituted bis(1H-pyrazol-1-yl)methane chelating agents, Figure 1a [3,4]. Synthetically, Trofimenko prepared the substituted pyrazole ligands with methylene bridges by autoclaving the pyrazole and pyrazole salt in the presence of methylene bromide or methylene iodide [4]. A second synthetic method using acetals of aldehydes and ketones was utilized for the preparation of a series of bis(1H-pyrazol-1-yl)methane ligands with substituted carbon linkers [4]. The preparation of poly(1H-pyrazol-1-yl)methanes utilizing multiple synthetic methods has been extensively studied and recently reviewed [3]. Recent applications of bis(1H-pyrazol-1-yl)methanes have highlighted its use in stabilizing chiral coordination complexes [5] and catalysis [6]. Structurally related to the bis(1H-pyrazol-1-yl)methane ligands, but less studied are the family of bis(1H-indazol-1-yl)methane ligands, Figure 1.
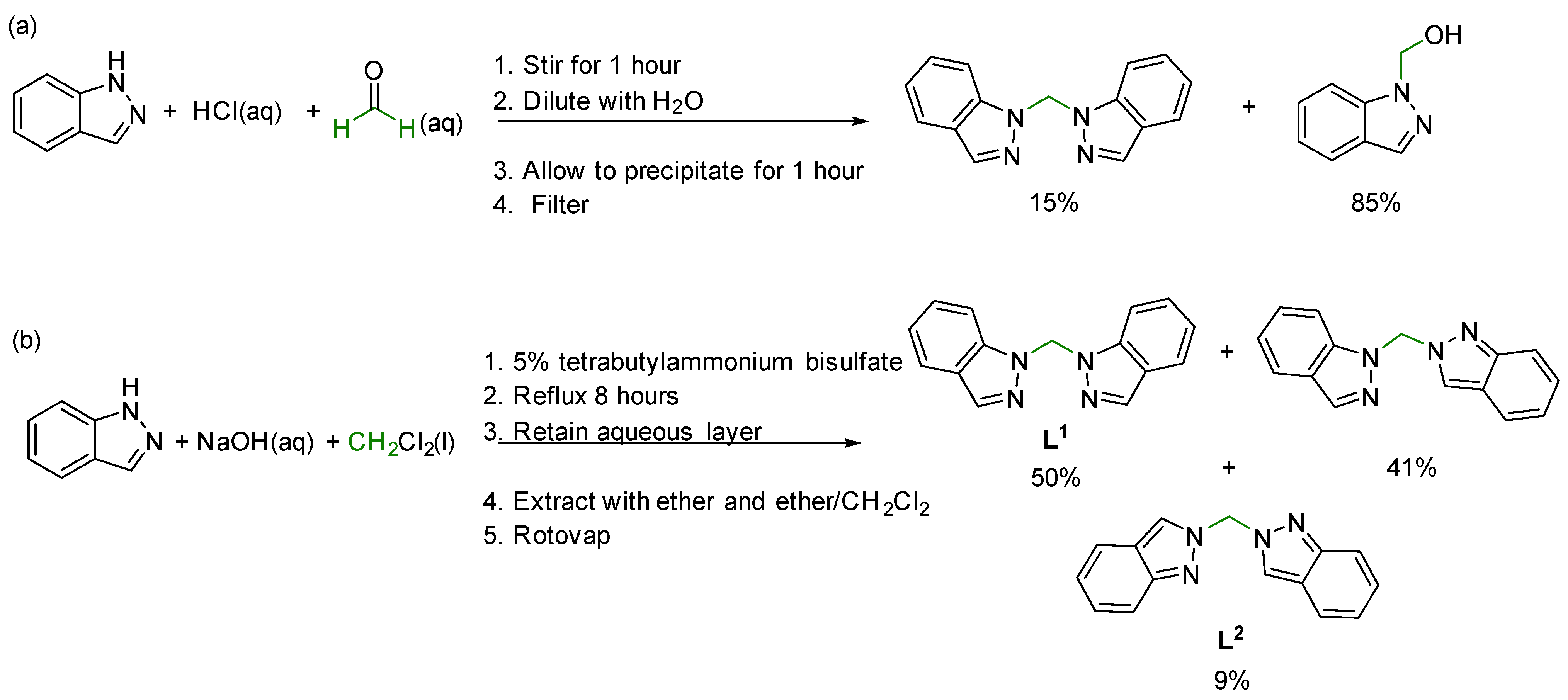
The synthesis of bis(1H-indazol-1-yl)methane was first reported in 1964 as a product mixture with 1-hydroxymethylindazole by combination of 1H-indazole with formalin and hydrochloric acid, Scheme 1a [7]. A mixture of bis(1H-indazol-1-yl)methane (L1), (1H-indazol-1-yl)(2H-indazol-2-yl)methane, and bis(2H-indazol-2-yl)methane (L2) was subsequently reported in 1982 utilizing methylene chloride and phase transfer conditions, Scheme 1b [8]. In 1999, two multi-step synthetic pathways that also required column separation to obtain L1 were reported through the formation of a 2-[(2-acetyoxyethoxy)methyl]indazole intermediate [9].
