
Spatial Contributions to 1H NMR Chemical Shifts of Free-Base Porphyrinoids


1
Institute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
2
Department of Chemistry, University of Helsinki, A.I. Virtanens Plats 1, P.O. Box 55, FI-00014 Helsinki, Finland
3
Chemistry of Materials, Paris-Lodron University of Salzburg, Jakob-Haringerstr. 2A, A-5020 Salzburg, Austria
*
Author to whom correspondence should be addressed.
Chemistry 2021, 3(3), 1005-1021; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030072
Submission received: 15 August 2021
/
Revised: 30 August 2021
/
Accepted: 1 September 2021
/
Published: 5 September 2021
(This article belongs to the Special Issue Current Density and Spectroscopy—A Themed Issue in Honor of Professor Riccardo Zanasi on the Occasion of His 70th Birthday)
Abstract
:A recently developed methodology for calculating, analyzing, and visualizing nuclear magnetic shielding densities is used for studying spatial contributions including ring-current contributions to H nuclear magnetic resonance (NMR) chemical shifts of aromatic and anti-aromatic free-base porphyrinoids. Our approach allows a visual inspection of the spatial origin of the positive (shielding) and negative (deshielding) contributions to the nuclear magnetic shielding constants. Diatropic and paratropic current-density fluxes yield both shielding and deshielding contributions implying that not merely the tropicity of the current density determines whether the contribution has a shielding or deshielding character. Instead the shielding or deshielding contribution is determined by the direction of the current-density flux with respect to the studied nucleus.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
This article is dedicated to Professor Riccardo Zanasi on the occasion of his 70th birthday. He graduated from the University of Modena, Italy in 1975, studying electric and magnetic properties of molecules. He is one of the pioneers in the field of current-density calculations with his first article from 1981 on the ring-current model of the cyclopropenyl cation [1]. The “continuous transformation of the origin of the current density (CTOCD) method” from 1994 [2], which Keith and Bader called “continuous set of gauge transformations (CSGT)”, was the starting point for the SYSMO and SYSMOIC program packages [3,4]. Professor Zanasi has published about 70 scientific articles on computational studies of magnetically induced current densities in molecules with “current density” in the title. He is the main developer of the SYSMO code. The topic of this article is closely related to the research interests of Professor Zanasi, since we have calculated and analyzed magnetic shielding densities (MSD) of free-base porphyrinoids. MSD is the spatial contribution to the chemical shifts in nuclear magnetic resonance (NMR) spectroscopy and calculated using the magnetically induced current density Figure S1.
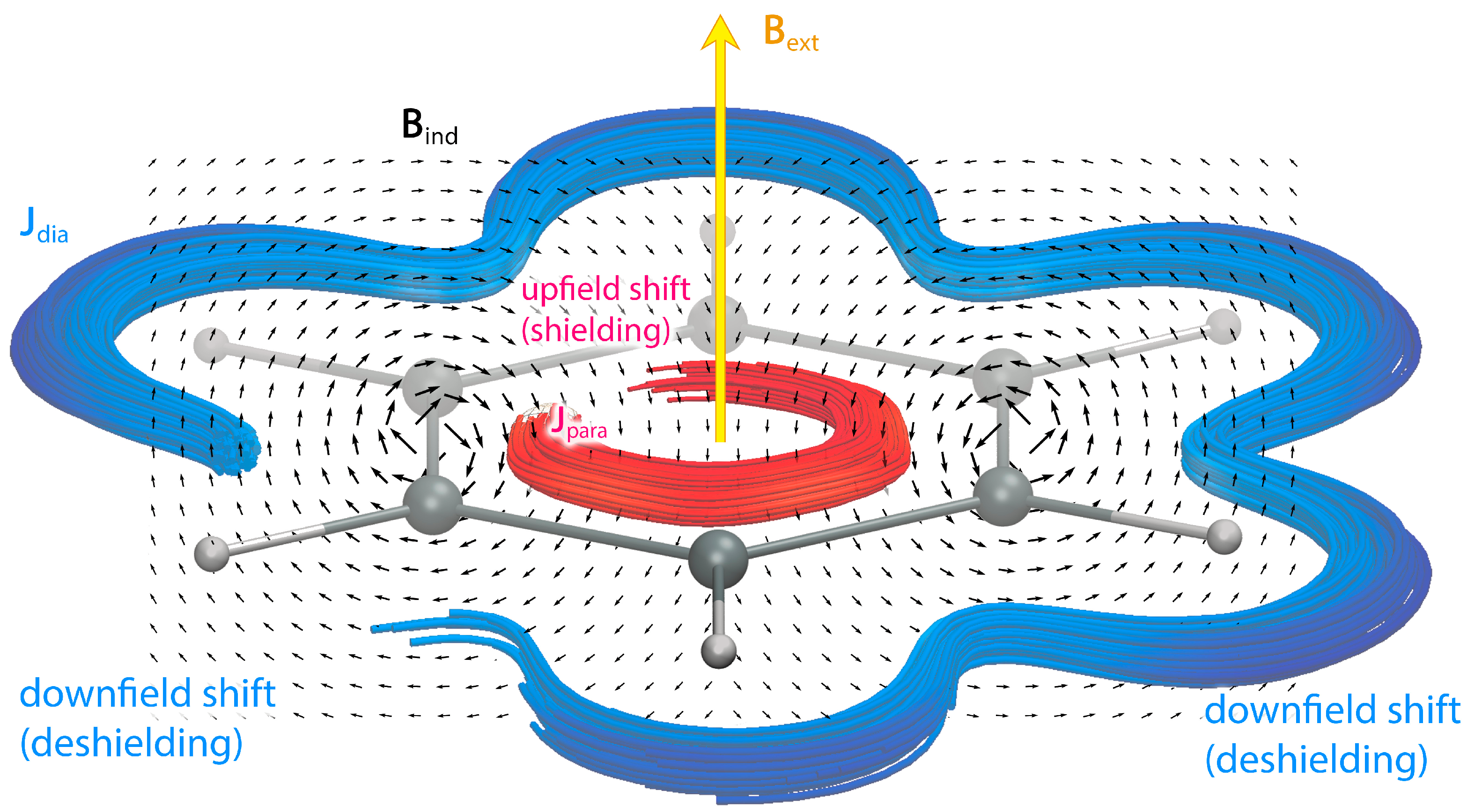
For the aromatic benzene molecule, it is well known that the magnetically induced ring current leads to a downfield shift of the H NMR signal, because the diatropic ring current induces a secondary magnetic field with field lines forming closed loops around the ring current. The field lines point in the opposite direction to the applied magnetic field inside the ring, while outside the ring they are parallel to the external field, see Figure 1. The hydrogen atoms of benzene outside the ring-current pathway are deshielded because the induced magnetic field enhances the applied one. However, a recent study of the MSD of benzene showed that the ring current leads to regions with significant shielding and deshielding contributions [5]. The ring-current contribution to the H NMR chemical shift of benzene can be estimated to be about 1.7 ppm from the experimental chemical shifts of benzene (5.6 ppm) and the vinylic hydrogen atoms of cyclohexene (7.3 ppm).
