1. Introduction
Synthesizing selective inhibitors for kinases belonging to the same family can be challenging, owing to the similarity in their sequences. One such family is the phosphatidylinositol 3-kinase-related kinases (PIKK) family of kinases, which comprises the DNA damage and response (DDR) pathway of kinases [
1,
2]. These kinases play a crucial role in mediating the detection and repair of damages imposed on DNA, thereby helping in the faithful transfer of genetic information from the parent cell to the daughter cell [
3,
4]. On the other hand, cancer cells exploit these kinases to repair their DNA damages, thereby eluding cell death. It is known that radiotherapy and most of the chemotherapeutic agents destroy cancer cells by causing DNA damage. Thus, the overexpression of the DDR kinases in cancer cells creates resistance to DNA damaging treatments [
5,
6,
7]. Therefore, it is essential that these kinases are inhibited to avoid resistance towards the existing modes of treatment.
The three major kinases involved in the DDR pathway are the ataxia telangiectasia mutated (ATM), the ataxia telangiectasia and rad3 related (ATR), and the DNA dependent protein kinase catalytic subunit (DNA-PKcs) [
1]. These three kinases belong to the PIKK family, along with the mammalian target of rapamycin (mTOR), the suppressor of morphogenesis in genitalia (SMG1), and the transformation/transcription domain-associated protein (TRRAP) [
8,
9]. We aimed to inhibit the ATM kinase, since it is involved in DNA double-stranded break (DSB) repair. On inhibiting ATM kinase, the DSBs remain unrepaired, causing genomic instability and finally leading to cell death [
10,
11,
12,
13].
We had designed and synthesized quinoline-3-carboxamide derivatives for the targeting of the ATM kinase [
14]. The synthesized inhibitors were tested for their cytotoxicity against cancer cell lines like HCT116, MDA-MB-468, and MDA-MB-231. KU60019, a known ATM kinase inhibitor, was used as the positive control. The structures of some of the synthesized compounds are shown in
Figure 1, and
Table 1 illustrates the GI
50 (µM) values of the compounds. Structure–activity relationship (SAR) studies were carried out for compound 6b by synthesizing its regioisomers. The results suggested that when the -OCH
3 is at the para position, the molecule has the best activity. Additionally, we have also done the molecular docking and molecular dynamics (MD) simulation studies for PI3Kγ, since it is also a close homologue of the ATM kinase [
15].
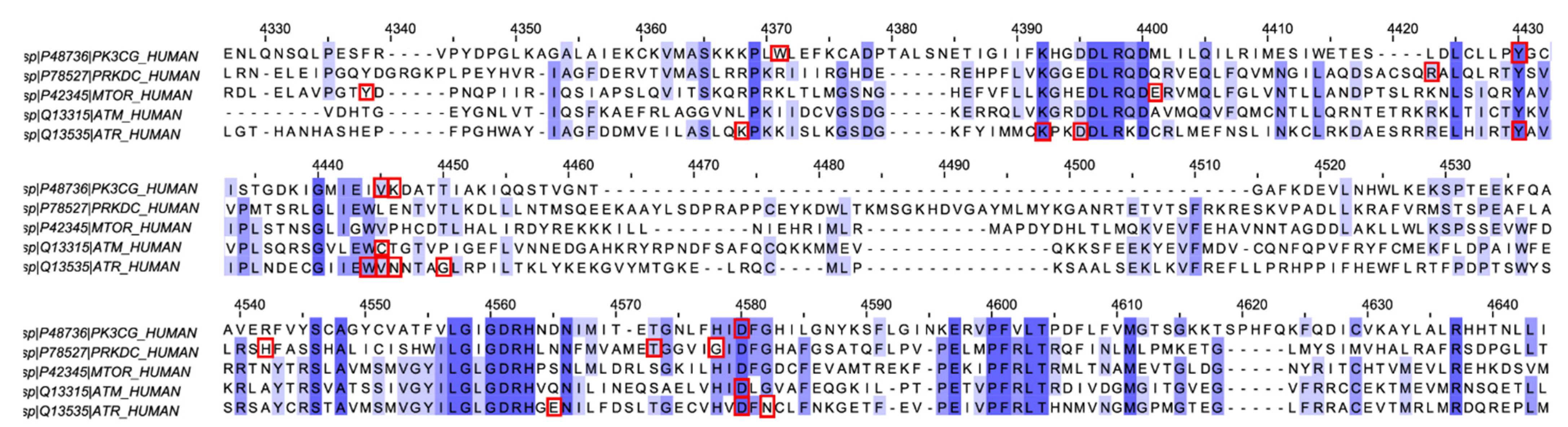
The kinase domain of ATM, ATR, DNA-PKcs, mTOR, and PI3Kγ share 25–40% of similarity in their sequence. Therefore, we decided to carry out molecular docking analysis for the synthesized molecules against all the mentioned kinases. In this article, we report the molecular docking and molecular dynamics simulation (MD simulation) studies of the synthesized molecules against the kinases of the DDR pathway (ATM, ATR, and DNA-PKcs), mTOR and PI3Kγ. The multiple sequence alignment (MSA) analysis for the proteins was also performed. Compound 6f showed the highest docking score against ATM, and thus, MD simulation was performed for the same with all the mentioned proteins. QikProp analysis was also done using Maestro 12.6 to quantify the absorption, distribution, metabolism, and excretion (ADME) properties of the molecules. The properties were all in the agreeable region. Finally, a brief analysis of the selectivity of the molecules towards the ATM kinase was also accomplished using the values obtained from the XP Visualizer module of Maestro 12.6.