There was a lapse in the literature for the utilization of bis(1H-indazol-1-yl)methane as a chelating ligand despite the significant indazole moiety that has been highly utilized in the pharmaceutical industry [10,11,12]. In 2010, Pettinari and coworkers utilized the 1982 synthetic method [8] (Scheme 1b) with the first reported separation of L1 and L2 [13]. While the spectroscopic evidence for the separation of L1 and L2 is compelling [13], an extensive search of the literature did not yield any experimental details or separation procedures. Reports with the L1 and L2 chelating ligands including structural and electronic characteristics of more than 50 new coordination complexes has been published since 2010 with binding to group 9, 10, 11, and 12 metal centers [13,14,15]. However, to our knowledge a high yielding, single step synthesis yielding exclusively the L1 ligand has not yet been reported. Herein, we report optimized reaction conditions for a metal catalyzed one-pot synthesis of bis(1H-indazol-1-yl)methane, L1, using dimethylsulfoxide as the methylene source. Characterization of L1 is detailed using 1H and 13C NMR, UV/Vis, FTIR, high resolution mass spectrometry, and single crystal X-ray diffraction.
2. Materials and Methods
All chemicals were used as received without further purification unless otherwise noted. Cobalt(II) chloride hexahydrate (98–102%) and iron(II) chloride (99.5%, anhydrous) were manufactured by Alfa Aesar (Ward Hill, MA, USA). 1H-indazole (98%), iron(II) chloride tetrahydrate (98%), zinc chloride (anhydrous, 99.999%), and nickel(II) chloride hexahydrate (99.9%) were obtained from Sigma-Aldrich (Milwaukee, WI, USA). Dimethyl sulfoxide (DMSO), diethyl ether, methylene chloride, methanol, and acetonitrile were ACS grade and purchased from Fisher Scientific (Waltham, MA, USA).
The 1H and 13C NMR spectra were collected using a Bruker Ascend spectrometer (Fällanden, Switzerland) operating at 400 and 100 MHz, respectively. DMSO-d6 (Acros; 99.9% D) was dried over activated molecular sieves and used for all NMR spectra collected. The residual solvent peak was used as the proton and carbon-13 reference. Mass spectra were collected in acetonitrile (anhydrous, 99.8%; Fisher Scientific) on a Thermo-Finnigan LTQ mass spectrometer (San Jose, CA, USA) with ions generated using an electrospray ionization (ESI) source and positive ion mode. UV-Vis solutions were prepared with 1 mM of L1 in methanol and collected on an Agilent Cary 60 UV-Vis spectrophotometer (Santa Clara, CA USA). FTIR of solid-state samples were collected at room temperature from 600–4000 cm−1 on a Thermo Scientific Nicolet iS10 with Smart iTR (Madison, WI USA).
2.1. Synthesis and Characterization of Bis(1H-indazol-1-yl)methane, L1
2.1.1. General Procedure for the Formation of L1
In a heavy wall cylindrical pressure vessel (15 mL; 150 psig rating, Ace Glass), 1.0 mmol of 1H-indazole, 0.01 mmol of metal catalyst, and 1.0 mL of DMSO were combined with a stir bar and sealed with a Teflon screw cap. The loaded reaction vessel was placed in an oil bath at 175 °C and stirred for 24 h. After cooling to room temperature, the crude product was precipitated with 20 mL of deionized water, extracted twice into 50 mL of ethyl acetate, and washed with 10 mL of brine. The organic solvent was removed under reduced pressure and the sample was dried under high vacuum overnight to produce the desired product. Further purification of the product can be accomplished using recrystallization in 50:50 methylene chloride and methanol resulting in a flaky off-white solid product.
2.1.2. Scale-Up Reaction Conditions
A macroscale synthesis of the ligand L1 was accomplished using 0.1 mol of 1H-indazole, 1 mmol of CoCl2·6H2O, and 10 mL of DMSO in a 50 mL round-bottom flask with a water cooled-condenser. A reflux temperature of 175 °C with increased reaction time was needed for complete conversion of 1H-indazole to L1 which can be monitored by TLC using 50:50 ethyl acetate and n-hexanes. The precipitation and workup of the product were consistent with the general procedure using a 10X scaling factor.
2.1.3. Characterization of L1
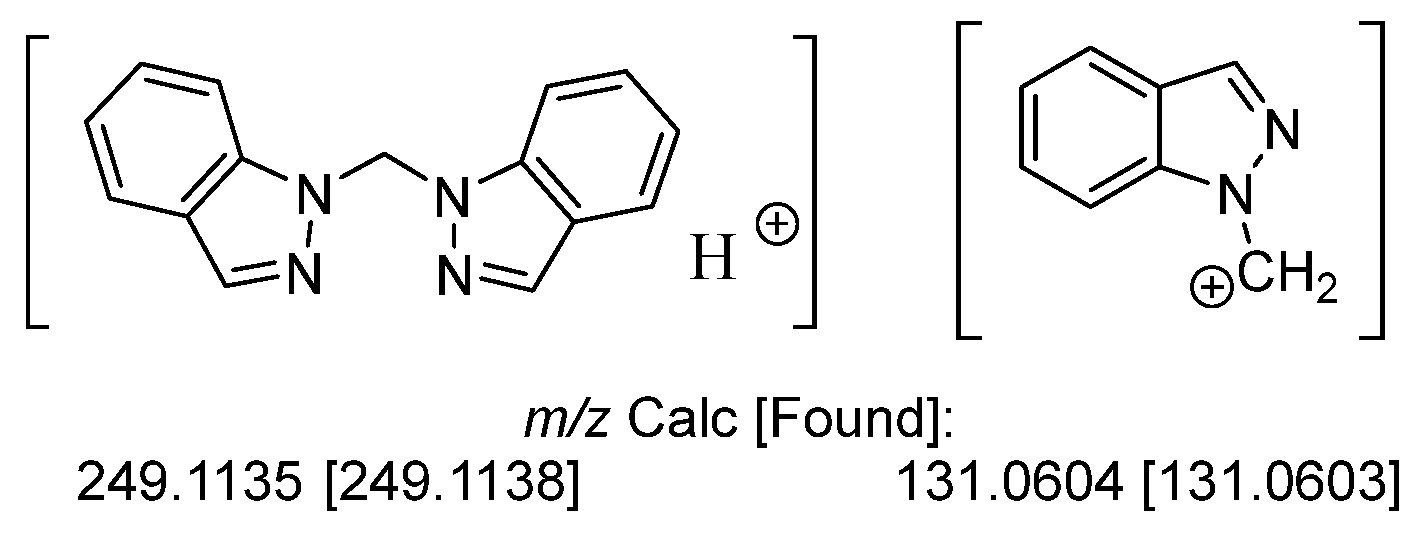