The origin of the H NMR chemical shifts is even more complex when studying aromatic or anti-aromatic molecules bearing hydrogen atoms inside the ring-current pathway. The aromatic free-base trans-porphyrin is an example of such molecules. The accepted notion is that the H NMR signal of the inner hydrogen is upfield shifted and the H NMR signals of the meso and hydrogen nuclei are downfield shifted because the direction of the induced magnetic field is in the opposite direction to the applied one inside the porphyrin ring and parallel to the external field outside the ring. The chemical shifts of the H NMR signals of anti-aromatic molecules are influenced in the opposite manner compared to aromatic ones because anti-aromatic molecules sustain strong paratropic ring currents. Measurements of the H NMR chemical shifts of the inner or outer hydrogen atoms of porphyrinoids are often used for assessing their aromatic character experimentally. Calculated H NMR chemical shifts can be used for estimating the direction and the strength of the magnetically induced ring current and for identifying porphyrinoid tautomers [6,7,8].
The ring-current model of aromatic and anti-aromatic molecules [9,10] with a current-density pathway forming a closed loop in the orbitals above and below the molecular ring might be a somewhat oversimplified explanation of the aromatic or anti-aromatic contribution to the H NMR chemical shifts. Since the vector potential of the nuclear magnetic moment declines rapidly with the distance from the nucleus, the largest contributions to the H NMR chemical shifts originate from the current density near the considered nucleus.
Instead of the well known nucleus-independent-chemical-shift (NICS) functions [11,12,13], we investigate the MSD calculated using the magnetically induced current density. The MSD yields detailed information about spatial contributions to the NMR chemical shifts. The MSD calculated using the Biot–Savart expression for NMR chemical shifts can be more useful for interpreting experimental NMR data than alternative approaches [14], since it is possible not only to quantify the amount of positive and negative contributions to the shielding constant of a specific nucleus but also to identify the spatial origin of the most relevant contributions.
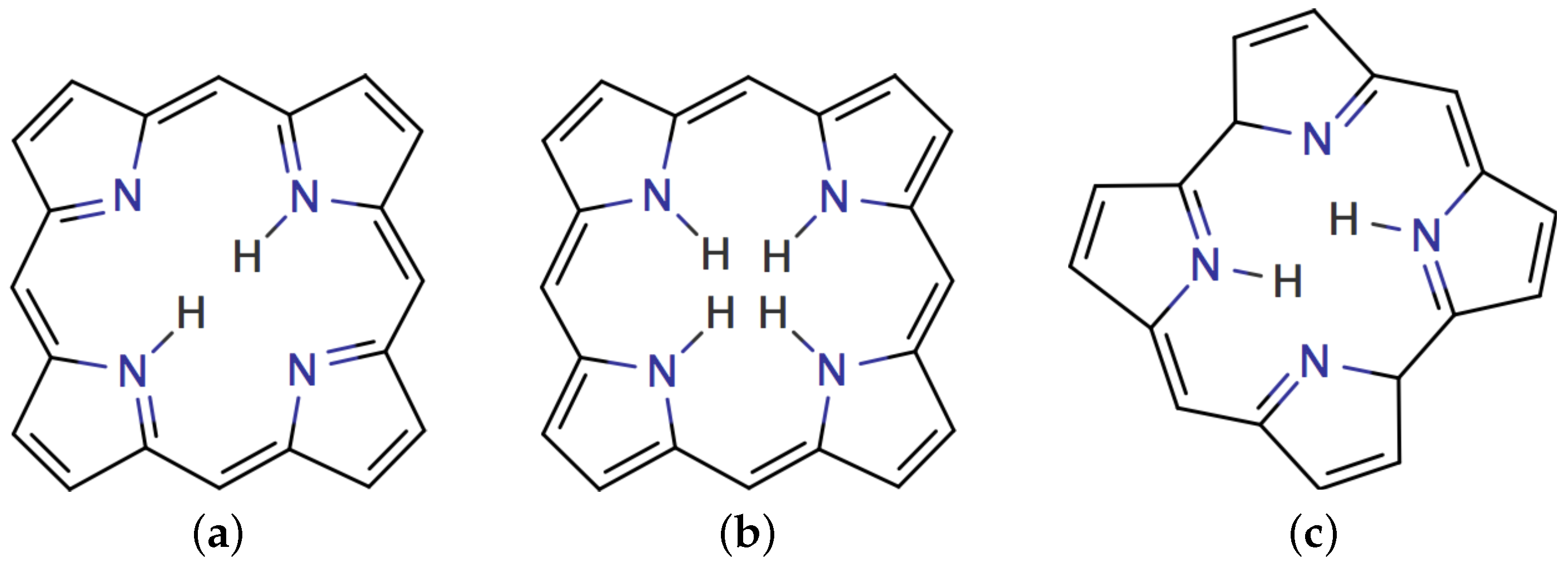
In this work, we apply our methodology for analyzing spatial contributions to the H NMR magnetic shieldings of inner and outer hydrogen atoms of free-base trans-porphyrin, free-base trans-isophlorin, and free-base trans-norcorrole (Figures S2 and S3). Free-base trans-porphyrin serves as the typical aromatic porphyrin. Free-base trans-isophlorin proposed by Woodward represents anti-aromatic porphyrins [15,16], whereas anti-aromatic norcorroles are the smallest tetrapyrrole porphyrinoids that have been synthesized [17,18,19]. The underlying theory is briefly described in Section 2. The computational levels are presented in Section 3.1 and the employed numerical methods for calculating the shielding density are described in Section 3.2. The obtained shielding densities are discussed in Section 4. Finally, the study is summarized and concluded in Section 5.