2. Materials and Methods
A Swiss modeling tool was used to model the structure of the kinase domain of DNA-PKcs. The template used to model DNA-PKcs was 7k0y.1, which corresponds to the Cryo-EM structure of the activated form of DNA-PK (complex VI) [
16]. The model was validated using the Ramachandran plot. The kinase domains of ATM and ATR were used from previously available data [
14,
17]. The protein data bank (PDB) files for mTOR and PI3Kγ are 5WBY and 5G55, respectively [
14,
17]. Molecular docking and the QikProp studies for all the synthesized inhibitors (
Figure 1) were performed using Maestro 12.6 (Schrödinger, LLC, New York, NY, 2020). Molecular dynamics simulations were performed using NAMD 2.13 [
18]. The key features of the proteins are highlighted in
Table S1 in the
Supplementary Materials.
5. Discussion
From the docking poses of 6f with ATM and PI3Kγ in
Figure 2, it is evident that PI3Kγ makes better contact with the ligand. However, the docking score for all the ligands is higher than those for ATM. Analyzing the values of the rewards and penalties obtained from the XP visualizer panel of Maestro yielded a possible cause for the molecules to have a better binding affinity towards ATM.
Table 6 gives a comparison of both the rewards and penalties for ATM and PI3Kγ. Similarly, the penalties and rewards for the remaining three proteins with 6f have been included in
Table S6 in the
Supplementary Materials. Even though the rewards for the protein−ligand Van der Waal energy and hydrogen bond interactions that are hydrophobically packed are almost similar for these two proteins, the penalties for buried polar atoms, intraligand contacts, and exposed hydrophobic ligand groups are much higher for PI3Kγ. Therefore, the ligands are more stabilized when interacting with ATM, leading to a significantly higher docking score.
