∞3[Cu2(mand)2(hmt)]–MOF: A Synergetic Effect between Cu(II) and Hexamethylenetetramine in the Henry Reaction


Abstract
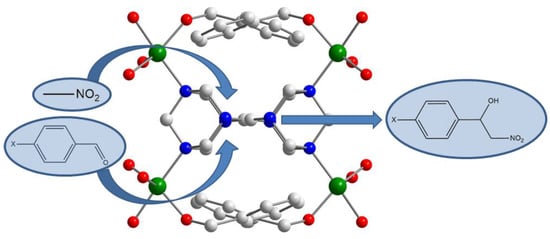
:
1. Introduction
2. Materials and Methods
3. Results
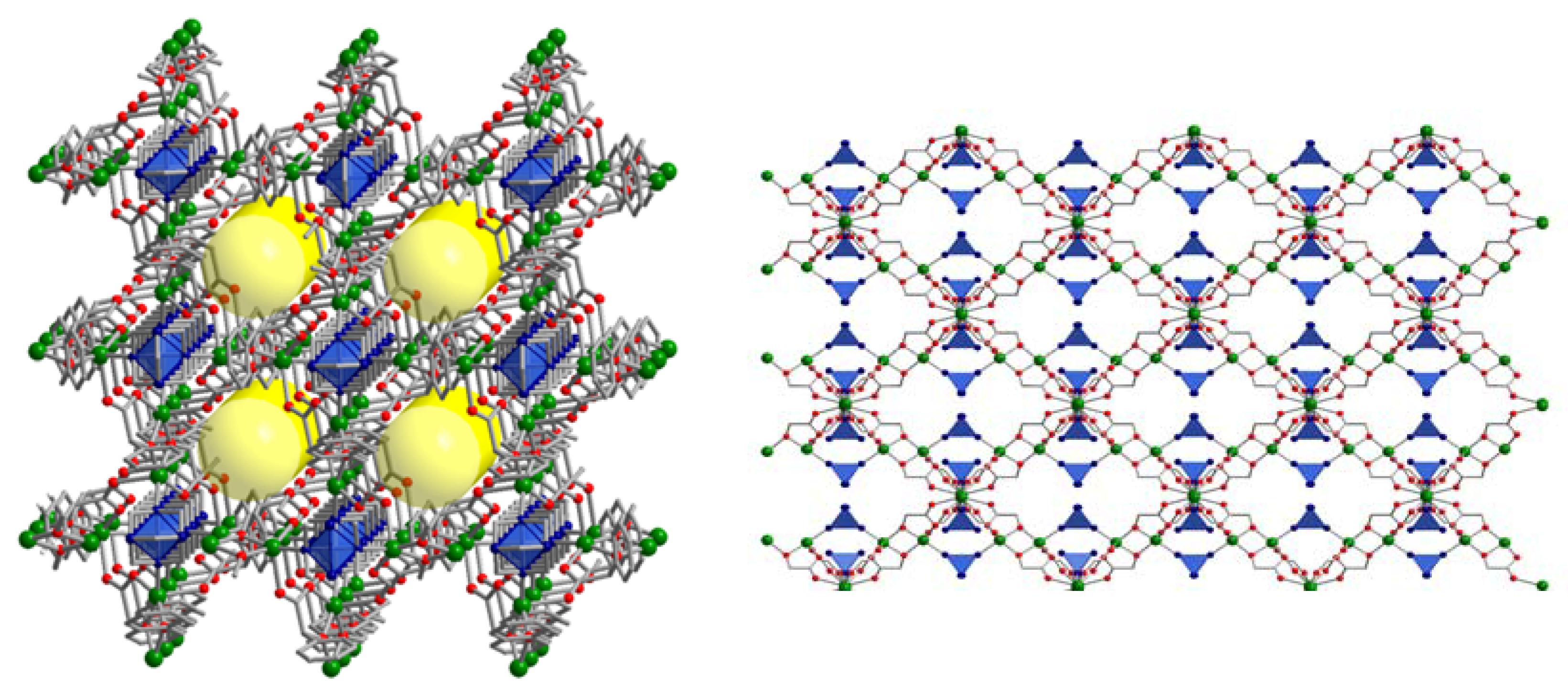
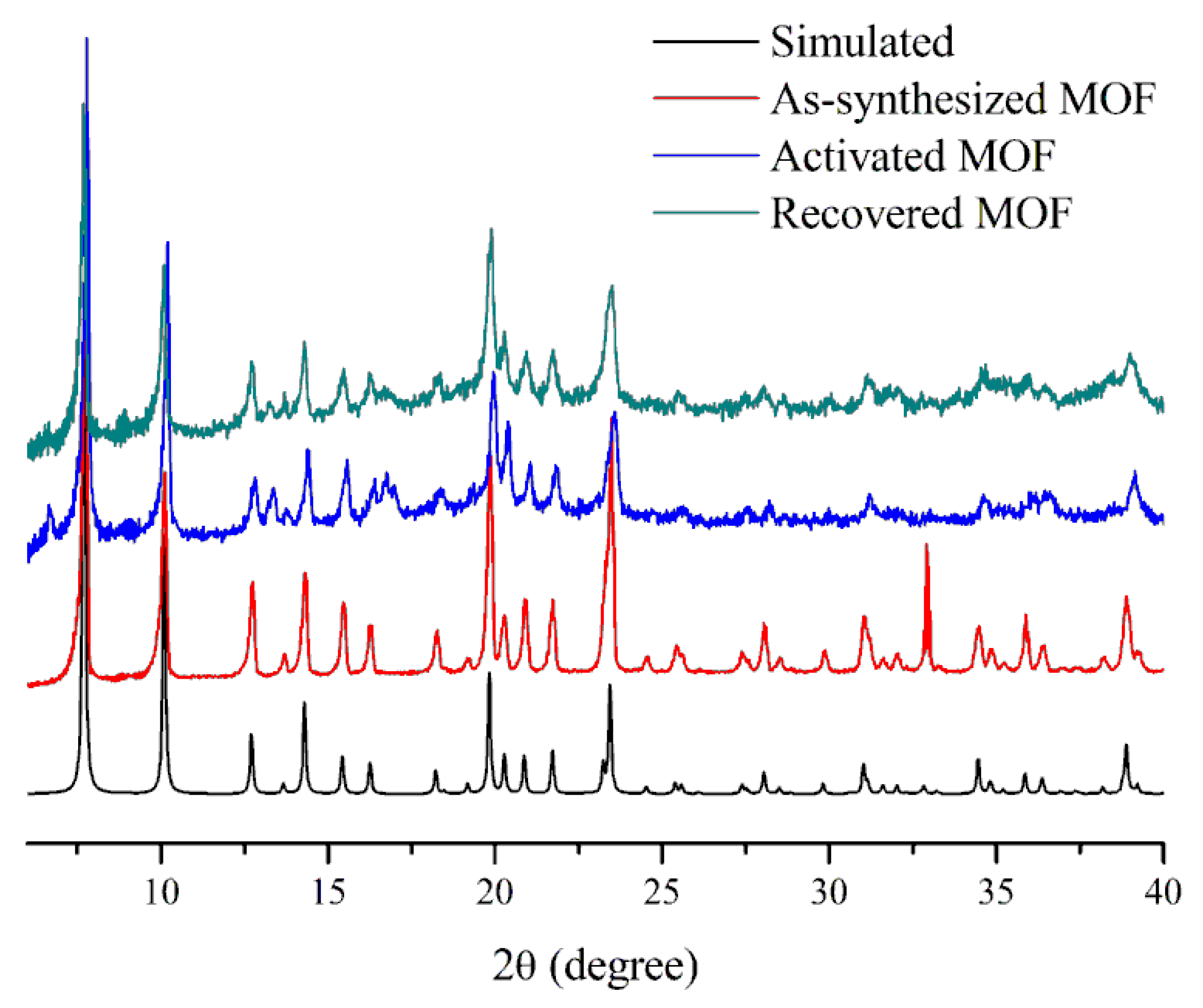
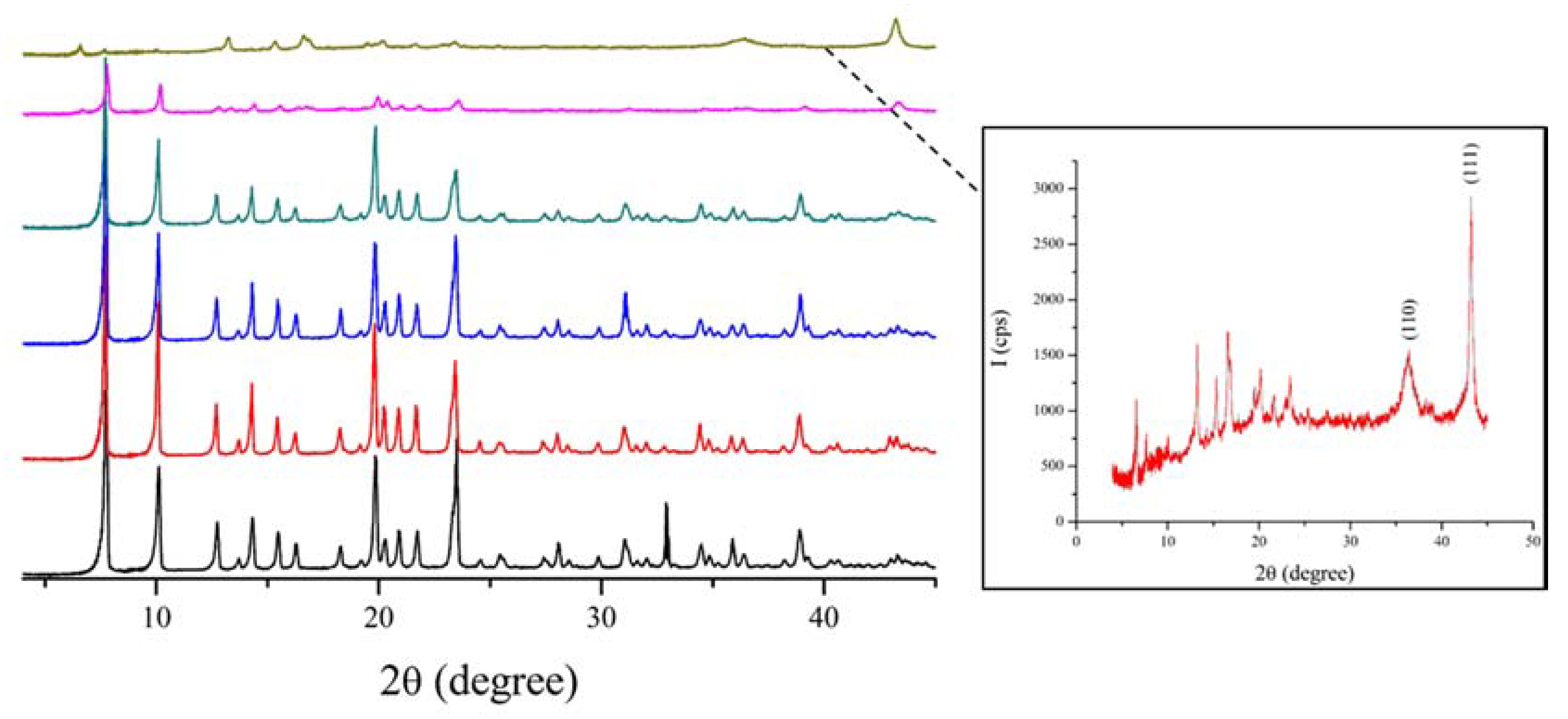
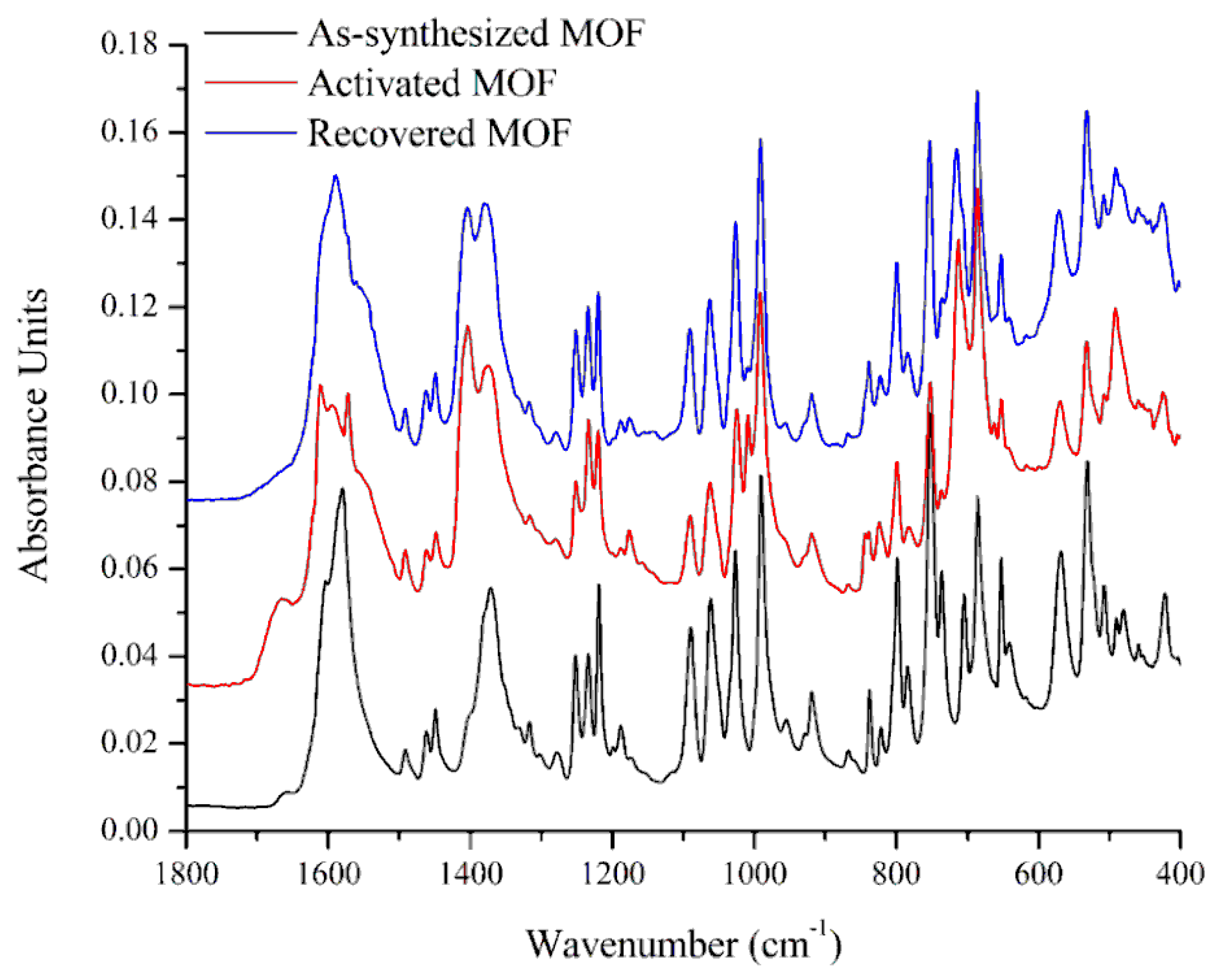
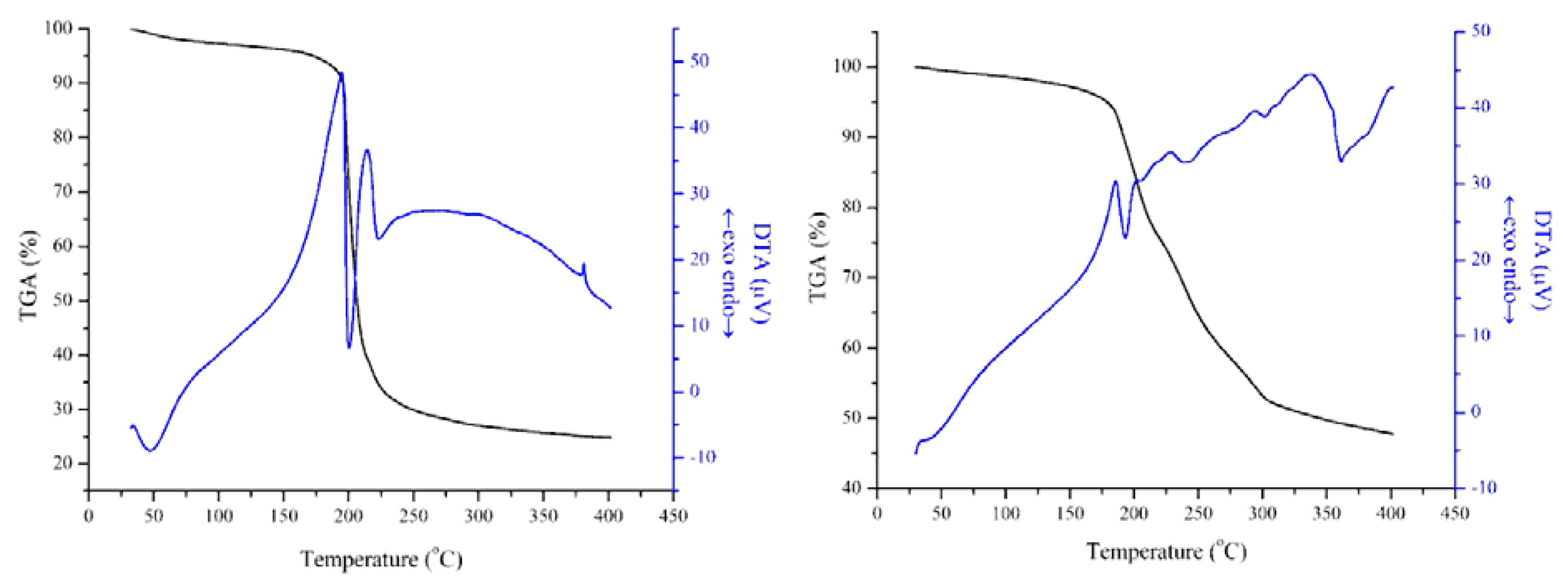
3.1. Characterization of the Catalysts
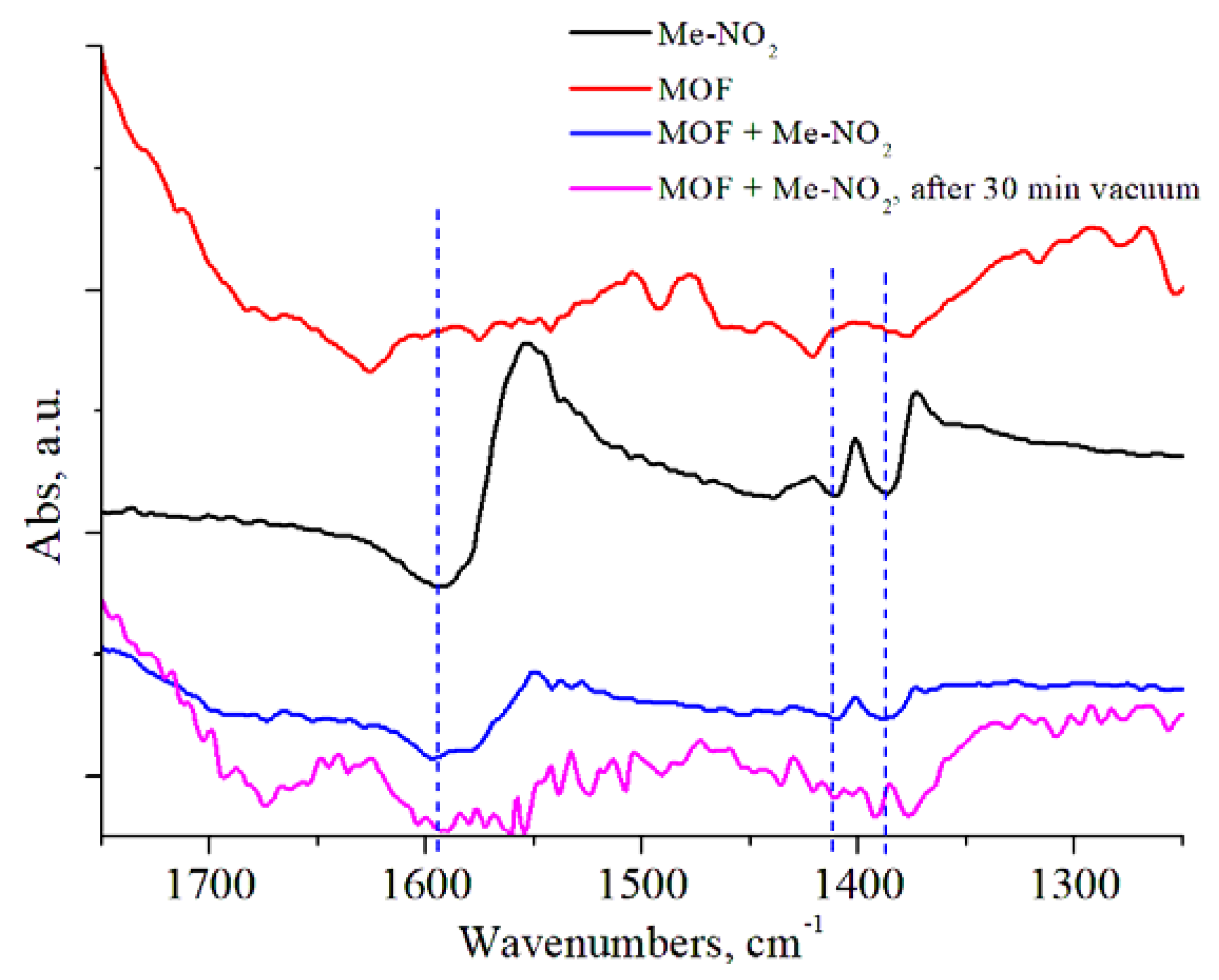
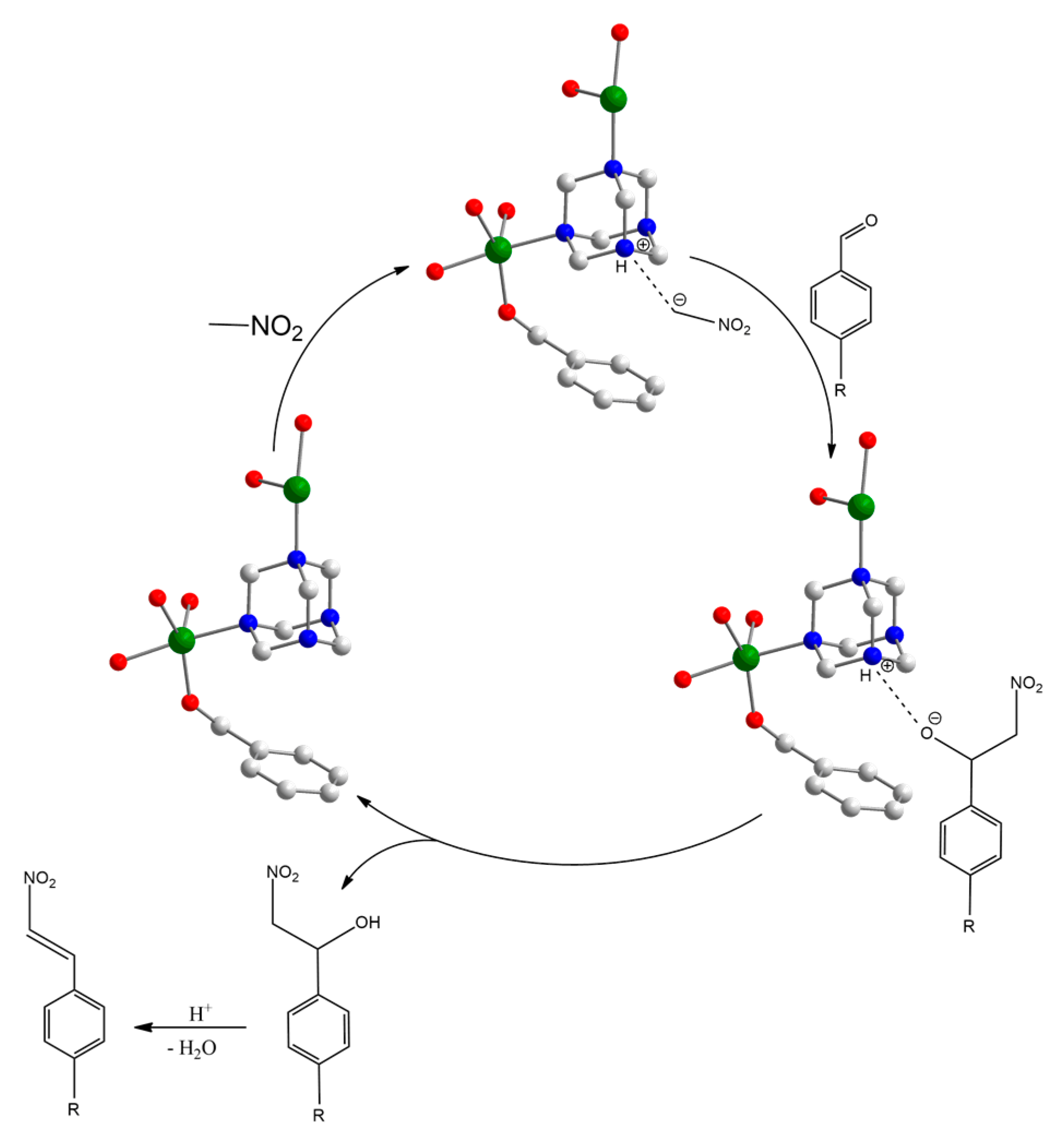
3.2. Catalytic Behaviour of MOF
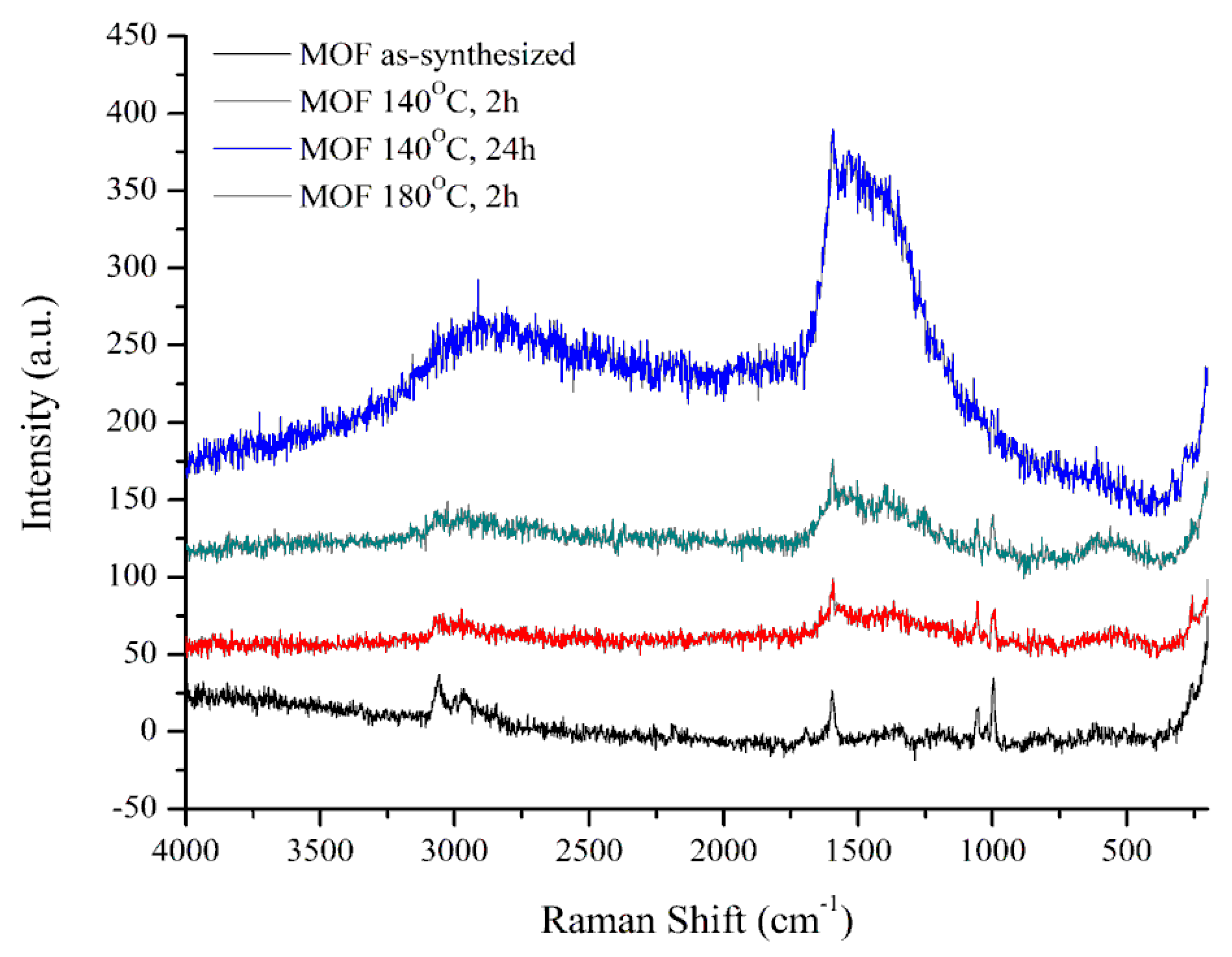
3.3. The Stability of the Catalyst
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grünker, R.; Bon, V.; Müller, P.; Stoeck, U.; Krause, S.; Mueller, U.; Senkovka, I.; Kaskel, S. A new metal–organic framework with ultra-high surface area. Chem. Commun. 2014, 50, 3450–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.O.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; et al. Ultrahigh Porosity in Metal-Organic Frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farha, O.K.; Yazaydin, A.O.; Eryazici, I.; Malliakas, C.D.; Hauser, B.G.; Kanatzidis, M.G.; Nguyen, S.T.; Snurr, R.Q.; Hupp, J.T. De novo synthesis of a metal–organic framework material featuring ultrahigh surface area and gas storage