Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O


Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556-5670, USA
*
Author to whom correspondence should be addressed.
Chemistry 2021, 3(1), 228-237; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010017
Submission received: 11 December 2020
/
Revised: 29 January 2021
/
Accepted: 2 February 2021
/
Published: 6 February 2021
(This article belongs to the Special Issue Memorial Issue Dedicated to Dr. Howard D. Flack: The Man behind the Flack Parameter)
Abstract
:The X-ray structure of racemic [Co(sep)][Co(edta)]Cl2·2H2O is reported and reveals hetero-chiral stereospecificity in the interactions of [Co(sep)]3+ with [Co(edta)]−. Hydrogen-bonding along the molecular C2-axes of both complexes accounts for the stereospecificity. The structure of Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O has been re-determined. Previous structural data for this compound were collected at room temperature and the model did not sufficiently describe the disorder in the structure. The cryogenic temperature used in the present study allows the disorder to be conformationally locked and modeled more reliably. A clearer inspection of other, structurally interesting, interactions is possible. Again, hydrogen-bonding along the molecular C2-axis of [Co(en)3]3+ and the equatorial carboxylates of [Co(edta)]− is the important interaction. The unique nature of the equatorial carboxylates and molecular C2-axis in [Co(edta)]−, straddled by two pseudo-C3-faces where the arrangement of the carboxylate groups conveys the same helicity, is highlighted. Implications of these structures in understanding stereoselectivity in ion-pairing and electron transfer reactions are discussed.
1. Introduction
Structural studies have played a critical role in the determination of absolute configuration [1], an essential component of investigations of the chiral discriminations involving transition metal complexes. For over forty years, there have been attempts to understand the underlying principles that govern the diastereoselectivity that results from hydrogen-bonded interactions between complex cations such as [Co(en)3]3+ (en = ethane-1,2-diamine) and complex anions such as [Co(edta)]− (edta4− = 2,2′,2″3,2‴-(ethane-1,2-diyldinitrilo)tetraacetate(−4)) [2,3,4,5]. Indeed, the first reported resolution of [Co(edta)]− involved formation of a diastereoselective salt with Λ-[Co(en)3]3+ as resolving agent [6].
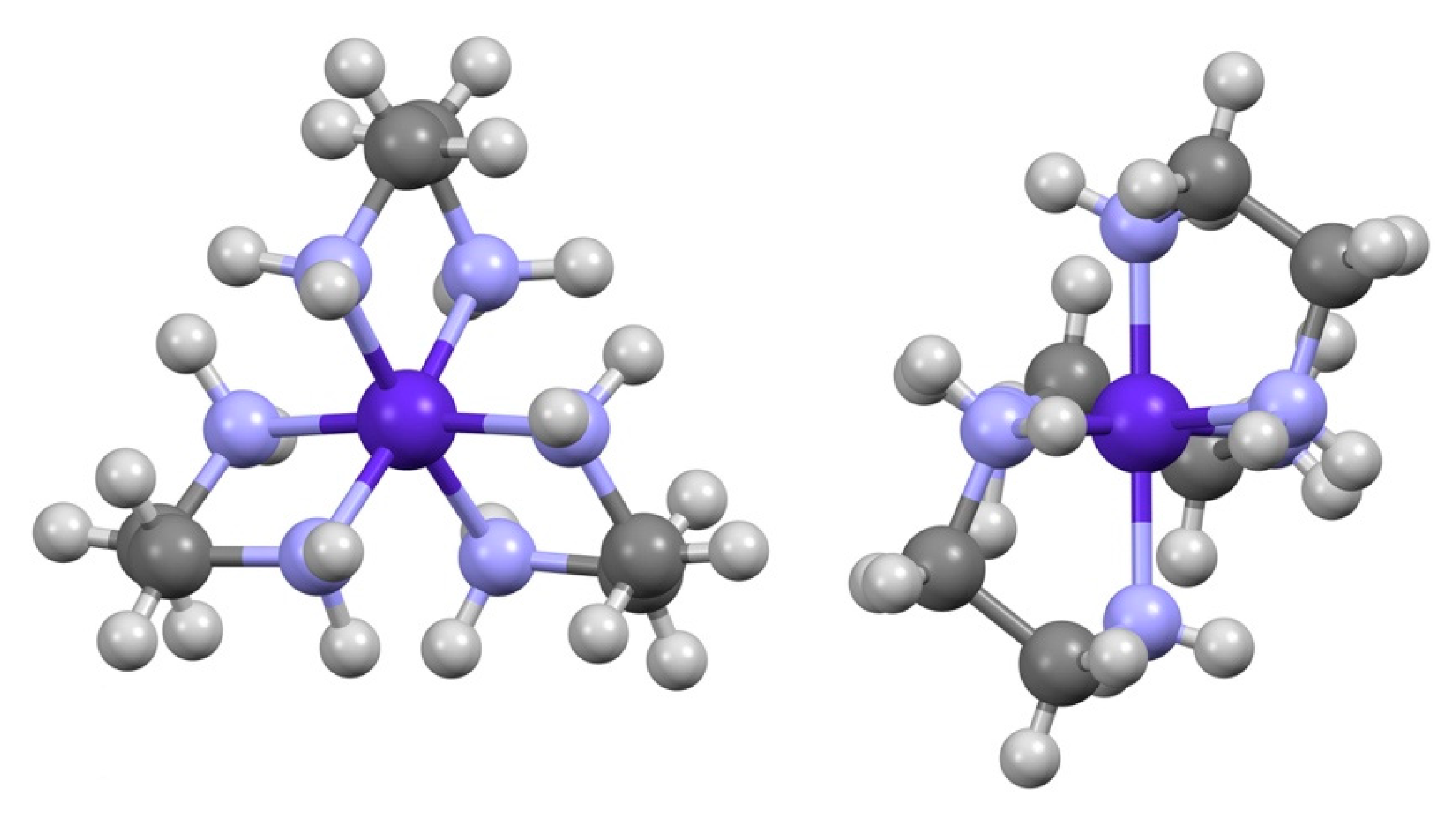
Yoneda and co-workers employed the structurally related cations ∆-[Co(en)3]3+, ∆-[Co(sep)]3+, and ∆(λ,λ,λ)-[Co((RR)-chxn)3]3+ (sep = (1,3,6,8,10,13,16,19-octaazabicyclo [6.6.6]icosane), chxn = trans-1,2-cyclohexanediamine) to infer orientation effects in a series of careful chromatographic experiments, and suggested the dominant interactions in solution align major symmetry axes in the participating complexes that are homochiral [2,3]. Thus, ∆-[Co(sep)]3+, where the P(C3)-axis is sterically inhibited, interacts predominantly through the M(C2)-axis and with an anion interacting through its C3-axis, will have a M(C2)M(C3) or ∆Λ-preference, while with an anion interacting through its C2-axis, the preference will be M(C2)M(C2) or ∆∆, Figure 1.
The complex ∆-[Co(en)3]3+, like ∆-[Co(sep)]3+, is found to interact through the M(C2)-axis while, conversely, the sterically more restricted ∆(λ,λ,λ)-[Co((RR)-chxn)3]3+ has a preference for interactions through the P(C3)-axis. It is through such arguments that [Co(ox)3]3−, [Co(gly)(ox)2]2−, and [Co(edta)]− (ox2− = oxalate(−2), gly− = glycinate(−1)) have been identified as using their C3 or pseudo-C3 carboxylate faces in hydrogen bonding with the cations, see Figure 2.