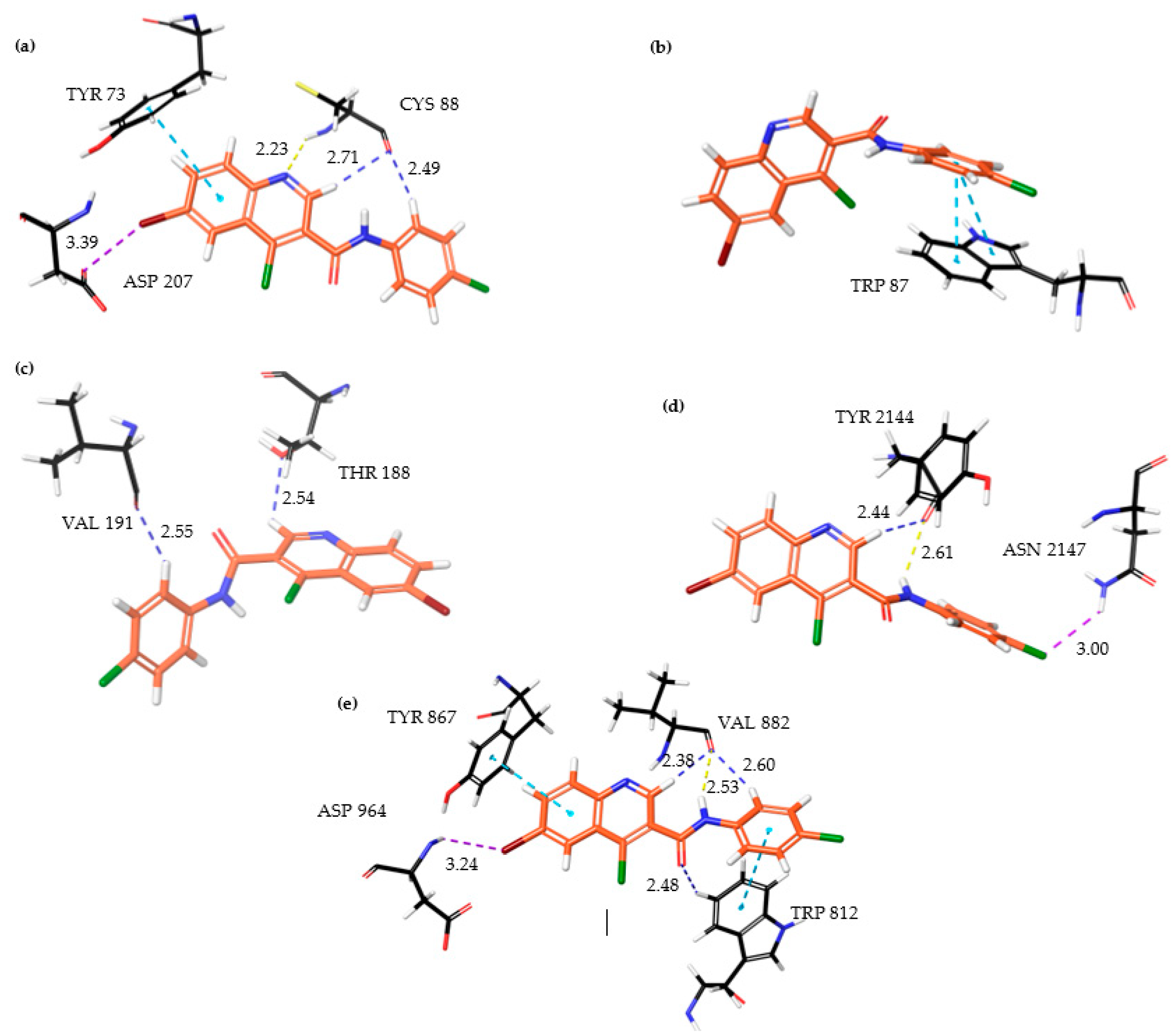
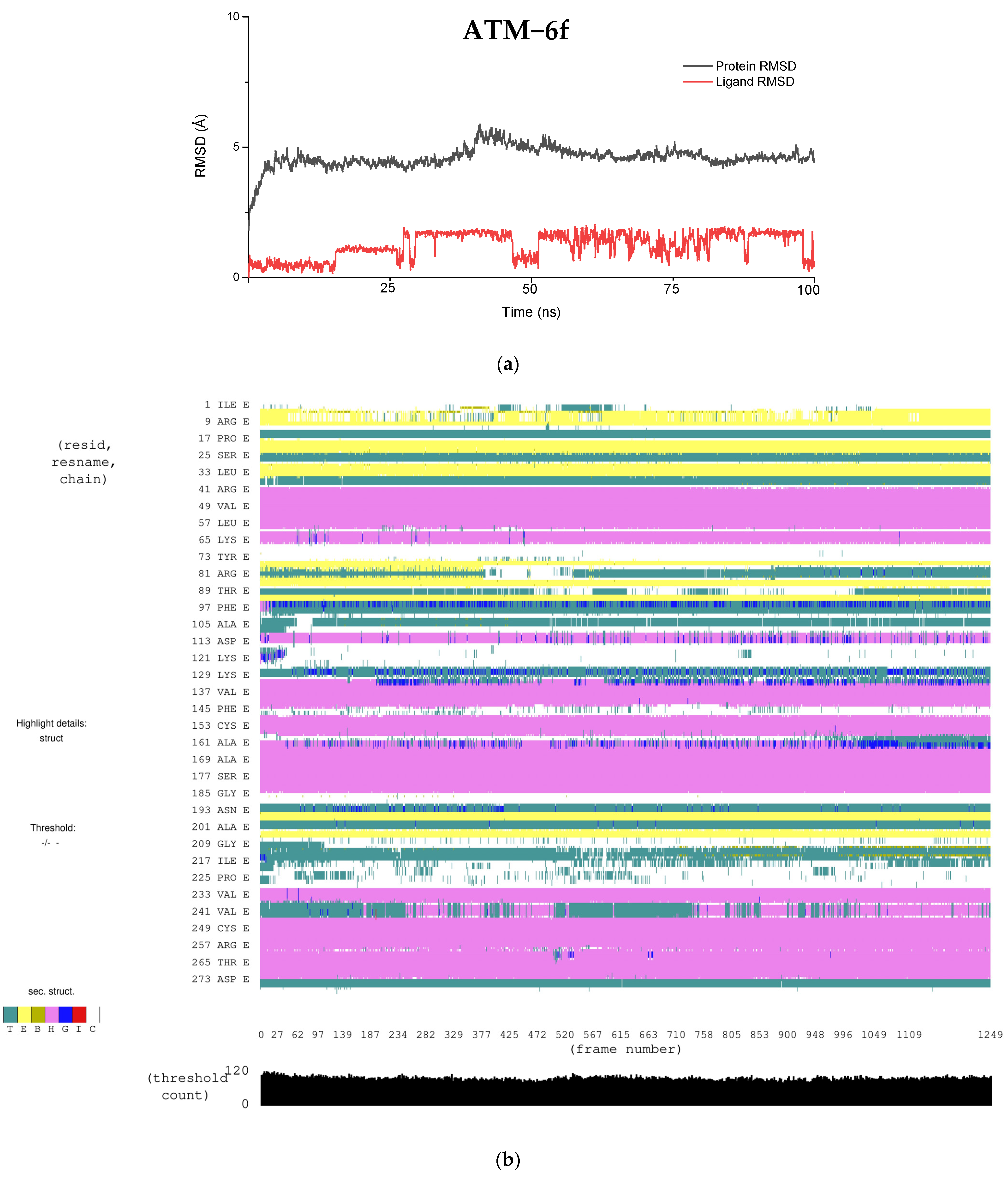
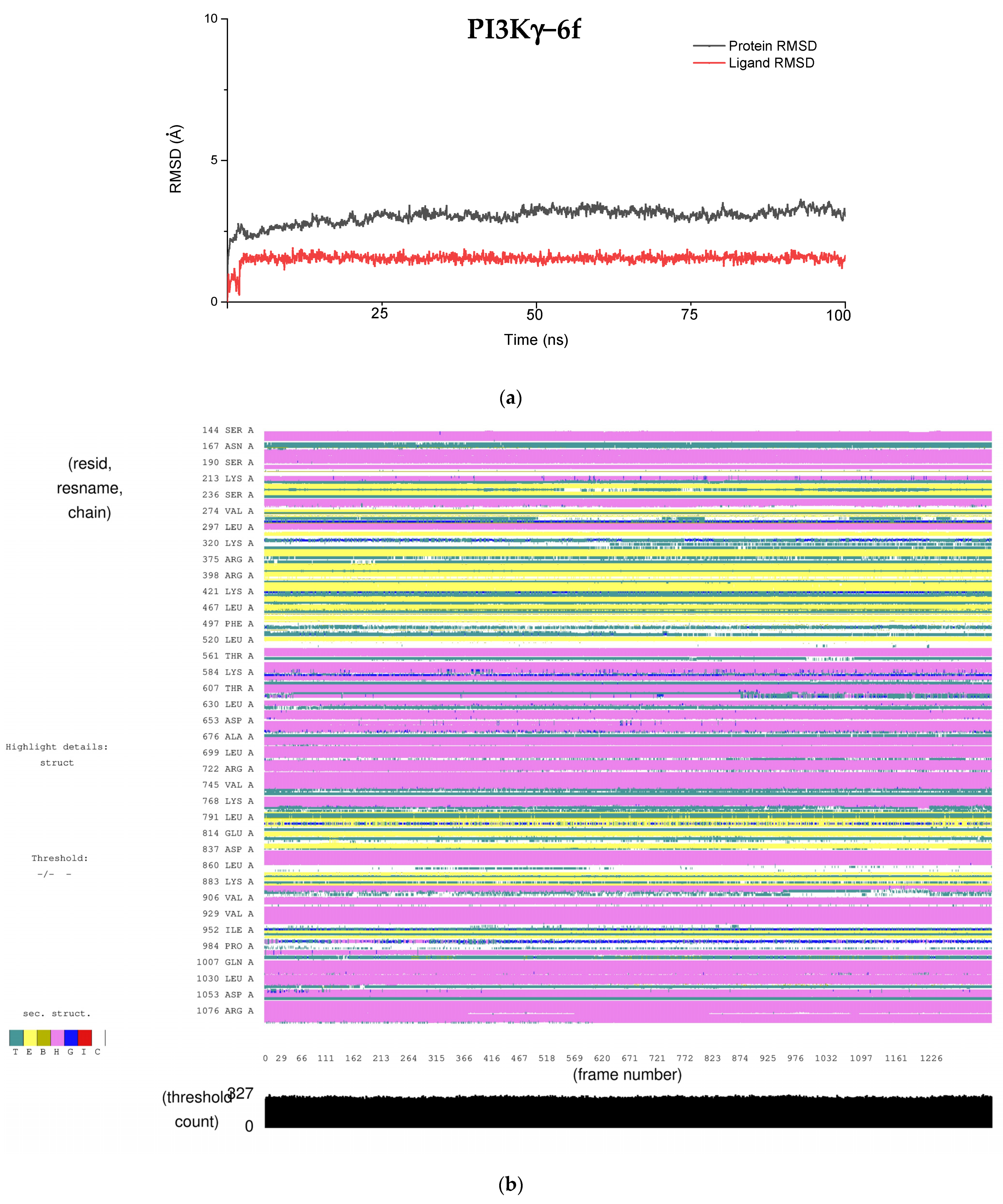
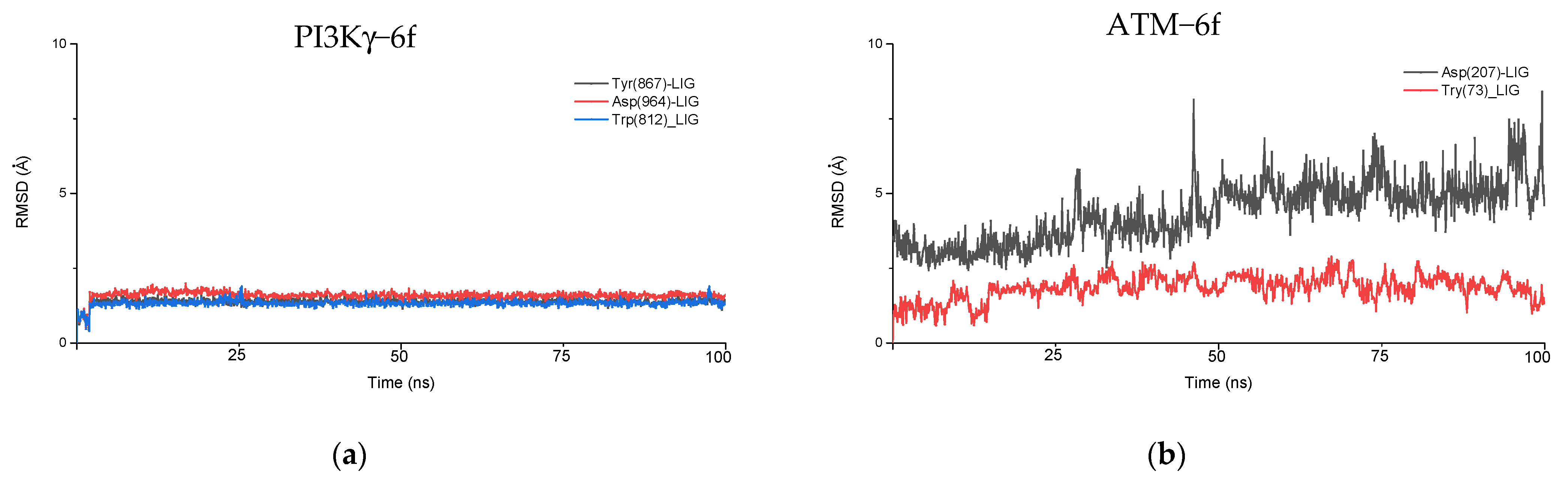
As discussed above, due to the large penalties resulting from intraligand contact, PI3Kγ shows a reduced binding affinity for the ligand, which results in a lower docking score. We analyzed the RMSD fluctuations of key binding site residues that interact with ligands to understand them further. In the PI3Kγ−6f complex, our docking study suggests that Trp(812), Tyr (867), and Asp(964) are the three key residues interacting with the ligand. The RMSD plot of these residues, along with ligand (6f), is shown in
Figure 6a. Interestingly, in all three cases, the RMSD remains constant at around 1.5 Å, implying a stronger intermolecular interaction between these residues and ligands. This observation is consistent with our docking results, where the rewards (stabilization energy) for PI3Kγ are nearly the same as those seen in ATM (
Table 6). However, such a shorter distance between active site residues and ligands may likely result in steric clashes between the atoms present, resulting in large penalties. Moreover, constant RMSD between key residues and ligands, with a magnitude of around 1.5 Å, exemplifies the rigidity of the PI3Kγ binding site. It is probably because of this rigidness that ligand molecules cannot undergo conformational interconversion, which was generally observed in the other four proteins.