Formation of L1 was monitored by 1H NMR in DMSO-d6 for all reactions screened. 1H NMR (400 MHz, DMSO-d6, 295 K) δ: 8.12 (d, 1H, H3/3′, J = 0.9 Hz), 7.96 (dd, 1H, H7/7′, J = 8.5 and 0.9 Hz), 7.74 (dt, 1H, H4/4′, J = 8.1 and 1.0 Hz), 7.46 (ddd, 1H, H6/6′, J = 7.7, 1.3, and 1.1 Hz), 7.17 (ddd, 1H, H5/5′, J = 7.5, 1.0, and 0.8 Hz), 7.10 (s, 1H, CH2), see Figure S1 of the Supporting Information. Further characterization was undertaken to verify the assigned structure to L1. 13C{1H} NMR (100 MHz, DMSO-d6, 295 K) δ: 139.7 (C7a), 134.9 (C3), 127.2 (C6), 124.5 (C3a), 121.7 (C5), 121.4 (C4), 110.8 (C7), and 60.6 (CH2); see Figure S2 of the Supporting Information. HRMS (ESI): m/z 249.1138 [L1 + H]+ and 131.0603 [C7H5N2 + CH2]+. UV/Vis λmax (log ε): 251 (3.54), 260 (3.51), 288 (3.52), 293 (3.52), 299 (3.50). IR (cm−1): 3106 (w), 3060 (m), 2957 (w), 2921 (w), 1617 (m), 1499 (m), 1463 (m), 1439 (w), 1353 (m), 1279 (m), 1197 (s), 1150 (w), 1005 (m), 906 (m), 828 (m), 758 (s), 736 (s).
2.2. X-ray Crystallography
2.2.1. Experimental Conditions for Single Crystals Growth
Reaction conditions consisting of 3.1 mmol of 1H-indazole and 0.31 mmol of trans-dichlorotetrakis(pyridine)cobalt(III) chloride [16] were dissolved in 2.6 mL of DMSO. After refluxing at 175 °C for 24 h, the reaction mixture was poured into a beaker and allowed to concentrate at room temperature. After one week, long needle-like crystals were observed in a dark green mother liquor. The crude product was filtered and washed with DMSO and diethyl ether. After overnight vacuum drying, single crystals suitable for X-ray diffraction were obtained from recrystallization in methylene chloride.
2.2.2. X-ray Crystallography Data Collection
The structure was determined for the compound listed in Table 1. Single crystals were coated with Paratone-N oil and mounted under a cold stream of dinitrogen gas. Single crystal X-ray diffraction data were acquired on a Bruker Kappa APEXII diffractometer (Karlsruhe, Germany) equipped with a CCD detector and a graphite monochromator using Mo Kα radiation (λ = 0.71073 Å). Initial lattice parameters were obtained from a least-squares analysis of more than 100 reflections; these parameters were later refined against all data. The crystals did not show significant decay during data collection. Data were integrated and corrected for Lorentz and polarization effects using Bruker APEX4 software [17], and semiempirical absorption corrections were applied using SCALE [18]. Space group assignments were based on systematic absences, E statistics, and successful refinement of the structures. Structures were solved using Direct Methods and were refined with the aid of successive Fourier difference maps against all data using the SHELXT 6.14 software package [19]. Thermal parameters for all non-hydrogen atoms were refined anisotropically. All hydrogen atoms were assigned to ideal positions and refined using a riding model with an isotropic thermal parameter 1.2 times that of the attached carbon atom (1.5 times for methyl hydrogens). All other metric parameters can be found in deposited crystal structure file.
3. Results
3.1. One-Pot Synthesis of Bis(1H-indazol-1-yl)methane, L1
The generalized synthetic pathway for the optimized high yielding, one-pot synthesis forming exclusively L1 from 1H-indazole and dimethylsulfoxide using either a commercially available 3d-metal salt catalyst or a synthetic Co(III) coordination complex is summarized in Scheme 2. Following the 24 h heating at 175 °C in a pressure tube, the desired product L1 was precipitated from the DMSO reaction solution with deionized water. L1 was then extracted into ethyl acetate and washed with brine. Collection of the organic layer allowed for concentration by evaporation followed by overnight drying on a high vacuum line to yield the desired, pure L1 product which was verified in each reaction by 1H NMR.
Milder reaction conditions were tested using several other potential methylene source solvents near their boiling points in the presence of 10% CoCl2·6H2O including dimethylformamide, toluene, dibromomethane, acetonitrile, ethanol and dichloromethane. However, no product formation was observed from any solvent other than DMSO and only the starting material 1H-indazole was detected at the end of the 24 h reaction. Attempts at addition one or two molar equivalents of DMSO in toluene were also unsuccessful in forming L1. For a full listing of solvents and reaction details of conditions tested, see Table S4 of the Supplemental Information.