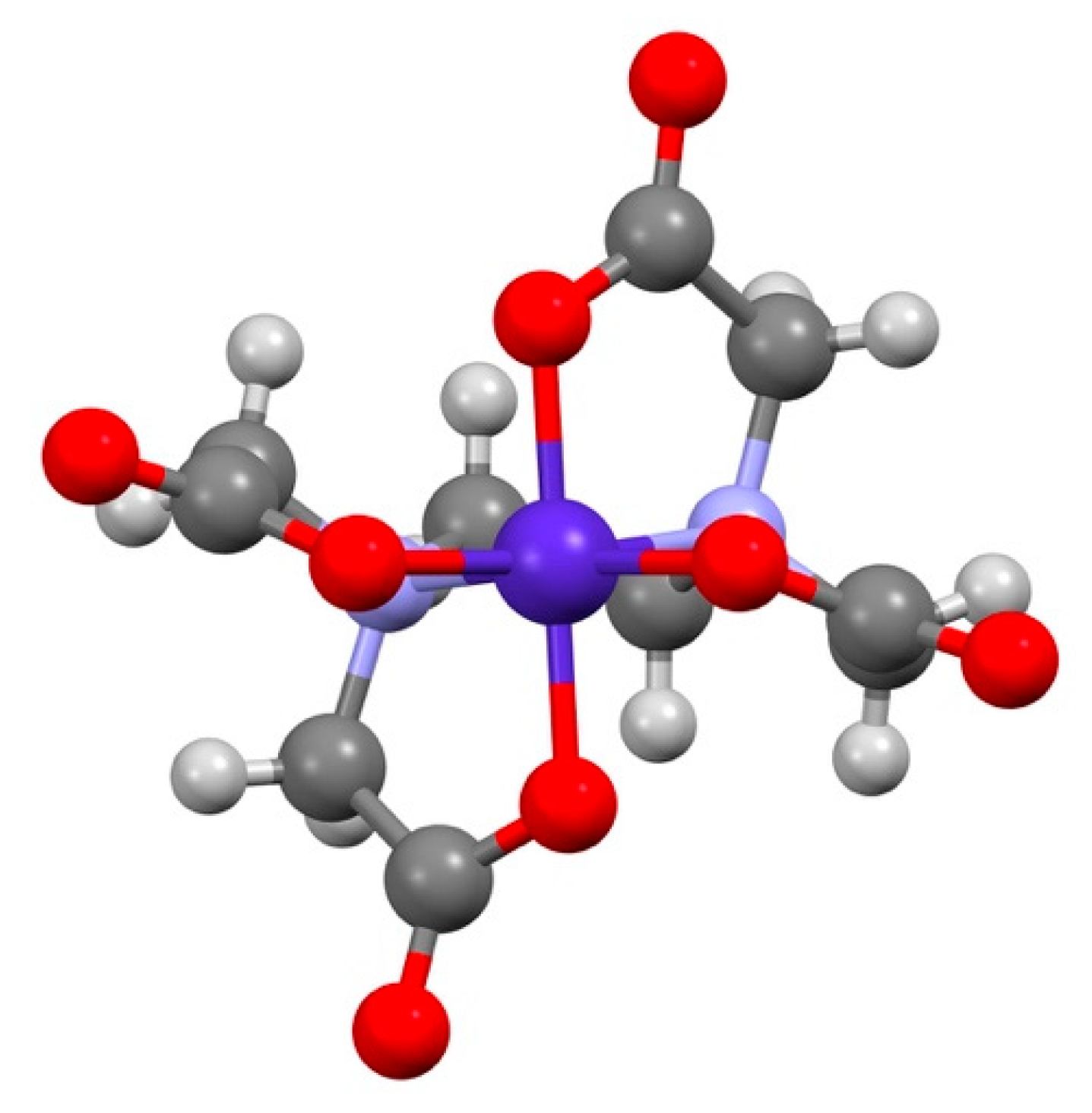
Quantitative studies of ion pairing by conductivity, notably by Tatehata and co-workers, are consistent with this simple explanation [4], although the discrimination is at the limit of detection. Thus, the ion pairing constant in solutions containing Λ-[Co(en)3]3+, ∆-[Co(edta)]−, is 125(5) M−1, whereas for the pair Λ-[Co(en)3]3+, Λ-[Co(edta)]−, it is 119(5) M−1 (25 °C, 0.01 M ionic strength (KI)) consistent with a Λ∆-preference. Limited solution structural data from NMR relaxation measurements in the presence of [Cr(en)3]3+, are also consistent and reveal [Co(edta)]− to use the pseudo-C3 carboxylate face in discriminations. However, in [Co(edta)]−, it must be noted that the chirality resulting from the arrangement of the carboxylate groups of the equivalent pseudo-C3 faces straddle the molecular C2 axis, see Figure 2. Consequently, unlike in the case of the tris-bidentate chelates, the helicity conveyed by the ligands along the principal C2-axis and the pseudo-C3 faces is the same [5].
Our interest derives from the useful correlation between the chiral recognition in these ion pairs and the chiral induction in the electron transfer reactions of [Co(edta)]− and other complexes with the pseudo-C3 face with [Co(en)3]2+ and derivatives [7,8,9,10,11,12,13]. A conclusion might be that the ion pairs serve as reasonable analogues for the precursor complex for the electron transfer process, despite the difference in the charge on the cation. The [Co(edta)]− system is particularly apt, as the complex has two very distinct sides; the carbon CH2-backbone of the ligand that is not capable of forming hydrogen-bonds, and the molecular C2-carboxylate axis surrounded by the two-pseudo-C3 faces, all, in the parlance of Yoneda, with the same handedness. Dipole considerations dictate that it is this latter side that should interact with hydrogen bonding cations.
Information derived from crystal structures on the interactions of [Co(edta)]− with [Co(en)3]3+ and its derivatives or their analogues is very limited. In an earlier study, the structure of Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O was reported [13]. In the present paper, this structure has been re-determined at cryogenic temperature to provide a better model for a chelate ring conformational disorder and improve an understanding of the hydrogen bonding between the complexes. Indeed, a characteristic of the chiral induction in electron transfer with [Co(edta)]− and diastereomeric derivatives of [Co(en)3]3+ is a dependence on chelate ring conformation. The structure of racemic [Co(sep)][Co(edta)]Cl2·2H2O has been determined for the first time. The relevance of these static structures in understanding electron transfer is discussed.
2. Materials and Methods
The compounds [Λ-(+)D-[Co(en)3]Cl3, Na[∆-(+)546Co(edta)]·4H2O, K[Co(edta)]·2H2O, and [Co(sep)]Cl3 [13,14] were prepared by literature methods. Crystals of Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O were obtained by diffusion of propan-2-ol into a solution prepared by the addition of 0.051 g (0.12 mmol) of Na[∆-(+)546Co(edta)]·4H2O, to a solution of 0.038 g (0.10 mmol) of [Λ-(+)D-[Co(en)3]Cl3, in 3 mL water as previously described [13]. An arbitrary sphere of data was collected on a violet rod-like crystal, having approximate dimensions of 0.267 × 0.062 × 0.040 mm, on a Bruker APEX-II diffractometer using a combination of ω- and φ-scans of 0.5° [15].
A sample of red-pink block microcrystals of [Co(sep)][Co(edta)]Cl2·2H2O (calc. (found): C 33.1 (33.5), H 5.81 (6.03); N 17.5 (17.2); Cl 8.87 (8.72) was obtained by slow evaporation of an aqueous solution containing 0.281 g (0.635 mM) of Na[Co(edta)]·4H2O and 0.10 g (0.212 mM) of [Co(sep)]Cl3·H2O. An arbitrary sphere of data was collected on a red-pink block-like crystal, having approximate dimensions of 0.031 × 0.018 × 0.010 mm, on a Bruker PHOTON-2 CMOS diffractometer using a combination of ω- and φ-scans of 0.5° [15].
For both structures, data were corrected for absorption and polarization effects, and analyzed for space group determination [16]. The structures were solved by dual-space methods and expanded routinely [17]. Models were refined by full-matrix least-squares analysis of F2 against all reflections [18]. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters.
Atomic displacement parameters for hydrogen atoms in Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O were modeled as a mixture of refined and constrained geometries. Hydrogen atoms on the Co complexes were modeled with atoms riding on the coordinates of the atom to which they are bonded with atomic displacement parameters tied to that of the atom to which they are bonded (Uiso(H) = 1.2 Ueq (C/N)). Water hydrogen atoms were included in positions located from a difference Fourier map. Most water hydrogen atoms were refined freely; several that did not model well were modeled with atomic displacement parameters tied to that of the oxygen to which they are bonded (Uiso(H) = 1.5 Ueq(O)).
In [Co(sep)][Co(edta)]Cl2·2H2O, one water was found to be disordered over two positions during refinement. In the final structure, this atom was modeled with two, half-occupancy oxygen atoms, and concomitant hydrogen atoms, at sites suggested as the loci of the original extended displacement parameters. Residual electron density (2.24 e−/Å3) is located near (0.87 Å) Co4 of one of the [Co(edta)]− anions. It is unclear what this residual density might be, and is likely due to Fourier ripple or a very small amount of otherwise unresolvable molecular disorder. Hydrogen atoms for [Co(sep)][Co(edta)]Cl2·2H2O were treated as a mixture of freely refined and geometrically constrained atoms. Hydrogen atoms bonded to carbon and nitrogen were treated as riding models with Uiso(H) = 1.2 Ueq(C). Water hydrogen atoms were modeled at locations initially located from a difference Fourier map and subsequently tied to the coordinates of the oxygen to which they are bonded. Atomic displacement parameters for water hydrogen atoms in this model were restrained to Uiso(H) = 1.5 Ueq(O).
The disorder in Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O was resolved more thoroughly with a cryogenic measurement of the data for the complex. The ethane-1,2-diamine was observed to be a twist disorder of the ethylene backbone, across the crystallographic two-fold axis that bisects the ethylene chain. Only carbon atom C3 is the unique atom in the model. The two sites were modeled from density observed in a Fourier difference map and the site occupancy ratios summed to unity yielding an approximately 0.77:0.23 ratio. The major component was refined with anisotropic displacement parameters and the minor component with an isotropic atom. Hydrogen atoms about the disorder (on nitrogen and the disordered carbon) were modeled using routine methodology (occupancies tied to the disorder component, riding atom positions, and displacement parameters).
3. Results
3.1. [Co(sep)][Co(edta)]Cl2·2H2O
The structure of [Co(sep)][Co(edta)]Cl2·2H2O (Supplementary Materials) consists of two crystallographically independent, yet chemically identical, [Co(sep)]3+ cations, two [Co(edta)]− anions, four chlorine anions, and four water molecules of crystallization in the asymmetric unit of the primitive, centrosymmetric, triclinic space group P-1. Crystal data are summarized for [Co(sep)][Co(edta)]Cl2·2H2O [19].