capacities. Nat Chem. 2010, 2, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Zhao, D.; Sun, D.; Zhou, H.C. An Isoreticular Series of Metal–Organic Frameworks with Dendritic Hexacarboxylate Ligands and Exceptionally High Gas-Uptake Capacity. Angew. Chem. Int. Ed. 2010, 49, 5357–5361. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Grunder, S.; Cordova, K.E.; Valente, C.; Furukawa, H.; Hmadeh, M.; Gandara, F.; Whalley, A.C.; Liu, Z.; Asahina, S.; et al. Large-Pore Apertures in a Series of Metal-Organic Frameworks. Science 2012, 336, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.X.; He, Y.P.; Zhang, J. Tuning MOF Stability and Porosity via Adding Rigid Pillars. Inorg. Chem. 2012, 51, 9649–9654. [Google Scholar] [CrossRef]
- Marx, S.; Kleist, W.; Huang, J.; Maciejewskia, M.; Baiker, A. Tuning functional sites and thermal stability of mixed-linker MOFs based on MIL-53(Al). Dalton Tran. 2010, 39, 3795–3798. [Google Scholar] [CrossRef]
- Wittmann, T.; Siegel, R.; Reimer, N.; Milius, W.; Stock, N.; Senker, J. Enhancing the Water Stability of Al-MIL-101-NH2 via Postsynthetic Modification. Chem. Eur. J. 2014, 20, 314–323. [Google Scholar]