2. Theory
Nuclear magnetic shieldings are usually calculated using the gradient theory of electronic structure calculations as the mixed second derivative of the electronic energy with respect to the strength of the external magnetic field and the size of the nuclear magnetic moment [20,21,22,23,24]. The components of the nuclear magnetic shielding tensor, , in the and Cartesian directions can also be calculated as the second derivative of the magnetic interaction energy expressed as a spatial integral over the scalar product of the magnetically induced current-density susceptibility () due to the external magnetic field () and the first derivative of the vector potential of the nuclear magnetic moment () with respect to the size of the nuclear magnetic moment ( calculated at vanishing and [14,25,26,27],
The vector potential of the nuclear magnetic dipole moment in international system of units (SI) is given by
where is the position of nucleus I and is the vacuum permeability [28]. This scheme is implemented in the gauge-including magnetically induced currents (Gimic) method [29,30,31,32,33,34] for calculating the current-density susceptibility tensor (CDT) [35,36] as described in Section 3.2. Similarly, the nuclear magnetic shielding tensor can be calculated from the current density induced by the nuclear magnetic moment multiplied with the vector potential of the external magnetic field [14,36,37,38]. This approach has been employed in other studies [39,40], however it has not been considered in this work.
Even though the NMR shieldings obtained with the gradient theory and the integration approaches are the same, the integration method can provide information about orbital and spatial contributions to a given NMR chemical shift [36,37,41]. When a numerical representation of the current density is gauge-origin independent, the calculated NMR chemical shifts obtained with the integration approach are also independent of the gauge origin. We use the Gimic method [29,30,31,32] for calculating current densities, however, the CTOCD approach also leads to gauge-origin independence [2,4,42,43,44]. The spatial distribution of the shielding density provides detailed information about the origin of the individual elements of the nuclear magnetic shielding tensor, as well as the shielding constants [5,44,45,46,47,48,49,50]. Dividing the magnetic shielding density into positive and negative parts yields the spatial origins of the shielding and deshielding contributions to the shielding tensor and the isotropic shielding constants, providing a rigorous physical basis for interpreting NMR chemical shifts.
3. Computational Methods
3.1. Electronic and Molecular Structure Calculations
The molecular structures of free-base trans-porphyrin, free-base isophlorin, free-base trans-norcorrole were optimized with Turbomole [51] version 7.5 employing the B3LYP density functional [52,53,54], the def2-TZVP basis set using the resolution of the identity (RI) approximation [55,56], and the m5 quadrature grid [57]. The molecular structure of isophlorin was assumed to be planar. The planar isophlorin structure is not a minimum on the potential energy surface but a saddle point with several imaginary vibrational frequencies due to out-of-plane motions of the inner hydrogens. The optimized molecular structures are shown in Figure 2 and the Cartesian coordinates are given in the electronic supporting information (ESI). The NMR shielding constants were calculated with Turbomole at the BHandHLYP (LIBXC ID 436) [53,58,59] level of theory using gauge-including atomic orbitals (GIAO) [20,60,61,62]. The BHandHLYP functional that has 50% Hartree–Fock exchange yielded accurate magnetizabilities in a recent benchmark study [41]. For all NMR calculations the pcseg-3 basis set has been employed which has been optimized for the calculation of NMR parameters [63]. The pcseg-3 basis sets have been obtained from the EMSL basis set exchange library [64,65,66]. For the visualizations, the smaller def2-TZVP basis set has been employed which was tested to be sufficient.
3.2. Magnetic Shielding-Density Calculations
The use of finite one-particle basis sets in the calculation of CDT introduces a gauge dependence even though the exact solution of the Schrödinger equation is gauge invariant. However, the calculated CDT can be made gauge-origin independent by using GIAOs. GIAOs also lead to a fast basis-set convergence of the current density and magnetic properties, because GIAOs are correct to first order in the magnetic response for any choice of the gauge origin, whereas ordinary basis functions are correct only to zeroth order [67]. The GIAOs are given by [20,60]
where is the imaginary unit and is a standard Gaussian-type basis function centered at . The gauge origin is eliminated from the CDT expression when using GIAOs [29,31,32]. The CDT expression of the Gimic method is
where is the density matrix and are the magnetically perturbed density matrices in the atomic-orbital basis, is the Levi–Civita symbol with . denotes that the singular denominator appearing in all terms is omitted. The non-singular magnetic interaction operators are
and
where is the position of nucleus I. All terms containing the gauge origin cancel in Equation (4), making the CDT and the Biot–Savart expression independent of the gauge origin. All nuclear-position terms in Equation (4) also cancel, eliminating the coordinates of the nucleus I from the CDT expression. The current density depends implicitly on the nuclear positions, since the basis functions are located at the nuclei.