The cations have a cobalt atom encapsulated in an octahedral fashion by a sepulchrate ligand. The cobalt is coordinated by the amine nitrogen atoms, that retain their hydrogen atoms. The “apical” nitrogen atoms of the sepulchrate are non-coordinating. The conformation of the complex is ∆(λ,λ,λ) or Λ(δ,δ,δ) (lel3). In the anion, the cobalt is chelated by edta4− in a six-coordinate coordination geometry with geometry ∆Λ∆ or Λ∆Λ, abbreviated ∆ and Λ, respectively.
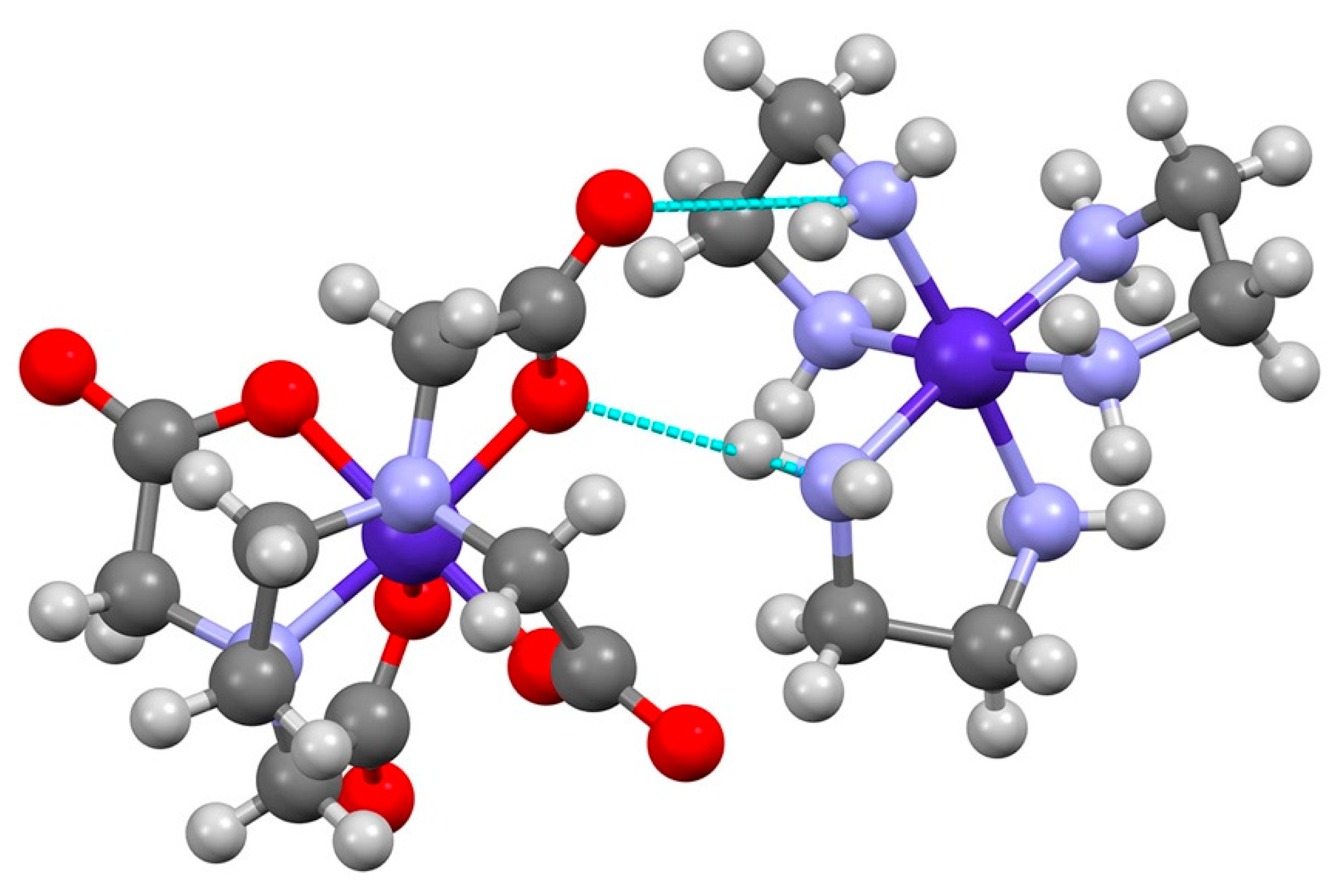
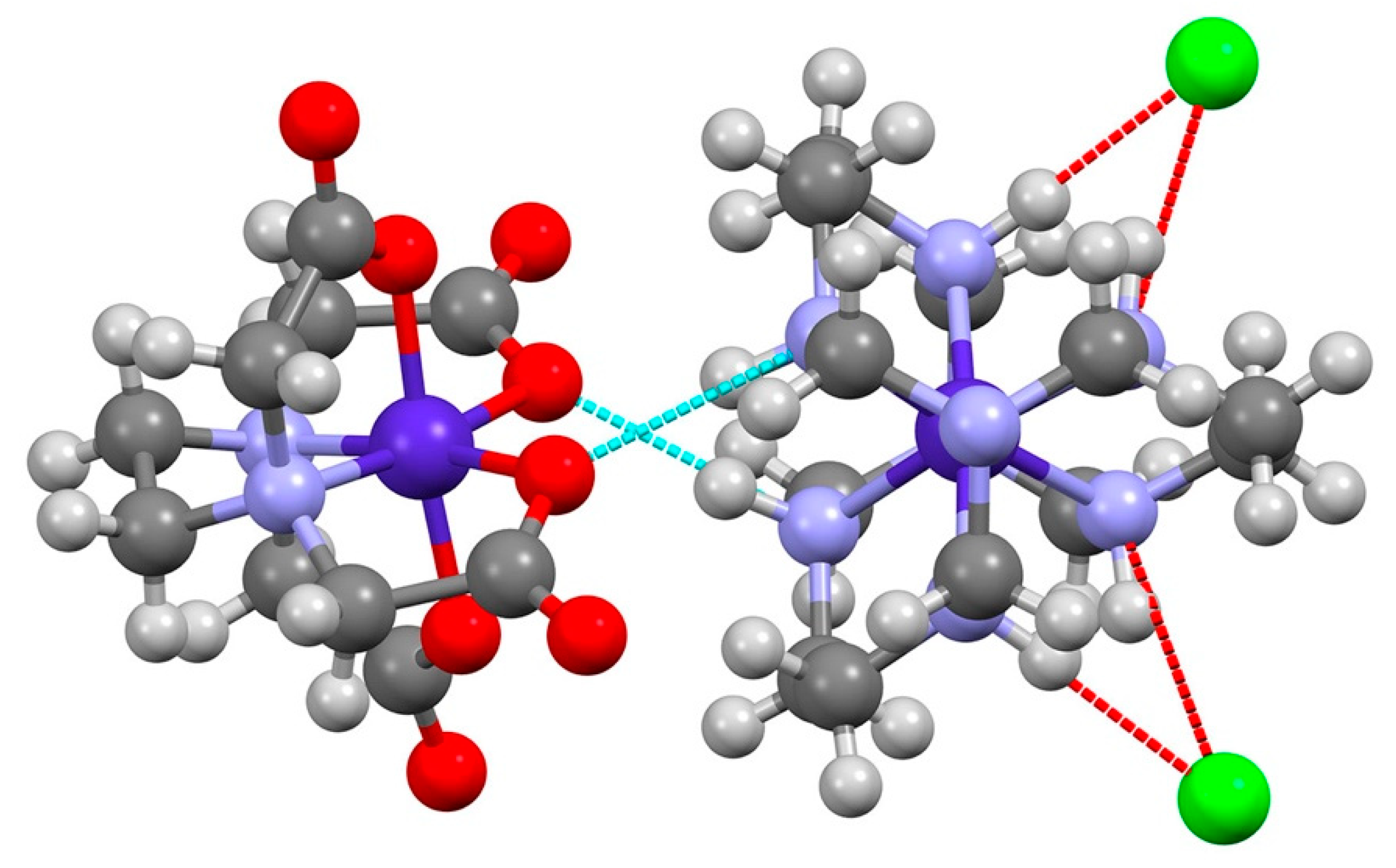
The complex ions are arranged in a hetero-chiral pairwise fashion ∆-[Co(sep)]Λ-[Co(edta)] or Λ-[Co(sep)]∆-[Co(edta)]. The Co-Co distances are 5.162(1) Å and 5.170(1) Å for the two independent pairs. The chlorine atoms form hydrogen bonds with two neighboring sepulchrate amide nitrogen atoms, Table 1, occupying two of the molecular C2-axes of the complex cation, but do not participate in further H-bonding. Each chloride atom is hydrogen bonded by two N-H atoms from a single sepulchrate. The third molecular C2-axis of the [Co(sep)]3+ is occupied in stereospecific fashion by [Co(edta)]− with a pair of N-H hydrogen bonds to the equatorially coordinated G-ring oxygens of its neighboring [Co(edta)]− anion (graph set notation ), see Figure 3. Thus, they also do not propagate the hydrogen bonded network.