Time optimization studies were also undertaken to find the minimum reaction time needed to fully convert 1H-indazole to the desired L1 in DMSO. Using a 1% catalyst loading of CoCl2·6H2O, reactions under our standard conditions were stopped and worked up at 8, 12, 16, 20, 24, and 48 h. The reactions at 16 h and earlier all showed no conversion to the desired L1 and only 1H-indazole was detected by 1H NMR. At 20 h, a mixture of 1H-indazole and L1 were present. Both the 24 and 48 h reactions showed complete conversion to the desired dimer product in similar yields giving the optimized reaction time of 24 h.
Moving forward with the DMSO solvent/methylene source and optimized reaction time, control reactions in the absence of a metal salt showed that dimer formation only partially occurs in the 24 h reaction window but a significant amount of 1H-indazole still remained, entry 1 of Table 2. Increasing the control reaction time does result in increased conversion of 1H-indazole into L1 along with multiple other products detected by 1H NMR that could not be easily separated (entry 2). Further reaction optimization revealed that a 1% catalyst loading of commercially available CoCl2, FeCl2, NiCl2, or ZnCl2 all resulted in high yields of the desired product L1 (entries 5, 7, 9–12) in 24 h. Regardless of the metal salt used, all the reactions took a minimum of 24 h for full conversion to L1. Hydrated as well as anhydrous forms of the cobalt(II) and iron(II) chloride salts both produced very similar results with the anhydrous forms giving slightly lower yields (entries 5 and 7; 9 and 10). Trans-[Co(pyridine)4Cl2]Cl was synthesized from the previous literature [16] and tested as a Co(III) catalyst (entry 8) which showed similar yields and product formation to the commercially available cobalt(II) chloride.
A macroscale reaction for the production of L1 was also tested for future applications. A high yielding gram-scale reaction was found with slight modification using a reflux setup to accommodate the increased amounts of starting materials and an increased reaction time which can be monitored by TLC for the complete conversion of 1H-indazole to L1. The scale-up tested had a consistently high yield of pure L1 product using a 1% CoCl2·6H2O catalyst providing evidence of scalability for this one-pot synthesis.
3.2. Characterization of Bis(1H-indazol-1-yl)methane, L1
1H and 13C NMR spectra have been previously reported for L1 [8,9,13] and were consistent with the chemical shifts and splitting patterns allowing for assignment of the methylene bridged dimer structure. A comparison table of the NMR data collected herein and previously reported in the literature is shown in Table S1 of the Supporting Information. UV/Vis data for L1 have also been previously determined [9] and were used to support the assigned structure of L1 as bis(1H-indazol-1-yl)methane. Key IR vibrations corresponding to the ν(C=C) and ν(C=N) at 1617 and 1499 cm−1 were observed for the L1 reported herein and are consistent with those previously reported [13]. Further support for the structure of L1 was obtained through high resolution ESI mass spectrometry which showed the expected parent ion as well as a monomeric fragment, Figure 2.
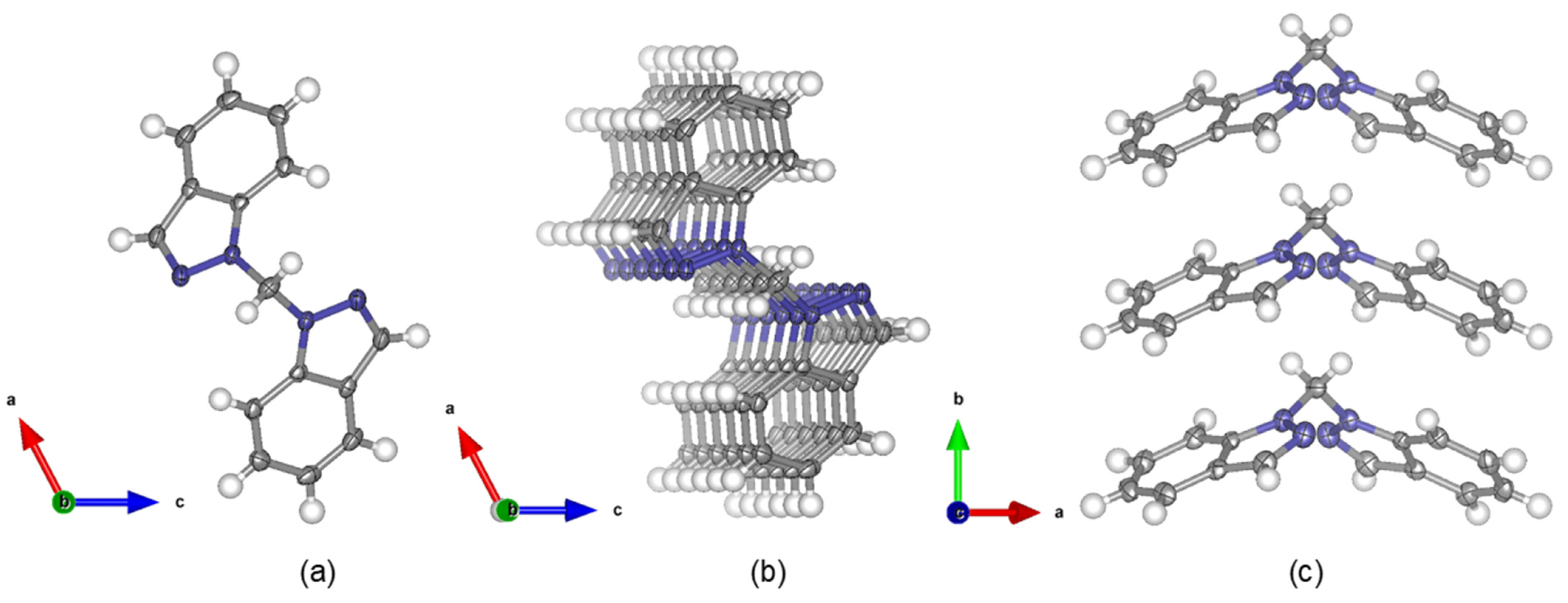
Single crystal X-ray structures with L1 bound to metal centers have been reported [12,13,14], but a single crystal structure of L1 could not be found in any crystallographic databases. Our structure determination revealed that L1 crystallizes in the monoclinic space group C2. No significant bond distances or bond angles within each indazole moiety of L1 differ significantly from the recently reported structure of 1H-indazole [20]. A 50% probability thermal ellipsoid plot of the single crystal structure of L1 is presented in Figure 3a with a N1-CH2-N1′ bond angle of 113.5(3)°. Extended pi-stacking in the crystallized structure can be viewed in Figure 3b,c with 4.0856 Å distances between the benzene rings.