The Biot–Savart expression for the nuclear magnetic shielding tensor of nucleus I, , is then
where is the vacuum permeability and . The integrand in Equation (7) is the magnetic shielding density (MSD) containing spatial contributions to the nuclear magnetic shielding of nucleus I [5,14,25,26,44,46,48,49,50]. Visualization of the MSD yields information about the spatial origin of the NMR chemical shift of nucleus I. NMR chemical shifts can be interpreted by plotting the positive and negative parts of the integrand separately. The visualization provides information about shielding and deshielding contributions originating from the relative direction of the current density with respect to the investigated atom I [5,32,68]. Contributions to NMR shieldings can be calculated by integrating the Biot–Savart expression in Equation (7) numerically using the CDT calculated in the integration points. We have implemented a numerical integration scheme into the Gimic program for calculating spatial contributions to nuclear magnetic shieldings and magnetizabilities [5,34,41]. Atomic contributions can be obtained by integrating atomic domains generated by the Numgrid library [69] using Becke’s multicenter scheme [70]. The atomic domains are constructed using the Becke partitioning scheme with the iteration order in the construction of the cut-off function [70]. Angular integration of the atom-centered domains is performed with Lebedev’s angular grid [71] and the radial integration grid is constructed as suggested by Lindh et al. [72]. The density matrix, the magnetically perturbed density matrices and the basis-set data are obtained from Turbomole [51] calculations of NMR shielding constants.
4. Results and Discussion
4.1. Free-Base trans-Porphyrin