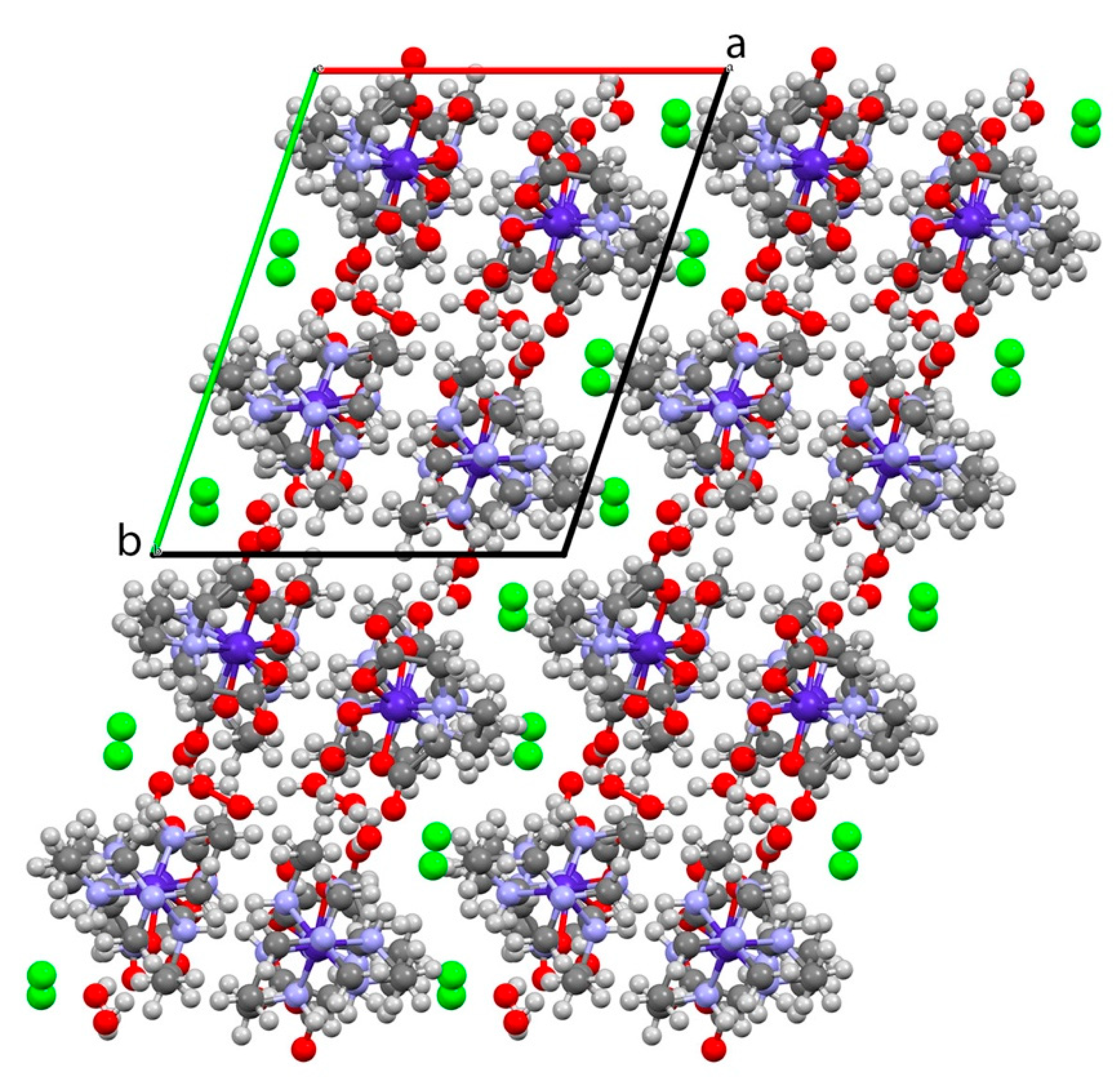
The hydrogen bonded network is extended through the structure with water molecules linking non-coordinated acetate oxygen atoms of [Co(edta)]−. The arrangement of molecules results in a 2D sheet of H-bonded molecules parallel to the b/c plane. Each [Co(edta)]− accepts four hydrogen bonds and is the “corner” of a 4-connected square. Located in the center of each square is a [Co(sep)]3+ from an adjacent sheet, related by inversion symmetry. Due to the orientation of the ligands, these sheets are bi-layers, with hydrophobic regions between layers, see Figure 4.
3.2. (Λ-[Co(en)3]∆-[Co(edta)]2Cl·10(H2O)
The complex, Λ-[Co(en)3]∆-[Co(edta)]2Cl·10(H2O) crystallizes as violet rod-like crystals from an aqueous solution. There are, collectively, two molecules of the cation, four molecules of [Co(edta)]− anion, two chloride anions, and 20 water molecules of crystallization in the unit cell of the primitive, acentric, orthorhombic space group P21212. The correct enantiomorph of the space group and absolute stereochemistry of the complex were determined both by comparison with the known configuration of the complex and by comparison of intensities of Friedel pairs of reflections. Friedel pair analysis (Flack x parameter = 0.006(6) [20] and Hooft y parameter = 0.001(4) [21] support the assignment. Crystal data are summarized for Λ-[Co(en)3]∆-[Co(edta)]2Cl·10(H2O) [22].
The [Co(en)3]3+ cation is located on the crystallographic two-fold axis at [0, 0.5, z] and the chloride anion on the two-fold axis at [1, 0, z], while the [Co(edta)]− anion is located in a general position. Thus, the charge carrying species in the asymmetric unit consist of one half [Co(en)3]3+ cation, one half chloride anion, and one [Co(edta)]− anion. One ethylene diamine ligand is disordered over two sites (the ligand in question bisects the two-fold axis) and was routinely modeled with partial occupancy atoms (0.77:0.23).
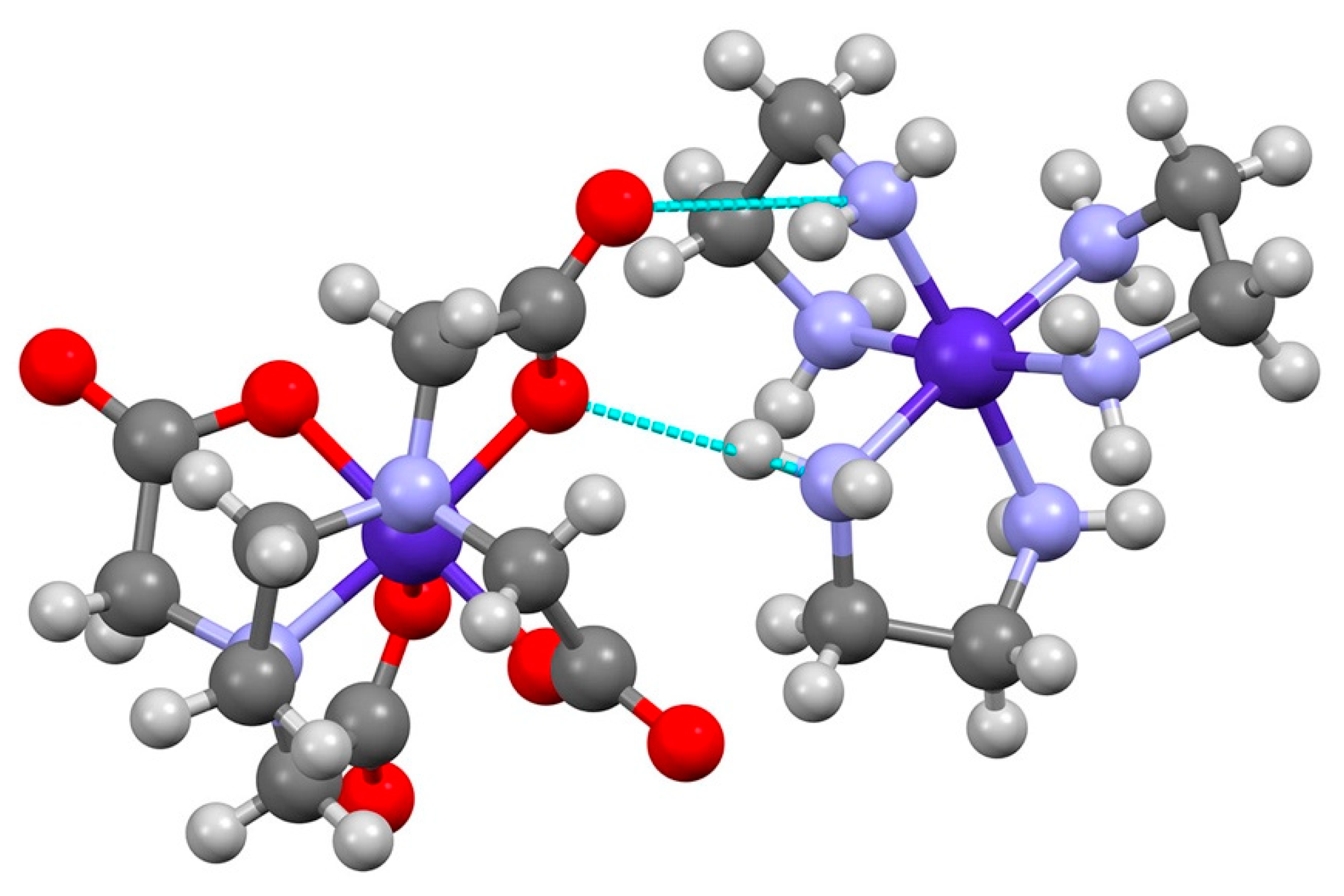
The chloride ions are coordinated by eight water molecules, part of an extensive hydrogen bonding network forming channels, parallel to the c-axis. The amine nitrogen atoms of the [Co(en)3]3+ cations also participate, with alternating [Co(en)3]3+ and Cl− in the ac-plane. The [Co(edta)]− anions alternate in orientation in a parallel ac-plane, completing a layered structure along the b-axis. The layers are held together by hydrogen bonding between the [Co(en)3]3+ and [Co(edta)]−. The two [Co(edta)]− anions interact with [Co(en)3]3+ in different fashion. One interaction, with a Co-Co distance of 5.844(1) Å lies roughly along a molecular C2-axis of [Co(en)3]3+. There is hydrogen-bonding between one N-H proton along the C2-axis, and a second nitrogen on the C3-face of [Co(en)3]3+ with the coordinated and un-coordinated oxygen atoms of the carboxylate group of one of the G-rings on [Co(edta)]− (graph set notation ). The interaction with the C2-axis nitrogen involves the disordered ethane-1,2-diamine ring on [Co(en)3]3+, and examination of the linearity of the hydrogen bonds for the two conformations reveals that the λ conformation is preferred, Table 1, and that the 5.844 Å interaction favors Λ(λ,λ,λ) (ob3), see Figure 5.