- Lillerud, K.P.; Olsbye, U.; Tilset, M. Designing Heterogeneous Catalysts by Incorporating Enzyme-Like Functionalities into MOFs. Top Catal. 2010, 53, 859–868. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Karagiaridi, O.; Farha, O.K.; Hupp, J.T. Activation of metal–organic framework materials. CrystEngComm. 2013, 15, 9258–9264. [Google Scholar] [CrossRef]
- Yuan, S.; Qin, J.S.; Lollar, C.T.; Zhou, H.C. Stable Metal–Organic Frameworks with Group 4 Metals: Current Status and Trends. ACS Cent. Sci. 2018, 4, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, I.J.; Khan, N.A.; Haque, E.; Jhung, S.H. Chemical and Thermal Stability of Isotypic Metal–Organic Frameworks: Effect of Metal Ions. Chem. Eur. J. 2011, 17, 6437–6442. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar] [CrossRef]
- Duan, J.; Jin, W.; Kitagawa, S. Water-resistant porous coordination polymers for gas separation. Coord. Chem. Rev. 2017, 332, 48–74. [Google Scholar] [CrossRef] [Green Version]
- Jasuja, H.; Walton, K.S. Effect of catenation and basicity of pillared ligands on the water stability of MOFs. Dalton Trans. 2013, 42, 15421–15426. [Google Scholar] [CrossRef]
- Canivet, J.; Vandichel, M.; Farrusseng, D. Origin of highly active metal–organic framework catalysts: Defects? Defects! Dalton Trans. 2016, 45, 4090–4099. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Burgoyne, A.R.; Meijboom, R. Knoevenagel Condensation Reactions Catalysed by Metal-Organic Frameworks. Catal. Lett. 2013, 143, 563–571. [Google Scholar] [CrossRef]