4. Discussion
4.1. One-Pot Synthesis of Bis(1H-indazol-1-yl)methane, L1
The optimized one-pot reaction conditions presented herein provide a high yielding, simple synthetic pathway for the formation of the single product L1 that can be utilized in chelating ligand applications. The dimerization and addition of a methylene group occur during the 24 h high temperature reaction where a commercially available metal(II) chloride salt or a Co(III) coordination complex catalyze the transformation of 1H-indazole. The use of DMSO as a methylene source in organic reactions has been recently reported [21,22,23,24,25,26,27], and this one-pot synthesis provides another example. Interestingly, no other solvent tested near its boiling point (DMF, toluene, CH2Br2, CH3CN, CH3CH2OH, CH2Cl2) in the presence of the CoCl2·6H2O catalyst was able to act as a methylene source and convert 1H-indazole to the desired L1 product. This suggests that the 175 °C temperature and/or high molar excess of DMSO along with a metal catalyst was necessary to remove protons from N1 and N1′, insert a CH2 group and dimerize 1H-indazole. Using the high temperature conditions over 24 h suggests that a thermodynamic product of exclusively L1 is formed and not the mixture of products that have been previously observed [7,8], Scheme 1. This observation is also consistent with the significantly more stable 1H- vs. 2H-indazole tautomer [28,29]. This inexpensive one-pot synthesis provides access to the L1 ligand for future development in chelating applications and potentially in the development of new medicinal compounds.
4.2. Crystal Structure of Bis(1H-indazol-1-yl)methane, L1
There are significant structural similarities between the known structure of the related bis(1H-pyrazol-1-yl)methane [30] and L1 as highlighted with selected bond lengths and angles in Figure 4. Full tables of bond lengths and angles are given in Tables S2 and S3 of the Supporting Information. Some differences between these ligands are of note. While the bond lengths between N1/N1′ and the methylene group are within experimental error between the two ligands, there is a 0.8° increase in bond angle between the N1—CH2—N1′ in L1 that is likely the result of increased steric hinderance and differing electronic properties. The indazole moiety causes extended pi-stacking networks in L1 as viewed in Figure 3b,c that are not present in bis(1H-pyrazol-1-yl)methane. In comparison to bis(1H-pyrazol-1-yl)methane, L1 also shows longer bond lengths throughout its pyrazole moiety except for N2/2′—C3/3′ where the bond lengths are compressed in L1. This trend within the pyrazole moiety may be explained by Clar’s aromatic π-sextet rule [31] and is consistent with the bond lengths observed in the crystal structures of 1H-pyrazole [32] and 1H-indazole [20].
Comparing the crystal structure of L1 to published structures of L1 coordinated to group 9, 10, 11 and 12 metal centers [13,14,15] reveals that significant rotation in solution around the C—N1/1′ bond is necessary to create bidentate bonding to metal centers through N2 and N2′. This could explain why L1 has been observed to act as a bridging ligand in bi- or poly-metallic Ag(I) coordination complexes [14]. It has also been observed that the decreased basicity [33] and steric differences of L1 in comparison to bis(1H-pyrazol-1-yl)methane allow for different stoichiometric coordination complexes to be formed [13] opening up new libraries of compounds and potentially new applications. Finally, the addition of the aromatic benzene ring to the pyrazole moiety in L1 shows that coordination complexes with L1 can also have extended pi-stacking networks [15] as was observed with the ligand alone. These steric and electronic differences for L1 could provide insights in future synthetic designs and application studies.
4.3. Mechanistic Studies and Future Applications of L1
The optimized reaction conditions provide some preliminary mechanistic evidence for the formation of L1 from 1H-indazole. The use of DMSO as the methylene source in similar reactions has been observed [21,22,23,24,25,26,27]. In some cases, the DMSO acts both as the methylene source and an oxidant in the presence of metals [24] and in other cases metal free catalysis is observed with DMSO and a strong oxidant [27]. In order to test if a strong oxidant might be involved in the conversion of 1H-indazole to L1 in DMSO, hydrogen peroxide was added in the absence of a metal catalyst (entry 3 of Table 2). However, there was no formation of the desired L1 under these reaction conditions. Even in the presence of a metal catalyst with H2O2, still no product formation was observed, entry 6 of Table 2. Our results indicate that a metal catalyst with DMSO at 175 °C was necessary to produce high yields of the desired L1. Time optimization studies also provide insight into the mechanism where a long induction period (16 < t < 20 h) was observed with only 1H-indazole being detected with the CoCl2 catalyst in DMSO at 16 h before production of the desired methylene dimer started to be detected at 20 h. A variety of metal catalysts in hydrated or anhydrous forms, in +2- or +3-oxidation states, and metals with more or less accessible redox states were all able to successfully complete the insertion of the methylene group and dimerization suggesting that the metal catalyst might act as a Lewis acid. Further support is given by the minimum reaction time for the formation of L1 being 24 h regardless of the 3d metal salt used. This finding of the metal salt acting as a Lewis acid is consistent with a previously postulated mechanism for the formation of methylenebisamides using DMSO as both a proposed methylene source and an oxidant in the presence of a NiCl2 catalyst for dimerization [24]. Further studies are needed to determine the mechanism of formation for L1 as well as related product formations considering the growth of literature utilizing DMSO as a methylene source in the presence of trace metal catalysts [23,24,26]. Further insights into the mechanism would continue to allow for reaction optimization for creating a family of substituted bis(1H-indazol-1-yl)methane complexes and exploring their potential applications.