The other interaction with a Co-Co distance of 7.509(1) Å shows a hydrogen bonding interaction between an uncoordinated carbonyl oxygen of an out-of-plane R-ring of [Co(edta)]− with an N-H proton from the disordered 1,2-diaminoethane ring on [Co(en)3]3+. Again, examination of the linearity of the hydrogen bonds for the two conformations reveals that the δ conformation is preferred, and that the 7.509 Å interaction favors Λ(δ,λ,λ) (lelob2).
4. Discussion
As expected, bond distances and angles within the molecular ions in both structures are comparable with related studies. However, the focus of this communication is the interactions between the metal-ion complexes.
It has been a generally accepted concept that the chiral discriminations of tris-bidentate chelate complexes such as [Co(en)3]3+ are the result of different orientations of hydrogen-bonding interactions since the C3 axis has the opposite helicity to the C2 axis [2,3,4]. Thus, ∆-[Co(en)3]3+ is P(C3)M(C2) using the nomenclature for P or positive helicity referring to a right-handed screw and M to the left-handed screw. Ion pairing discrimination studies with metal-complex carboxylate liganded anions carried out by chromatography and conductivity measurements have highlighted the importance of the hydrogen-bonded match of the C3 and C2-axes of [Co(en)3]3+ with a pseudo-C3 carboxylate face where three carboxylate groups form a facial motif that is not subtended by a chelate ring. The helicity of the pseudo-C3 carboxylate face in the anion projects to the axis with the same helicity in the cation. Thus the pseudo-C3 carboxylate face of ∆-[Co(edta)]− interacts preferentially with the N-H hydrogens on the C3-axes of ∆-[Co(chxn)3]3+ where the C2-axes are sterically encumbered, but with the N-H hydrogens on C2-axes of Λ-[Co(sep)]3+ where the C3-axes are encumbered.
However, the hexa-coordinated complex ion, [Co(edta)]−, differs in symmetry from a tris-bidentate chelate. The two pseudo-C3 carboxylate faces flank the C2 axis, and the ligand arrangement is such that all three present the same overall helicity. What is notable in both of the structures reported here is that the interactions involving the closest Co-Co distances involve the C2 axis or the in-plane G-rings of the anion, strongly suggesting a more important role for the helicity conveyed along the C2-axis of [Co(edta)]− in determining the discriminations. This observation is also consistent with the solution NMR structure of the ion pair, {[Cr(en)3]3+[Co(edta)]−}, where by symmetry, the paramagnetic cation straddles the C2-axis of [Co(edta)]− [5].
There is a structural comparison with ∆-[Ni(en)3]∆-[Ni(edta)]·4H2O that is also relevant [23]. The cation and anion share two interactions. The closest Ni-Ni distance is 5.40 Å and reveals a direct hydrogen bond formed between the non-coordinated G-ring oxygen of [Ni(edta)]2− and an N-H on the C3-axis of [Ni(en)3]2+ with a second interaction involving the coordinated O of the other G-ring, a bridging water molecule, and a second N-H on the C3-axis of [Ni(en)3]2+ (Graph set notation ). There is no direct pseudo-C3 carboxylate interaction involving the three N-H groups of the C3-axis of [Ni(en)3]2+. Instead, it is the in-plane G-ring carboxylates that again play a dominant role. A longer Ni-Ni distance at 6.14 Å involves a non-coordinated R-ring oxygen of [Ni(edta)]2− with two N-H protons on the C3-axis of [Ni(en)3]2+ (Graph set notation ).
While the distinction between the ligand arrangement conveying the same helicity through the C2-axis and pseudo-C3 faces in [Co(edta)]− with the same helicity may seem semantic, it should be noted that the dipole moment of [Co(edta)]− projects along the C2-axis. The idea that discriminations by [Co(edta)]− can be projected through the C2-axis and not only by the pseudo-C3-faces has implications in the interpretation of the extensive data on related outer-sphere stereoselective electron transfer. The reductions of [Co(edta)]− with both [Co(en)3]2+ and [Co(sep)]2+ have been shown to occur by an outer-sphere mechanism [7,9].
Computational work on the effects of distance and orientation in outer-sphere electron-transfer reactions between metal ion complexes has focused predominantly on the [Fe(OH2)6]3+/2+ and [Ru(OH2)6]3+/2+ self-exchange reactions [24,25,26]. Increasingly, sophisticated calculations [27,28,29,30,31] have not markedly changed the conclusions first reached that face-to-face interactions along the C3-axes represent the closest approach of the two metal centers, at distances roughly 5–6 Å, and the most favorable configuration for overlap of the donor and acceptor orbitals resulting in electron transfer. Other orientations over a range of distances provide less favorable pathways with super-exchange mechanisms involving the ligands more likely at distances in excess of 6 Å.
Unlike the self-exchange reactions, the outer-sphere oxidations of [Co(en)3]2+ and [Co(sep)]2+ by [Co(edta)]− involve complexes with opposite charges, and the additional electrostatic attraction can provide a more intimate interaction, generally at hydrogen-bonded distances. An attractive model for a C3-C3 interaction is provided by the structure of [Cr(en)3]3+[Cr(ox)3]3+ where the metal-metal distance is 4.98 Å [32], shorter than the closest approach distances found in the present structural studies with [Co(en)3]3+ (5.84 Å) and [Co(sep)]3+ (5.17 Å). Although the electron transfer precursor is dynamic and comparisons with static structures are fraught with problems, were distance the only factor, then for [Co(edta)]− projecting discrimination through the pseudo-C3-face, one might well expect the reaction stereoselectivity to reflect a dominant C3-C3 interaction and hence a homochiral, ∆∆ or ΛΛ, preference. That is not what is observed, providing a further piece of evidence that the discrimination by [Co(edta)]− is more complex.
Further, a detailed analysis of structural, charge, and dipolar effects on the stereoselective electron transfer data also revealed [33] that there is a distinction between [Co(edta)]− and the oxalate containing reagents that possess the pseudo-C3 motif such as C1-cis(N)-[Co(gly)2(ox)]− (gly− = glycinate(−1)). Stereoselectivity and chiral discriminations involving [Co(edta)]− should be considered in a class of their own.
It is noted that the chelate ring conformations of [Co(en)3]3+ (ob3 or lelob2) and [Co(sep)]3+ (lel3) differ in the two structures presented. Both cations employ the C2-axis in the closest interaction, and it is not unreasonable to expect that [Co(en)3]3+ (lel3) would interact in similar fashion to [Co(sep)3]3+ (lel3) since they differ mainly along the C3-axis. In stereoselective electron-transfer studies where [Co(edta)]− is used as an oxidant for [Co(R,S-pn)3]2+, (R,S-pn = R,S-propane-1,2-diamine), [Co(RR,SS-bn)3]2+ (RR,SS-bn = RR,SS-butane-2,3-diamine), and [Co(RR,SS-chxn)3]2+, the individual stereoselectivities for the conformation isomers can be determined and show a trend from homo-chiral to hetero-chiral with increasing ob-character, previously ascribed to differences in hydrogen-bonding [9,11]. The weighted average stereoselectivities are [Co(RR,SS-chxn)3]2+ (8% homo-), [Co(RR,SS-bn)3]2+ (3% homo-), [Co(R,S-pn)3]2+ (4% hetero-), and can be compared with those of the conformationally labile [Co(en)3]2+ (10% hetero-), and [Co(sep)]2+ (17% hetero-), reflecting the clear trend in discrimination in the tris-bidentate derivatives of [Co(en)3]2+. However, this does not provide insight into the discriminating nature of the oxidant [Co(edta)]−. The most apt comparison is with the oxidant Λ-[Co((R)-tacntp)] ((R)-tacntp3− = 1,4,7-tri-aza-cyclo-nonane-1,4,7-tris [2′(R)-2′-propionate](-3)), which has 3-fold symmetry with a strong dipole, but no C2-axis [34]. While the selectivity in the oxidation of [Co(en)3]2+ (11% hetero-) is comparable in sense and magnitude with the value for reaction with [Co(edta)]−, the weighted average selectivity with [Co(RR,SS-chxn)3]2+ (31% homo-) reflects a much stronger C3-C3 preference.