- Cai, J.; Wang, H.; Wang, X.; Duan, X.; Wang, Z.; Cui, Y.; Yang, Y.; Chen, B.; Qian, G. An amino-decorated NbO-type metal–organic framework for high C2H2 storage and selective CO2 capture. RSC Adv. 2015, 5, 77417–77422. [Google Scholar] [CrossRef]
- An, J.; Geib, S.J.; Rosi, N.L. High and Selective CO2 Uptake in a Cobalt Adeninate Metal−Organic Framework Exhibiting Pyrimidine- and Amino-Decorated Pores. J. Am. Chem. Soc. 2010, 132, 38–39. [Google Scholar] [CrossRef]
- Rostamnia, S.; Xin, H. Basic isoreticular metal–organic framework (IRMOF-3) porous nanomaterial as a suitable and green catalyst for selective unsymmetrical Hantzsch coupling reaction. Appl. Organometal. Chem. 2014, 28, 359–363. [Google Scholar] [CrossRef]
- Rostamnia, S.; Morsali, A. Basic isoreticular nanoporous metal–organic framework for Biginelli and Hantzsch coupling: IRMOF-3 as a green and recoverable heterogeneous catalyst in solvent-free conditions. RSC Adv. 2014, 4, 10514–10518. [Google Scholar] [CrossRef]
- Pérez-Mayoral, E.; Cejka, J. [Cu3(BTC)2]: A Metal–Organic Framework Catalyst for the Friedländer Reaction. ChemCatChem. 2011, 3, 157–159. [Google Scholar] [CrossRef]
- Gascon, J.; Aktav, U.; Hernandez-Alonso, M.D.; van Klink, G.P.M.; Kapteijin, F. Amino-based metal-organic frameworks as stable, highly active basic catalysts. J. Catal. 2009, 261, 75–87. [Google Scholar] [CrossRef]