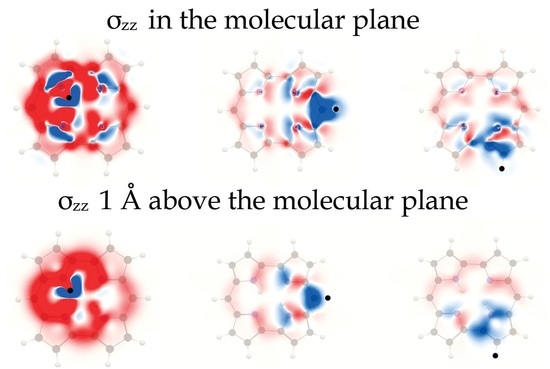
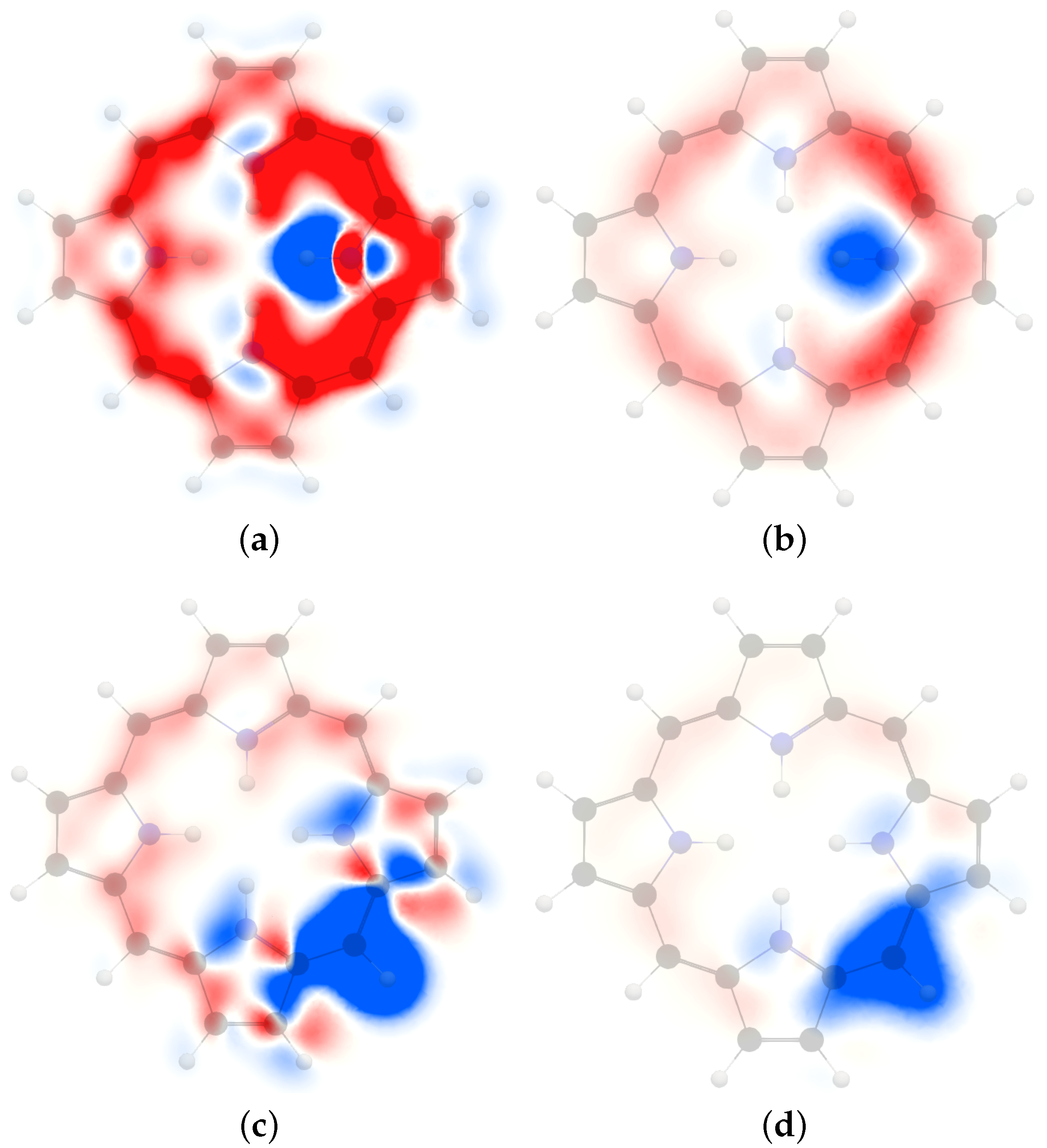
We have analyzed the ring-current contribution to the H NMR shielding constants by plotting the spatial distribution of its isotropic shielding constant (). The H NMR shielding density of trans-porphyrin in Figure 3 shows that the inner hydrogen atoms have shielding contributions in the molecular plane from the ring current along the outer edge of the molecule [73]. However, a stronger shielding contribution originates from the innermost pathway via the four pyrrole nitrogen atoms. The largest shielding contribution appears near the studied hydrogen, because of the singular denominator of the vector potential of the magnetic moment of the inner hydrogen. The nitrogen atom also sustains a local diatropic current-density vortex extending around the N–H bond. The paratropic ring current inside the porphyrin ring and the diatropic current-density vortex of the nitrogen of the adjacent pyrrole rings cause deshielding. A deshielding area is also seen at the other inner hydrogen where the paratropic ring-current flux inside the porphyrin ring is closest to the studied inner hydrogen. The MSD of of the inner hydrogen is seen 1 Å above the molecular plane in Figure 3. The effect of the ring current is weaker there than in the molecular plane. The deshielding contributions are also very small at a distance of 1 Å from the molecular plane because the paratropic ring current vanishes there.
The MSD of the meso hydrogen in the molecular plane shown in Figure 3 reveal a strong shielding contribution near the hydrogen. A deshielding contribution caused by the diatropic ring current flux in the vicinity of the ipso carbon and the nearest carbon atoms of the adjacent pyrrole