5. Conclusions
In conclusion, the structures presented highlight an important role for hydrogen bonding involving the unique C2-axis of [Co(edta)]− in chiral discriminations with [Co(en)3]3+ and derivatives. Stereoselectivity and chiral discriminations involving [Co(edta)]− should be considered in a class of their own. This has implications in the interpretation of data for related stereoselective electron transfer reactions and suggest that generalizations should be avoided.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2624-8549/3/1/17/s1, Figure S1: Labelling diagram for [Co(sep)][Co(edta)]Cl2·2H2O; Figure S2: Packing diagram for [Co(sep)][Co(edta)]Cl2·2H2O; Figure S3: View along c-axis for [Co(sep)][Co(edta)]Cl2·2H2O; Figure S4: Labelling diagram for Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O. X-Ray crystallographic files of [Co(sep)][Co(edta)]Cl2·2H2O and Λ-[Co(en)3]∆-[Co(edta)]2Cl·10H2O, CCDC 2049761, 2049762.
Author Contributions
Conceptualization, P.O. and A.G.L.; methodology, P.O., A.O., and A.G.L.; validation, P.O., A.O., and A.G.L.; formal analysis, A.O. and A.G.L.; investigation, P.O., A.O., and A.G.L.; resources, P.O., A.O., and A.G.L.; data curation, A.O., and A.G.L.; writing—original draft preparation, A.G.L.; writing—review and editing, A.G.L.; visualization, P.O., A.O., and A.G.L.; supervision, A.G.L.; project administration, A.G.L.; funding acquisition, A.O., and A.G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded under National Science Foundation (Grant No. CHE84-06113). The ALS is supported by the U.S. Dept. of Energy, Office of Energy Sciences, under contract DE-AC02-05CH11231.
Acknowledgments
Samples for synchrotron crystallographic analysis were submitted through the SCrALS (Service Crystallography at Advanced Light Source) program. Crystallographic data were collected at Beamline 12.2.1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Flack, H.D. On Enantiomorph-Polarity Estimation. Acta Crystallogr. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yamamoto, I.; Izumoto, S.; Yoneda, H. The Mode of Stereoselective Association between Complex Cation and Complex Anion. Bull. Chem. Soc. Jpn. 1983, 56, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Sakamoto, Y.; Ohguni, A.; Yoneda, H. Stereoselective Interaction of Chiral Metal Complexes in Solution as Studied by Chromatography. i. Modes of Chiral Discrimination and Optical Resolution of anion Complexes. Bull. Chem. Soc. Jpn. 1985, 58, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Tatehata, A.; Fujita, M.; Ando, K.; Asaba, Y. Chiral Recognition in Ion Pairing of an Optically Active Tris(Ethylenediamine)Cobalt(Iii) Cation and Applications to Chromatographic Resolution of Metal Complex Anions. J. Chem Soc. Dalton Trans. 1987, 8, 1977–1982. [Google Scholar] [CrossRef]
- Marusak, R.A.; Lappin, A.G. Observations on the Structure of the Ion Pair Formed between [Co(en)3]3+ and [Co(edta)]–. J. Phys. Chem. 1989, 93, 6856–6859. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Gyarfas, E.C.; Mellor, D.P. The Resolution and Racemization of Potassium Ethylenediaminetetraacetatocobaltate (III). J. Phys Chem. 1955, 59, 296–297. [Google Scholar] [CrossRef]
- Geselowitz, D.A.; Taube, H. Stereoselectivity in Electron-Transfer Reactions. J. Am. Chem Soc. 1980, 102, 4525–4526. [Google Scholar] [CrossRef]
- Osvath, P.; Lappin, A.G. Conformational Effects in Stereoselective Electron Transfer between Metal Ion Complexes. J. Chem. Soc. Chem. Commun. 1986, 14, 1056–1057. [Google Scholar] [CrossRef]
- Osvath, P.; Lappin, A.G. Stereoselectivity as a Probe of Reaction Mechanism in the Oxidation of [Co(en)3]2+ and Its Derivatives by [Co(edta)]–. Inorg. Chem. 1987, 26, 195–202. [Google Scholar] [CrossRef]
- Geselowitz, D.A.; Hammershøi, A.; Taube, H. Stereoselective Electron-Transfer Reactions of (Ethylenediaminetetraacetato)cobaltate(III), (Propylenediaminetetraacetato)cobaltate(III), and (1,2-Cyclohexanediaminetetraacetato)cobaltate(III) with Tris(ethylenediamine)cobalt(II). Inorg. Chem. 1987, 26, 1842–1845. [Google Scholar] [CrossRef]
- Tatehata, A.; Mitani, T. Stereoselectivity in Electron-Transfer Reactions of Tris(ethylenediamine)cobalt(II) with Several Anionic Cobalt(III) Complexes. Chem. Lett. 1989, 18, 1167–1170. [Google Scholar] [CrossRef]
- Tatehata, A.; Muraida, A. Temperature Dependence of Chiral Recognition in the Oxidation of Tris(ethylenediamine)cobalt(II) Complex by Optically Active Anionic Cobalt(III) Complexes. Chem. Lett. 1996, 25, 461–462. [Google Scholar] [CrossRef]
- Warren, R.M.L.; Haller, K.J.; Tatehata, A.; Lappin, A.G. Chiral Discrimination in the Reduction of [Co(edta)]– by [Co(en)3]2+ and [Ru(en)3]2+. X-ray Structure of [Λ-Co(en)3][Δ-Co(edta)]2Cl.10H2O. Inorg. Chem. 1994, 33, 227–232. [Google Scholar] [CrossRef]
- Harrowfield, J.M.; Herlt, A.J.; Sargeson, A.M. Caged Metal Ions: Cobalt Sepulchrates. Inorg. Synth. 1980, 20, 85–86. [Google Scholar]
- APEX-3; Bruker AXS: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Crystal data for C22H46Cl2Co2N10O10; Mr = 799.45; Triclinic; space group P-1; a = 13.0187(8) Å; b = 15.4433(10) Å; c = 17.2326(11) Å; α = 90.310(2)°; β = 108.030(2)°; γ = 107.509(2)°; V = 3123.3(3) Å3; Z = 4; T = 150(2) K; λ(synchrotron) = 0.72880 Å; μ(synchrotron) = 1.387 mm−1; dcalc = 1.700 g.cm−3; 162884 reflections collected; 13890 unique (Rint = 0.1250); giving R1 = 0.0561, wR2 = 0.1480 for 10514 data with [I > 2σ(I)] and R1 = 0.0768, wR2 = 0.1643 for all 13890 data. Residual electron density (e–·Å−3) max/min: 2.239/-0.632. Available online: https://rruff.geo.arizona.edu/AMS/amcsd.php (accessed on 6 February 2021).
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Cryst. 2013, B69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Cryst. 2008, 41, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Crystal data for C26H68ClCo3N10O26; Mr = 1149.14; Orthorhombic; space group P21212; a = 12.8265(17) Å; b = 21.055(3) Å; c = 8.1536(11) Å; α = 90°; β = 90°; γ = 90°; V = 2202.0(5) Å3; Z = 2; T = 120(2) K; λ(Mo-Kα) = 0.71073 Å; μ(Mo-Kα) = 1.280 mm−1; dcalc = 1.733 g.cm−3; 42751 reflections collected; 5478 unique (Rint = 0.0550); giving R1 = 0.0271, wR2 = 0.0642 for 5032 data with [I > 2σ(I)] and R1 = 0.0323, wR2 = 0.0662 for all 5478 data. Residual electron density (e–·Å−3) max/min: 0.500/-0.533. Available online: https://rruff.geo.arizona.edu/AMS/amcsd.php (accessed on 6 February 2021).