- Valvekensa, P.; Vandichelb, M.; Waroquierb, M.; Van Speybroeck, V.; De Vos, D. Metal-dioxidoterephthalate MOFs of the MOF-74 type: Microporous basic catalysts with well-defined active sites. J. Catal. 2014, 317, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Qi, Y.; Gao, H.; Andriamitantsoa, R.S.; Zhenga, N.; Wang, G. A general post-synthetic modification approach of amino-tagged metal–organic frameworks to access efficient catalysts for the Knoevenagel condensation reaction. J. Mater. Chem. A 2015, 3, 17320–17331. [Google Scholar] [CrossRef]
- Savonnet, M.; Aguado, S.; Ravon, U.; Bazer-Bachi, D.; Lecocq, V.; Bats, N.; Pinela, C.; Farrusseng, D. Solvent free base catalysis and transesterification over basic functionalised Metal-Organic Frameworks. Green Chem. 2009, 11, 1729–1732. [Google Scholar] [CrossRef]
- Grupta, A.K.; Dea, D.; Bharadwaj, P.K. A NbO type Cu(II) metal–organic framework showing efficient catalytic activity in the Friedländer and Henry reactions. Dalton Trans. 2017, 46, 7782–7790. [Google Scholar] [CrossRef]
- Yu, H.; Xie, J.; Zhong, Y.; Zhang, F.; Zhu, W. One-pot synthesis of nitroalkenes via the Henry reaction over amino-functionalized MIL-101 catalysts. Catal. Commun. 2012, 29, 101–104. [Google Scholar] [CrossRef]