rings. The more distant part of the diatropic ring current shields the nucleus of the meso hydrogen. The paratropic ring current inside the porphyrin results in a shielding contribution inside the meso carbon. One can also see alternating shielding and deshielding contributions from the paratropic ring current inside the porphyrin ring due to its relative direction with respect to the nucleus of the studied meso hydrogen. The MSD of the meso hydrogen 1 Å above the molecular plane shown in Figure 3 has a similar pattern as in the molecular plane. However, the contributions away from the molecular plane are much smaller.
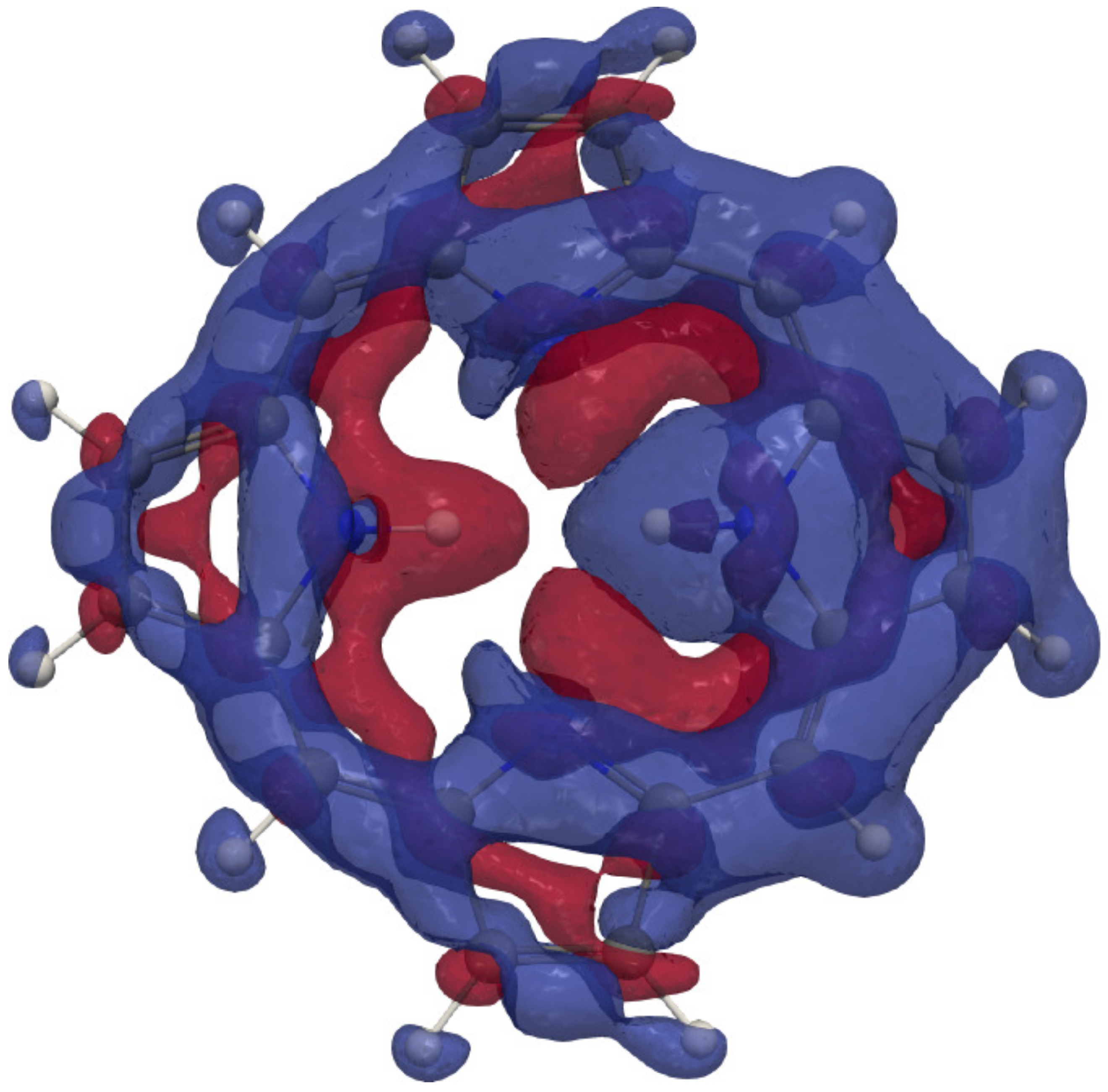
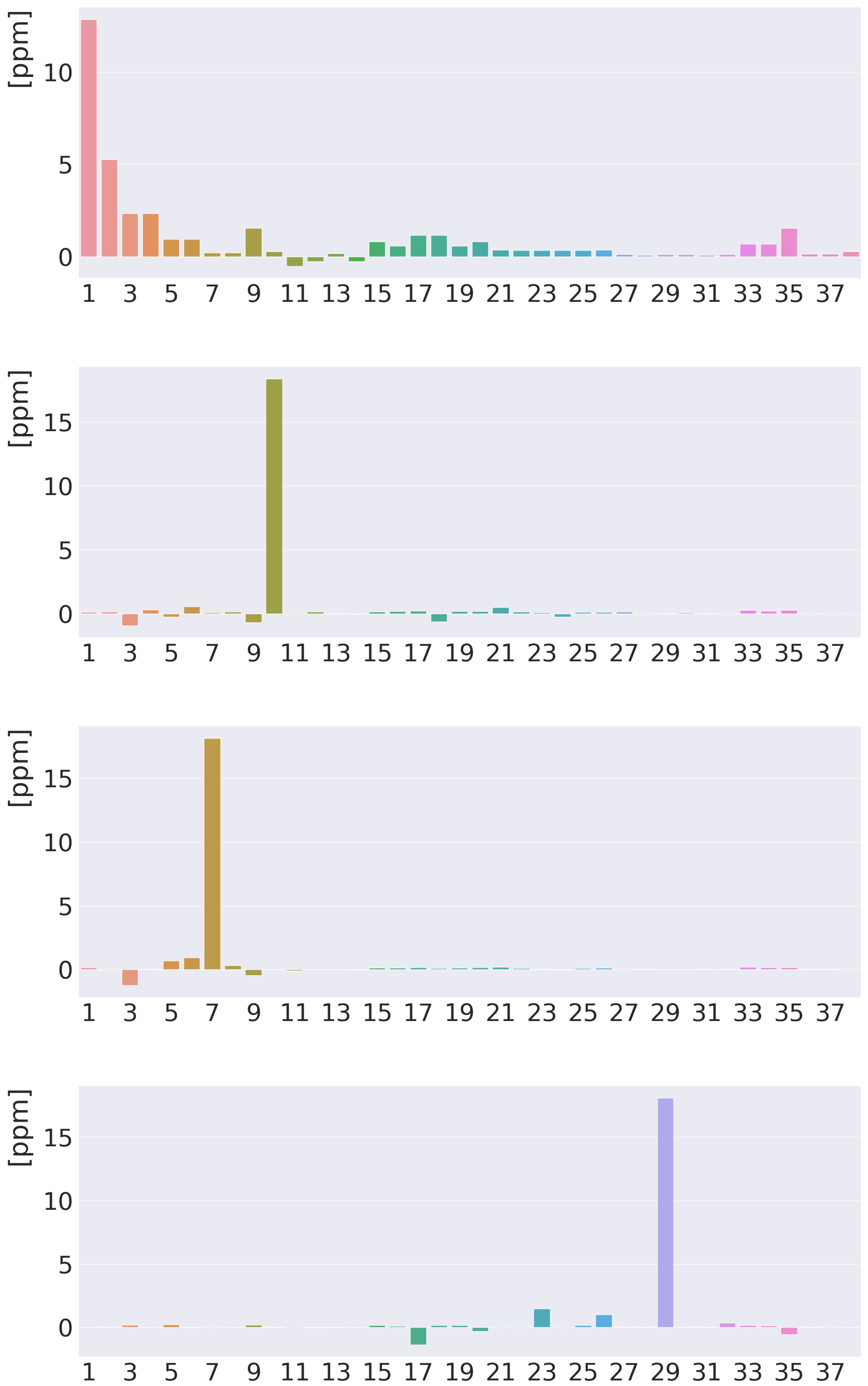
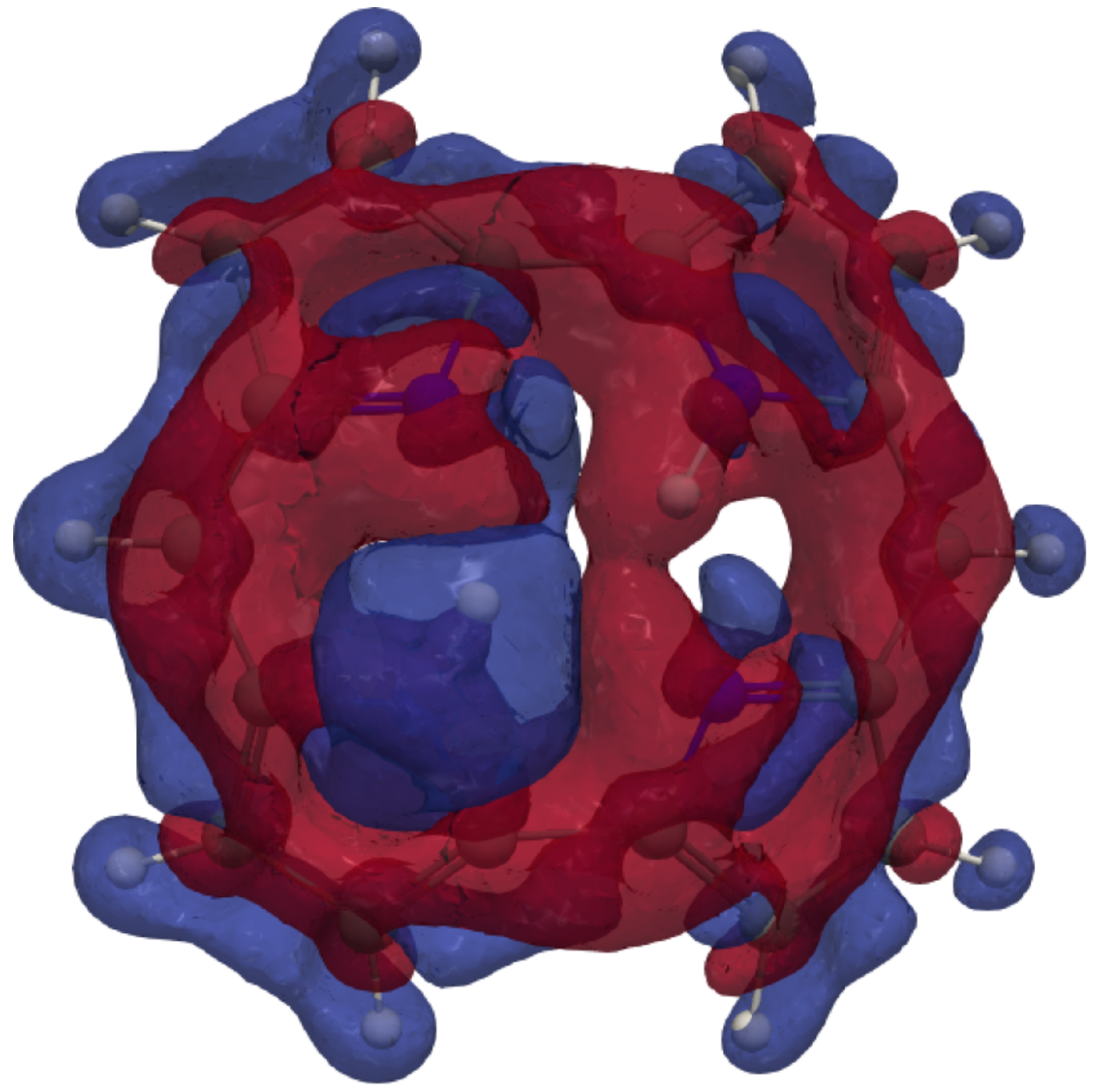
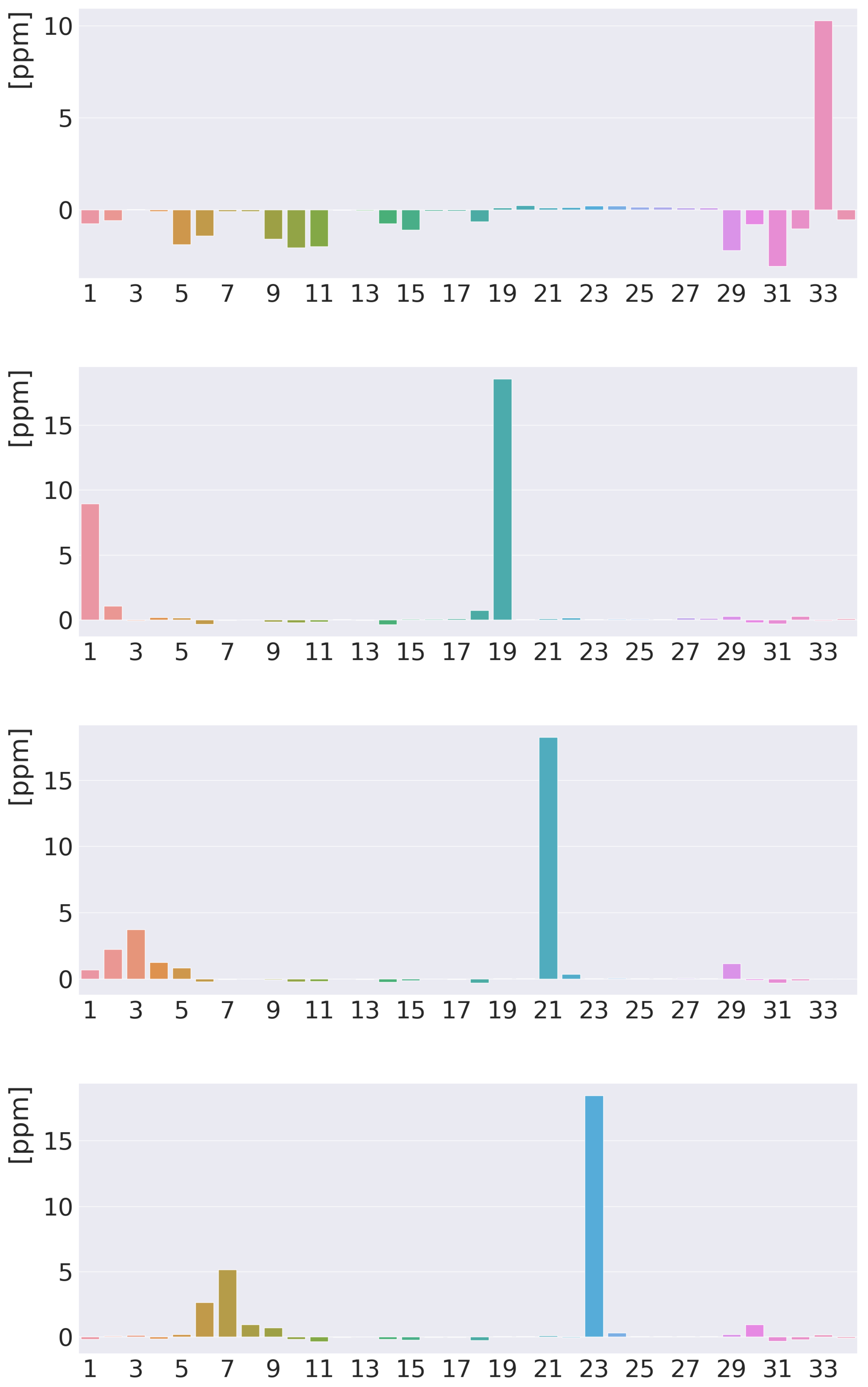
The 3D picture of the MSD of the inner hydrogen in Figure 4 shows the shielding contribution from the diatropic ring current along the outer edge of the molecule and the deshielding contribution from the paratropic ring current inside the porphyrin ring. Lots of atoms contribute significantly to the isotropic shielding constant of the inner hydrogen of 37.52 ppm, because the distances from it to many of the other atoms are relatively short. The local contribution from the inner H is only 12.85 ppm. The contributions from N, C of the same pyrrole ring, the nearest C, and the nearest C atoms of the adjacent pyrrole rings are 5.28, 2.35, 1.53, and 1.16 ppm, respectively. The rest of the atomic contributions to of the inner hydrogen is in the range of ppm. Contributions from each atom to the of the hydrogen atoms are reported in the ESI and summarized in Figure 5. The 3D pictures of the MSD of the meso and hydrogen atoms are reported in the ESI.
4.2. Free-Base Isophlorin