- Sysoeva, T.F.; Agre, V.M.; Trunov, V.K.; Dyatlova, N.M.; Fridman, A.Y. Crystal structure of complex compound of nickel(II) with ethylenediamine and ethylenediaminetetraacetic acid, Ni2en3A.4H2O. J. Struct. Chem. (Engl. Transl.) 1986, 27, 97–103. [Google Scholar] [CrossRef]
- Tembe, B.L.; Friedman, H.L.; Newton, M.D. The Theory of Fe2+–Fe3+ Electron Exchange in Water. J. Chem. Phys. 1982, 76, 1490–4367. [Google Scholar] [CrossRef]
- Logan, J.; Newton, M.D. Ab initio study of electronic coupling in the aqueous Fe2+-Fe3+ electron exchange process. J. Chem. Phys. 1983, 78, 4086–4091. [Google Scholar] [CrossRef]
- Newton, M.D. Electronic Structure Analysis of Electron-Transfer Matrix Elements for Transition-Metal Redox Pairs. J. Phys. Chem. 1988, 92, 3049–3056. [Google Scholar] [CrossRef]
- Kumar, P.V.; Tembe, B.L. Solvation structure and dynamics of the Fe2+–Fe3+ ion pair in water. J. Chem. Phys. 1992, 97, 4356–4367. [Google Scholar] [CrossRef]
- Babu, C.S.; Madhusoodanan, M.; Sridhar, G.; Tembe, B.L. Orientations of [Fe(H2O)6]2+ and [Fe(H2O)6]3+ Complexes at a Reactive Separation in Water. J. Am. Chem. Soc. 1997, 119, 5679–5681. [Google Scholar] [CrossRef]
- Migliore, A.; Sit, P.H.-L.; Klein, M.L. Evaluation of Electronic Coupling in Transition-Metal Systems Using DFT: Application to the Hexa-Aquo Ferric-Ferrous Redox Couple. J. Chem. Theory Comput. 2009, 5, 307–323. [Google Scholar] [CrossRef]
- Oberhofer, H.; Blumberger, J. Insight into the Mechanism of the Ru2+–Ru3+ Electron Self-Exchange Reaction from Quantitative Rate Calculations. Angew. Chem. Int. Ed. 2010, 49, 3631–3634. [Google Scholar] [CrossRef]
- Miliordos, E.; Xantheas, S.S. Ground and Excited States of the [Fe(H2O)6]2+ and [Fe(H2O)6]3+ Clusters: Insight into the Electronic Structure of the [Fe(H2O)6]2+-[Fe(H2O)6]3+ Complex. J. Chem. Theory Comput. 2015, 11, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Larsson, K.; Neal, T.J.; Wyllie, G.R.A.; Shang, M.; Lappin, A.G. Structure and magnetic properties of [Cr(en)2(ox)][Cr(en)(ox)2]·2H2O, Δ-[Cr(en)2(ox)]Δ-[Cr(en)(ox)2] and Δ-[Cr(en)2(ox)]Δ-[Cr(en)(ox)2]. Inorg. Chem. Commun. 2001, 4, 635–639. [Google Scholar] [CrossRef]
- Mitani, T.; Honma, N.; Tatehata, A.; Lappin, A.G. Shape selectivity in outer-sphere electron transfer reactions. Inorg. Chim. Acta 2002, 331, 39–71. [Google Scholar] [CrossRef]
- Saenz, G.L.; Warren, R.M.L.; Shang, M.; Lappin, A.G. Stereoselectivity in the Reduction of Λ-[1,4,7-Triazacyclononane-1,4,7-tris[2’(R)-2’-propionate]cobalt(III)]. J. Coord Chem. 1995, 34, 129–137. [Google Scholar] [CrossRef]
Figure 1.
View down the molecular C3- and C2-axes of ∆(λ,λ,λ)-[Co(en)3]3+ showing the different helicities, P and M respectively, established by the arrangement of the chelating ligands. In ∆(λ,λ,λ)-[Co(sep)]3+, the molecular C3-axis is capped.
Figure 1.
View down the molecular C3- and C2-axes of ∆(λ,λ,λ)-[Co(en)3]3+ showing the different helicities, P and M respectively, established by the arrangement of the chelating ligands. In ∆(λ,λ,λ)-[Co(sep)]3+, the molecular C3-axis is capped.
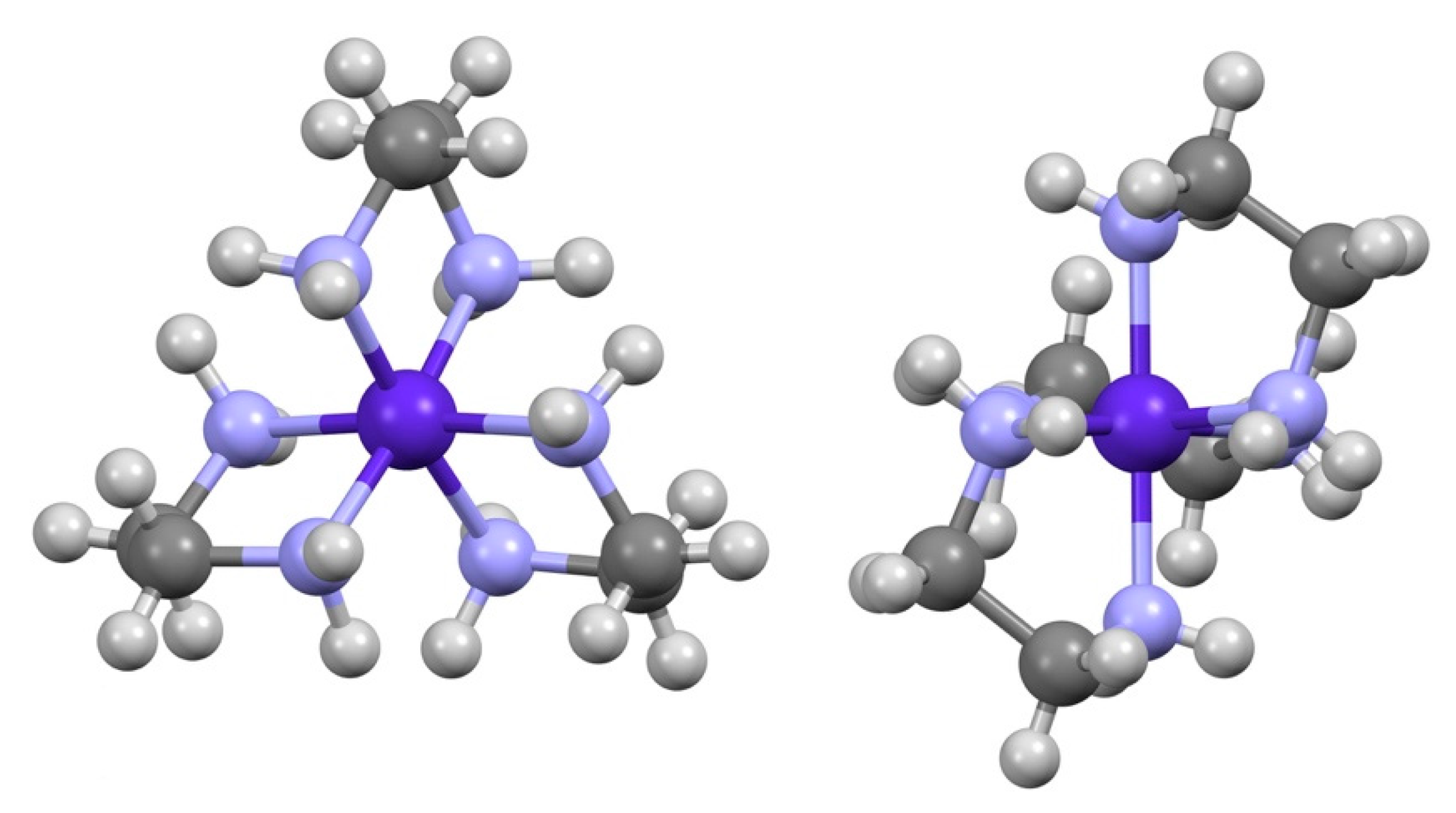
Figure 2.
View down the molecular C2-axis of ∆Λ∆-[Co(edta)]− showing how it is flanked by the carboxylate pseudo-C3-faces.
Figure 2.
View down the molecular C2-axis of ∆Λ∆-[Co(edta)]− showing how it is flanked by the carboxylate pseudo-C3-faces.