5. Conclusions
This article presents on a simple, high yielding one-pot synthesis producing exclusively bis(1H-indazol-1-yl)methane (L1) from 1H-indazole and DMSO catalyzed by 1% of any of the following metal salts: CoCl2, FeCl2, NiCl2, ZnCl2, or trans-[Co(pyridine)4Cl2]Cl. Complete characterization of L1 is presented including 1H and 13C NMR, UV/Vis, FTIR, high resolution mass spectrometry, and for the first time single crystal X-ray diffraction. Creating synthetic access and diffraction data will allow for expansion of our knowledge on the bis(1H-indazol-1-yl)methane family of ligands to be utilized in future synthetic, mechanistic, chelating and pharmaceutical applications.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/reactions3010005/s1, Table S1: 1H and 13C NMR data collected in this study for L1 and compared to the previous literature. All measurements in DMSO-d6 at 100 and 400 MHz for 13C and 1H, respectively, unless otherwise noted., Figure S1: 1H NMR of bis(1H-indazol-1-yl)methane (L1) in DMSO-d6, Figure S2: 13C NMR of bis(1H-indazol-1-yl)methane (L1) in DMSO-d6, Table S2: Crystallographic Table of Bond Lengths for L1, Table S3: Crystallographic Table of Bond Angles for L1. Table S4: Testing solvents for conversion of 1H-indazole to bis(indazol-1-yl)methane using 10% CoCl2·6H2O and 24 h near the boiling point of each solvent as listed. Structural data for L1 were deposited in CCDC under the deposit number 2127100 and can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 7 December 2021).
Author Contributions
Conceptualization, A.M.M.; methodology, N.M.L., N.S.J. and A.M.M.; formal analysis, B.S.N. and A.M.M.; investigation, N.M.L., N.S.J. and A.M.M.; data curation, N.M.L., N.S.J., B.S.N. and A.M.M.; writing—original draft preparation, A.M.M.; writing—review and editing, N.M.L., N.S.J., B.S.N. and A.M.M.; visualization, B.S.N. and A.M.M.; supervision, A.M.M.; project administration, A.M.M.; funding acquisition, N.S.J. and A.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institute of Health, grant number 5T34GM092711-04 as well as supported by the NIH Common Fund, through the Office of Strategic Coordination, Office of the NIH Director with the linked awards: TL4GM118992, RL5GM118990, & UL1GM118991.
Data Availability Statement
The data presented in this study are available in the main text and Supplementary Materials.
Acknowledgments
The authors gratefully acknowledge Donald Dick at Colorado State University for the collection of the high resolution mass spectrometry data.
Conflicts of Interest
The authors declare no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Pettinari, C.; Pettinari, R. Metal derivatives of poly(pyrazolyl)alkanes☆II. Bis(pyrazolyl)alkanes and related systems. Coord. Chem. Rev. 2004, 249, 663–691. [Google Scholar] [CrossRef]
- Hückel, W.; Chneider, H.B. N-Tripyrazolyl-methan. Berichte Dtsch. Chem. Ges. A B Ser. 1937, 70, 2024–2026. [Google Scholar] [CrossRef]
- Alkorta, I.; Claramunt, R.M.; Díez-Barra, E.; Elguero, J.; de la Hoz, A.; López, C. The organic chemistry of poly(1H-pyrazol-1-yl)methanes. Coord. Chem. Rev. 2017, 339, 153–182. [Google Scholar] [CrossRef]
- Trofimenko, S. Geminal poly(1-pyrazolyl)alkanes and their coordination chemistry. J. Am. Chem. Soc. 1970, 92, 5118–5126. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Tejeda, J.; Sánchez-Barba, L.F. Recent Advances in the Design and Coordination Chemistry of Heteroscorpionate Ligands Bearing Stereogenic Centres. Eur. J. Inorg. Chem. 2008, 2008, 5309–5326. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Sánchez, A.L.; Sánchez-Barba, L.F. Metal complexes with heteroscorpionate ligands based on the bis(pyrazol-1-yl)methane moiety: Catalytic chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Pozharskii, F.T.; Kazanbieva, M.A.; Tertov, B.A. 1-Hydroxymethyl Derivates of Indazole. Zhurnal Obs. Khimii. 1964, 34, 3367–3370. [Google Scholar]
- Juliá, S.; Sala, P.; Del Mazo, J.; Sancho, M.; Ochoa, C.; Elguero, J.; Fayet, J.-P.; Vertut, M.-C. N-Polyazolylmethanes. 1. Synthesis and NMR Study of N,N′-Diazolylmethanes. J. Heterocycl. Chem. 1982, 19, 1141–1145. [Google Scholar] [CrossRef]
- Boryski, J. ChemInform Abstract: Acyclonucleosides of Indazole and Their Rearrangements. ChemInform 2010, 30, 1019–1027. [Google Scholar] [CrossRef]
- Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, W. Product Class 2: 1H- and 2H-Indazoles. In Science of Synthesis: Houben-Weyl Methods for Molecular Transformations; Neier, R., Ed.; Georg Thieme Verlag: New York, NY, USA, 2002; pp. 227–324. [Google Scholar]