- Karmakar, A.; Martins, L.M.D.R.S.; Hazra, S.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Metal−Organic Frameworks with Pyridyl-Based Isophthalic Acid and Their Catalytic Applications in Microwave Assisted Peroxidative Oxidation of Alcohols and Henry Reaction. Cryst. Growth Des. 2016, 16, 1837–1849. [Google Scholar] [CrossRef]
- Ilyes, E.; Florea, M.; Madalan, A.M.; Haiduc, I.; Parvulescu, V.I.; Andruh, M. A Robust Metal–Organic Framework Constructed from Alkoxo-Bridged Binuclear Nodes and Hexamethylenetetramine Spacers: Crystal Structure and Sorption Studies. Inorg. Chem. 2012, 51, 7954–7956. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhu, J.J. Spherical hollow assembly composed of Cu2O nanoparticles. J. Cryst. Growth 2003, 256, 134–138. [Google Scholar] [CrossRef]
- Thamann, T.J.; Loehr, T.M. Raman spectra and normal coordinate analysis of the copper(II) and copper(III) complexes of biuret and oxamide. Spectrochim. Acta A 1980, 36, 751–760. [Google Scholar] [CrossRef]
- Dhumal, N.R.; Singh, M.P.; Anderson, J.A.; Kiefer, J.; Kim, H.J. Molecular Interactions of a Cu-Based Metal–Organic Framework with a Confined Imidazolium-Based Ionic Liquid: A Combined Density Functional Theory and Experimental Vibrational Spectroscopy Study. J. Phys. Chem. C 2016, 120, 3295–3304. [Google Scholar] [CrossRef] [Green Version]