The spatial distribution of the isotropic MSD of free-base isophlorin in Figure 6 shows that the inner hydrogen atoms and the meso hydrogen atoms have deshielding contributions from the paratropic ring current in the molecular plane along the outer edge of the molecule. The shielding contributions originate mainly from the current density in the vicinity of the studied inner hydrogen.
The 3D contour of the ring-current contribution to of the inner hydrogen is shown in Figure 7. The ring current following the inner route is stronger than the one via the carbons which means that its contribution to the shielding density is bigger. Furthermore, the vector potential of the nuclear magnetic moment, declines rapidly with increasing distance from the nucleus I.
Dividing the MSD of the inner hydrogen into atomic domains that are integrated separately shows that many atoms contribute significantly to its value of −6.28 ppm. Since the distances from the inner hydrogen to adjacent atoms are short, the vector potential of the nuclear magnetic moment of the inner hydrogen has a significant amplitude at the atomic domains of the neighboring atoms leading to large atomic contributions to of the inner hydrogen.
The local contribution from the atomic domain of the inner hydrogen is 16.46 ppm, whereas contributions from adjacent atoms are negative. The contributions from the adjacent and trans inner hydrogen atoms are −0.79 ppm and −0.57 ppm, respectively. The contributions from N, C of the same pyrrole ring, the nearest C, and the C atoms of the neighboring pyrrole ring are 0.96, −2.18, −2.38, and −1.85 ppm, respectively. The rest of the atomic contributions are in the range of ppm. Contributions from each atom to the of the inner hydrogen are summarized in Figure 8 and the atomic contributions from all atoms to of the inner hydrogen are reported in the ESI.