- Pettinari, C.; Marinelli, A.; Marchetti, F.; Ngoune, J.; Galindo, A.; Álvarez, E.; Gómez, M. Synthesis and Coordination Chemistry of Two N2-Donor Chelating Di(indazolyl)methane Ligands: Structural Characterization and Comparison of Their Metal Chelation Aptitudes. Inorg. Chem. 2010, 49, 10543–10556. [Google Scholar] [CrossRef] [PubMed]
- Pettinari, C.; Marchetti, F.; Orbisaglia, S.; Pettinari, R.; Ngoune, J.; Gómez, M.; Santos, C.; Álvarez, E. Group 11 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane) ligands. CrystEngComm 2013, 15, 3892–3907. [Google Scholar] [CrossRef]
- Santos, C.; Gómez, M.; Álvarez, E.; Ngoune, J.; Marchetti, F.; Pettinari, R.; Pettinari, C. Group 9 and 10 complexes with the bidentate di(1H-indazol-1-yl)methane and di(2H-indazol-2-yl)methane ligands: Synthesis and structural characterization. New J. Chem. 2016, 40, 5695–5703. [Google Scholar] [CrossRef]
- Hart, K.F.; Joe, N.S.; Miller, R.M.; Nash, H.P.; Blake, D.J.; Morris, A.M. Synthesis and Characterization of trans-Dichlorotetrakis(imidazole)cobalt(III) Chloride: A New Cobalt(III) Coordination Complex with Potential Prodrug Properties. Bioinorg. Chem. Appl. 2018, 2018, 4560757. [Google Scholar] [CrossRef]
- Bruker. APEX4; Bruker AXS: Madison, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. SADABS—A Program for Area Detector Absorption Correction; University of Gottingen: Gottingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL, version 6.14; Bruker AXS: Madison, WI, USA, 1999. [Google Scholar]
- González, J.J.L.; Ureña, F.P.; Moreno, J.R.A.; Mata, I.; Molins, E.; Claramunt, R.M.; López, C.; Alkorta, I.; Elguero, J. The chiral structure of 1H-indazoles in the solid state: A crystallographic, vibrational circular dichroism and computational study. New J. Chem. 2012, 36, 749–758. [Google Scholar] [CrossRef]
- Mohammadi, B.; Khorrami, B.R. A simple and one-pot multi-component reaction to the synthesis of methylenebisamides. Mon. Für Chem.-Chem. Mon. 2017, 149, 1089–1092. [Google Scholar] [CrossRef]
- Patel, O.P.S.; Anand, D.; Maurya, R.K.; Yadav, P.P. H2O2/DMSO-Promoted Regioselective Synthesis of 3,3′-Bisimidazopyridinylmethanes via Intermolecular Oxidative C(sp2)–H Bond Activation of Imidazoheterocycles. J. Org. Chem. 2016, 81, 7626–7634. [Google Scholar] [CrossRef]
- Ai, H.-J.; Qi, X.; Peng, J.-B.; Ying, J.; Wu, X.-F. Palladium-Catalyzed Cross-Coupling of Arylboronic Acid and Benzonitriles Using DMSO as the Methylene Source. Asian J. Org. Chem. 2018, 7, 2045–2048. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Tavakoli, G.; Saeednia, B. Ni-Catalyzed Synthesis of Methylenebisamides: Dual Role of DMSO Both as Methylene Source and Oxidant. ChemistrySelect 2017, 2, 1316–1322. [Google Scholar] [CrossRef]
- Zheng, K.; Zhuang, S.; Shu, W.; Wu, Y.; Yang, C.; Wu, A. Molecular iodine-mediated formal [2+1+1+1] cycloaddition access to pyrrolo[2,1-a]isoquinolines with DMSO as the methylene source. Chem. Commun. 2018, 54, 11897–11900. [Google Scholar] [CrossRef]
- Tashrifi, Z.; Khanaposhtani, M.M.; Larijani, B.; Mahdavi, M. Dimethyl Sulfoxide: Yesterday’s Solvent, Today’s Reagent. Adv. Synth. Catal. 2020, 362, 65–86. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Shen, Z.; Yuan, Y.; Sun, P. Synthesis of symmetrical methylene-bridged imidazoheterocycles using DMSO as methylene source under metal-free conditions. Org. Biomol. Chem. 2016, 14, 6523–6530. [Google Scholar] [CrossRef]
- Catalan, J.; de Paz, J.; Elguero, L.G. Importance of aromaticity on the relative stabilities of indazole annular tautomers: An ab initio study. J. Chem. Soc. Perkin Trans. 1996, 2, 57–60. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Theoretical estimation of the annular tautomerism of indazoles. J. Phys. Org. Chem. 2005, 18, 719–724. [Google Scholar] [CrossRef]
- Churchill, M.R.; Churchill, D.G.; Huynh, M.H.V.; Takeuchi, K.J.; Castellano, R.K.; Jameson, D.L. Crystal and molecular structure of di(1-pyrazolyl)methane, CH2(C3N2H3)2. J. Chem. Crystallogr. 1996, 26, 93–97. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Larsen, F.K.; Lehmann, M.S.; Søtofte, I.; Rasmussen, S.E.; Shimizu, A. A Neutron Diffraction Study of the Crystal and Molecular Structure of Pyrazole, C3H4N2. Acta Chem. Scand. 1970, 24, 3248–3258. [Google Scholar] [CrossRef] [Green Version]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
Figure 1.