- Escamilla-Roa, E.; Cartwright, J.H.E.; Sainz-Diaz, C.I. Chemobrionic Fabrication of Hierarchical Self-Assembling Nanostructures of Copper Oxide and Hydroxide. ChemSystemsChem 2019, 1, e190001. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman Spectrum of Graphene and Graphene Layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Ballini, R.; Palmieri, A. Formation of Carbon-Carbon Double Bonds: Recent Developments via Nitrous Acid Elimination (NAE) from Aliphatic Nitro Compounds. Adv. Synth. Catal. 2019, 361, 5070–5097. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Solvent | Conversion (%) | Selectivity for Nitroaldol (%) | Selectivity for Nitroalkene (%) |
|---|---|---|---|
| H2O (deionized) | None | None | None |
| Isopropyl alcohol | None | None | None |
| Dichloromethane | None | None | None |
| Dichloroethane | None | None | None |
| Chloroform | None | None | None |
| Acetonitrile | 40 | 91 | 9 |
| 1,4-dioxane | 30 | 83 | 17 |

| Catalyst | Solvent | Carbonyl Substrate | Conversion (%) | Selectivity for Nitroaldol (%) | Selectivity for Nitroalkene |
|---|---|---|---|---|---|
| No catalyst | 1,4-Dioxane |  | None | None | None |
| Mandelic Acid (5.21 mg) | Ethanol |  | None | None | None |
| HMT (2.4 mg) | Ethanol |  | 31 | 4 | 96 |
| Cu(ClO4)2·6H2O (12.25 mg) | Ethanol |  | 64 | 23 | 76 |