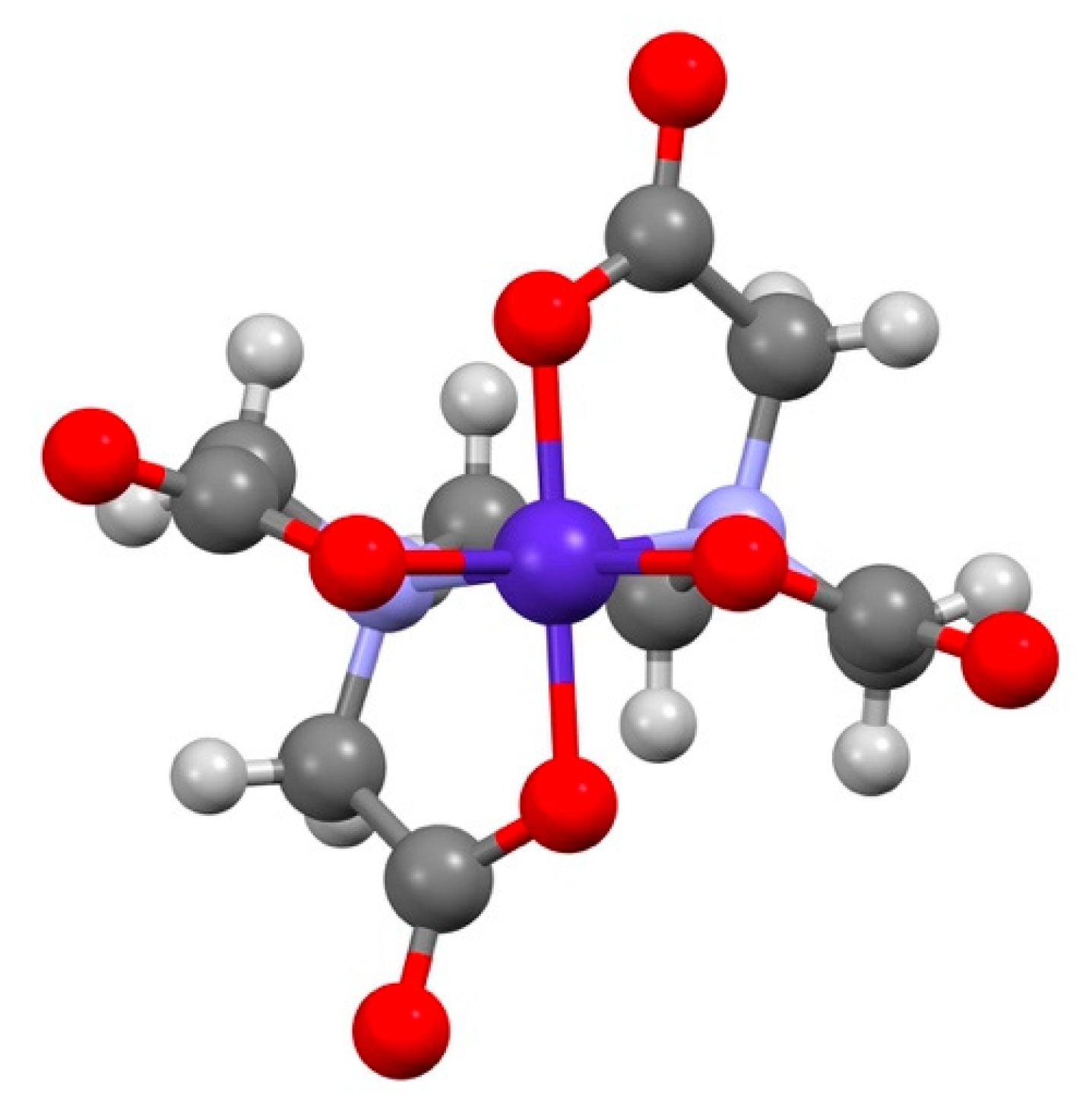
Figure 3.
Hydrogen bonding between ∆-[Co(sep)]3+ and Λ-[Co(edta)]−. The view is down the molecular C3-axis of [Co(sep)]3+ and shows the interaction with Cl− on the molecular C2-axes. Bond distances for complexes (Å): Co-N (∆-[Co(sep)]3+) 1.973(3), 1.974(3) 1.979(3) 1.987(3) 1.987(3) 1.993(3); Co-N (Λ-[Co(edta)]−) 1.921(3), 1.930(3); Co-O (G-ring) 1.898(3), 1.911(3); Co-O (R-ring) 1.879(3), 1.898(3).
Figure 3.
Hydrogen bonding between ∆-[Co(sep)]3+ and Λ-[Co(edta)]−. The view is down the molecular C3-axis of [Co(sep)]3+ and shows the interaction with Cl− on the molecular C2-axes. Bond distances for complexes (Å): Co-N (∆-[Co(sep)]3+) 1.973(3), 1.974(3) 1.979(3) 1.987(3) 1.987(3) 1.993(3); Co-N (Λ-[Co(edta)]−) 1.921(3), 1.930(3); Co-O (G-ring) 1.898(3), 1.911(3); Co-O (R-ring) 1.879(3), 1.898(3).

Figure 4.
Packing diagram for [Co(sep)][Co(edta)]Cl2·2H2O looking down the crystallographic c-axis. Note the hydrophobic channels parallel to the b/c plane alternating with the hydrogen-bonded channels containing the Cl− ions.
Figure 4.
Packing diagram for [Co(sep)][Co(edta)]Cl2·2H2O looking down the crystallographic c-axis. Note the hydrophobic channels parallel to the b/c plane alternating with the hydrogen-bonded channels containing the Cl− ions.

Figure 5.
Hydrogen bonding between Λ-[Co(en)3]3+ and ∆-[Co(edta)]− with a Co-Co distance of 5.844 (1) Å. The view is roughly down the molecular C3-axis of Λ-[Co(en)3]3+ and shows the Λ(λ,λ,λ) (ob3) conformation. Bond distances for complexes (Å): Co-N (Λ-[Co(en)3]3+) 1.955(2), 1.965(2), 1.960(3); Co-N (∆-[Co(edta)]−) 1.921(3), 1.927(2); Co-O (G-ring) 1.914(2), 1.921(3); Co-O (R-ring) 1.888(2), 1.877(2).
Figure 5.
Hydrogen bonding between Λ-[Co(en)3]3+ and ∆-[Co(edta)]− with a Co-Co distance of 5.844 (1) Å. The view is roughly down the molecular C3-axis of Λ-[Co(en)3]3+ and shows the Λ(λ,λ,λ) (ob3) conformation. Bond distances for complexes (Å): Co-N (Λ-[Co(en)3]3+) 1.955(2), 1.965(2), 1.960(3); Co-N (∆-[Co(edta)]−) 1.921(3), 1.927(2); Co-O (G-ring) 1.914(2), 1.921(3); Co-O (R-ring) 1.888(2), 1.877(2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Hydrogen bonds between metal-ion complexes [Å and °].
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| Λ-[Co(sep)]∆-[Co(edta)]Cl2 | ||||
| N(7)-H(7)···Cl(1) | 1.00 | 2.21 | 3.204(3) | 175.2 |
| N(3)-H(3)···Cl(1) | 1.00 | 2.27 | 3.215(3) | 157.5 |
| N(2)-H(2)···Cl(2) | 1.00 | 2.19 | 3.138(3) | 158.0 |
| N(6)-H(6)···Cl(2) | 1.00 | 2.17 | 3.171(3) | 174.9 |
| N(5)-H(5)···O(1) | 1.00 | 2.03 | 2.919(4) | 146.8 |
| N(8)-H(8)···O(5) | 1.00 | 2.03 | 2.947(4) | 151.7 |
| ∆-[Co(sep)]Λ-[Co(edta)]Cl2 | ||||
| N(10)-H(10)···Cl(3) | 1.00 | 2.11 | 3.075(3) | 162.9 |
| N(16)-H(16)···Cl(3) | 1.00 | 2.36 | 3.346(3) | 168.1 |
| N(11)-H(11)···Cl(4) | 1.00 | 2.17 | 3.139(3) | 163.6 |
| N(13)-H(13)···Cl(4) | 1.00 | 2.25 | 3.238(3) | 169.4 |
| N(14)-H(14)···O(15) | 1.00 | 2.02 | 2.908(4) | 147.4 |
| N(15)-H(15)···O(9) | 1.00 | 1.97 | 2.883(4) | 151.0 |
| Λ-[Co(en)3]∆-[Co(edta)]2Cl | ||||
| G-ring interactions | ||||
| N(1)-H(1NA)···O(5)#2 a | 0.87(4) | 3.07(4) | 3.757(4) | 137(3) |
| N(1)-H(1NA)···O(6)#2 b | 0.87(4) | 2.23(4) | 3.009(3) | 149(3) |
| N(3)-H(3NB)···O(5)#4(ob) a | 0.91 | 2.10 | 2.985(3) | 162.8 |
| N(3)-H(3NB)···O(6)#4(ob) b | 0.91 | 3.13 | 3.783(3) | 129.9 |
| N(1)-H(1NA)···O(5)#2 a | 0.87(4) | 3.07(4) | 3.757(4) | 137(3) |
| N(1)-H(1NA)···O(6)#2 b | 0.87(4) | 2.23(4) | 3.009(3) | 149(3) |
| R-ring interactions | ||||
| N(1)-H(1NA)···O(2)#1 b | 0.87(4) | 3.02(4) | 3.623(4) | 129(3) |
| N(1)-H(1NB)···O(8)#3 b | 0.77(5) | 3.08(5) | 3.558(4) | 123(4) |
| N(3)-H(3NC)···O(2)(lel) b | 0.91 | 1.96 | 2.850(3) | 165.1 |
| N(3)-H(3NA)···O(8)#4(ob) b | 0.91 | 3.39 | 3.820(3) | 111.4 |
| N(3)-H(3ND)···O(7)#4(lel) a | 0.91 | 3.02 | 3.489(3) | 114.1 |
a Coordinated oxygen, b terminal oxygen.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Osvath, P.; Oliver, A.; Lappin, A.G. Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O. Chemistry 2021, 3, 228-237. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010017
AMA Style
Osvath P, Oliver A, Lappin AG. Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O. Chemistry. 2021; 3(1):228-237. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010017
Chicago/Turabian StyleOsvath, Peter, Allen Oliver, and A. Graham Lappin. 2021. "Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O" Chemistry 3, no. 1: 228-237. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3010017