Substitution positions and structural similarities for neutral (a) bis(1H-pyrazol-1-yl)methane and (b) bis(1H-indazol-1-yl)methane chelating ligands shown arranged for bidentate binding through the two nitrogen atoms colored in blue. Upon bidentate binding to a metal center, these ligands form six-membered rings with boat conformations.
Figure 1.
Substitution positions and structural similarities for neutral (a) bis(1H-pyrazol-1-yl)methane and (b) bis(1H-indazol-1-yl)methane chelating ligands shown arranged for bidentate binding through the two nitrogen atoms colored in blue. Upon bidentate binding to a metal center, these ligands form six-membered rings with boat conformations.

Scheme 1.
Previously reported one-step synthetic pathways for the formation of bis(1H-indazole)methane from 1H-indazole with relative amounts of multiple products obtained [7,8]. (a) The first synthesis reported in 1964 used formalin, hydrochloric acid and 1H-indazole [7] and (b) a subsequent synthesis reported in 1982 used methylene chloride, sodium hydroxide and 1H-indazole [8]. Both previous syntheses result in a mixture of products.
Scheme 1.
Previously reported one-step synthetic pathways for the formation of bis(1H-indazole)methane from 1H-indazole with relative amounts of multiple products obtained [7,8]. (a) The first synthesis reported in 1964 used formalin, hydrochloric acid and 1H-indazole [7] and (b) a subsequent synthesis reported in 1982 used methylene chloride, sodium hydroxide and 1H-indazole [8]. Both previous syntheses result in a mixture of products.

Scheme 2.
One-pot synthetic pathway and workup for the formation of L1.

Figure 2.
High resolution ESI mass spectrometry parent and fragment ions observed for L1.

Figure 3.
Crystal structure of bis(1H-indazol-1-yl)methane, L1 with thermal ellipsoids displayed at 50% probability (a) viewed down the b-axis. Extended packing viewed (b) with a 5° rotation about the crystallographic b-axis and (c) down the crystallographic c-axis.
Figure 3.
Crystal structure of bis(1H-indazol-1-yl)methane, L1 with thermal ellipsoids displayed at 50% probability (a) viewed down the b-axis. Extended packing viewed (b) with a 5° rotation about the crystallographic b-axis and (c) down the crystallographic c-axis.

Figure 4.
Select bond lengths and angles of (a) bis(1H-pyrazol-1-yl)methane [30] and (b) L1.
Figure 4.
Select bond lengths and angles of (a) bis(1H-pyrazol-1-yl)methane [30] and (b) L1.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal data and details of refinement for L1.
| Parameters | L1 |
|---|---|
| Formula | C15H12N4 |
| Formula Weight (g/mol) | 248.29 |
| Temperature | 100(2) K |
| Crystal system | Monoclinic |
| space group, Z | C2, 4 |
| a (Å) | 23.6129(19) |
| b (Å) | 4.0856(4) |
| c (Å) | 13.9334(12) |
| α (°) | 90 |
| β (°) | 117.743(5) |
| γ (°) | 90 |
| Volume (Å3) | 1189.67(19) |
| (g/cm3) | 1.3806 |
| R1a | 0.0863 |
| wR2 a | 0.0909 |
| GOF | 1.037 |
a Definitions of R1 and wR2. R1 = Σ ||Fo| − |Fc||/Σ |Fo|; wR2 = {[w(Fo2 − Fc2)2]/[w(Fo2)2]}1/2.
Table 2.
Reaction screening conditions for the formation of L1 from 1H-indazole and DMSO as the methylene source. All reactions were run at 175 °C in high pressure reaction test tubes.
Table 2.
Reaction screening conditions for the formation of L1 from 1H-indazole and DMSO as the methylene source. All reactions were run at 175 °C in high pressure reaction test tubes.
| Entry | Time (h) | Metal Catalyst | Additive | Dimer Formation? | Isolated Yield a |
|---|---|---|---|---|---|
| 1 | 24 | - | - | Partial | - |
| 2 | 72 | - | - | Yes b | - b |
| 3 | 24 | - | H2O2 | No | - |
| 4 | 24 | 10% CoCl2·6H2O | - | Yes | 94% |
| 5 | 24 | 1% CoCl2·6H2O | - | Yes | 96% |
| 6 | 24 | 1% CoCl2·6H2O | H2O2 | No | - |
| 7 | 24 | 1% CoCl2 | - | Yes | 92% |
| 8 | 24 | 1% [Co(pyridine)4Cl2]Cl | - | Yes | 96% |
| 9 | 24 | 1% FeCl2·4H2O | - | Yes | 98% |
| 10 | 24 | 1% FeCl2 | - | Yes | 96% |
| 11 | 24 | 1% NiCl2·6H2O | - | Yes | 96% |
| 12 | 24 | 1% ZnCl2 | - | Yes | 94% |
a Average isolated yield of duplicate experiments. b Evidence of dimer formation is present in the 1H NMR. However, there is a mixture of products present and attempts at isolation of the dimer were unsuccessful.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lind, N.M.; Joe, N.S.; Newell, B.S.; Morris, A.M. High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts. Reactions 2022, 3, 59-69. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions3010005
AMA Style
Lind NM, Joe NS, Newell BS, Morris AM. High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts. Reactions. 2022; 3(1):59-69. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions3010005
Chicago/Turabian StyleLind, Natalie M., Natalie S. Joe, Brian S. Newell, and Aimee M. Morris. 2022. "High Yielding, One-Pot Synthesis of Bis(1H-indazol-1-yl)methane Catalyzed by 3d-Metal Salts" Reactions 3, no. 1: 59-69. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions3010005