| Activated MOF (10 mg) | 1,4-Dioxane |  | 30 | 83 | 17 |
| Activated MOF (10 mg) | Acetonitrile |  | 40 | 91 | 9 |
| Activated MOF (10 mg) | Acetonitrile |  | 48 | 98 | 2 |
| Activated MOF (10 mg) | Acetonitrile |  | 66 | 16 | 71 |
| Activated MOF (10 mg) | Acetonitrile |  | 100 | 0 | 0 |
| Activated MOF (10 mg) | Acetonitrile |  | 12 | 100 | 0 |
| Activated MOF (10 mg) | Acetonitrile |  | 39 | 89 | 11 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szalad, H.; Candu, N.; Cojocaru, B.; Păsătoiu, T.D.; Andruh, M.; Pârvulescu, V.I. ∞3[Cu2(mand)2(hmt)]–MOF: A Synergetic Effect between Cu(II) and Hexamethylenetetramine in the Henry Reaction. Chemistry 2020, 2, 50-62. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010006
Szalad H, Candu N, Cojocaru B, Păsătoiu TD, Andruh M, Pârvulescu VI. ∞3[Cu2(mand)2(hmt)]–MOF: A Synergetic Effect between Cu(II) and Hexamethylenetetramine in the Henry Reaction. Chemistry. 2020; 2(1):50-62. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010006
Chicago/Turabian StyleSzalad, Horațiu, Natalia Candu, Bogdan Cojocaru, Traian D. Păsătoiu, Marius Andruh, and Vasile I. Pârvulescu. 2020. "∞3[Cu2(mand)2(hmt)]–MOF: A Synergetic Effect between Cu(II) and Hexamethylenetetramine in the Henry Reaction" Chemistry 2, no. 1: 50-62. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010006