In the ATM−6f complex, two interacting residues are Tyr (73) and Asp (207), forming π−π stacking and halogen bonding with the ligand, respectively. RMSD fluctuations for Tyr(73)−6f (ligand) and Asp(207)−6f(ligand) are shown in
Figure 6b. For Tyr(73)−6f (ligand), the RMSD value shows a variation in the range of 1.5−2 Å, indicating a strong π−π stacking which remains stable during the simulation. Meanwhile, in the case of Asp(207)−6f(ligand), the RMSD shows larger variation, implying the dynamic formation and breakage of weaker halogen bonds, probably due to conformational interconversion of the ligand. Overall, the RMSD fluctuations between these residues and the ligand suggest a flexibility of binding sites, large enough to allow for the free conformational interconversion of the ligand. It is due to the flexibility of the binding site; the penalties due to intraligand contacts are negligible in the ATM−6f complex.
Similarly, for the ATR−6f complex and the mTOR−6f complex, the critical binding site residue shows similar fluctuations in RMSD (
Figure S9), implying binding site flexibility. This observation is also consistent with the ligands’ conformational switching observed in all four proteins (except PI3Kγ, as discussed earlier). Furthermore, MSA analysis demonstrated that though the binding sites of all the proteins are confined to the kinase domain and its nearby region, the interacting residues were not a part of the conserved region. This factor can contribute to the differential selectivity of the inhibitors even though the nature of some of the residues were similar.
In summary, the docking affinity of 6f (ligand) with PIKK family kinases is determined by a subtle interplay of the natures of active site residues, stabilizing interactions, and penalties resulting from the rigidity/flexibility of the binding site.
6. Conclusions
One of the major problems in kinase drug discovery is the off−target kinase inhibition. Therefore, predicting the off−target binding of the inhibitors would help in fast−tracking the process of drug development. In this study, we analyzed the binding affinity of compound 6f against five different kinases sharing a reasonable sequence similarity. 6f was shown to bind selectively to ATM in comparison with the other proteins of interest. MD simulation showed that all of the protein−ligand complexes were stable and that secondary structure variations during the simulation were minimal. Interestingly, the trajectory analysis of the ligand (6f) in all cases suggested the ligand’s conformational interconversion (except for in the case of the PI3Kγ−6f complex). The conformational changes observed in the ligand were attributed to the free rotation of the C–N bond and the binding site flexibility of the proteins. In the case of the PI3Kγ−6f complex, the binding site was found to be very rigidly packed, thus prohibiting the free rotation of the C–N bond. Moreover, the binding site rigidity in the PI3Kγ−6f complex resulted in steric clashes between active site residues and ligands, thus yielding a higher penalty and a lower docking score. Finally, MSA analysis also aided us in understanding the residue conservation across the proteins and rationalizing the selectivity of the molecules towards ATM.
To conclude, the molecules synthesized have higher binding affinity and selectivity towards ATM compared to the other kinases of interest. The proteins share a reasonable sequence similarity in the kinase domain where the designed inhibitors are speculated to bind. The interacting residues do not fall in the conserved region, thereby resulting in selectivity towards ATM. This result can be considered as a preliminary investigation, which warrants further validation using in vitro assays.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}