The paratropic ring current along the outer edge of the molecule in the molecular plane causes deshielding of the meso hydrogen, which has also shielding and deshielding contributions along the inner ring-current pathway at the adjacent pyrrole rings due to the relative direction of the ring current with respect to the studied nucleus. The MSD of the meso hydrogen in Figure 6 has shielding contributions near the hydrogen and the ipso carbon originating from the paratropic ring current inside the hydrogen and from the diatropic current-density flux outside the hydrogen. The MSD contributions at the molecular plane and 1 Å away from it has a similar shape but is weaker as the distance from the molecular plane increases.
Calculations of the atomic contributions to the MSD show that the shielding domain of the meso hydrogen atoms includes the nearest carbons. Since the distances from the meso hydrogen to other atoms are longer than for the inner hydrogen, the contributions from the adjacent atoms are somewhat smaller than for the carbons.
The largest atomic contributions to of the meso hydrogen are 18.80, 12.40, 2.70, and 1.66 ppm from the meso hydrogen, C, and from the two closest C atoms. The largest atomic contributions to of the hydrogen are 18.48, 7.06, 2.94, and 1.23 ppm from the studied hydrogen, C, the adjacent C and C, respectively. Contributions from each atom to the of the hydrogen atoms are summarized in Figure 8. The atomic contributions to of the meso and hydrogen atoms from all atoms are reported in the ESI. The 3D pictures of the MSD of the meso and hydrogen atoms are reported in the ESI.
4.3. Free-Base trans-Norcorrole
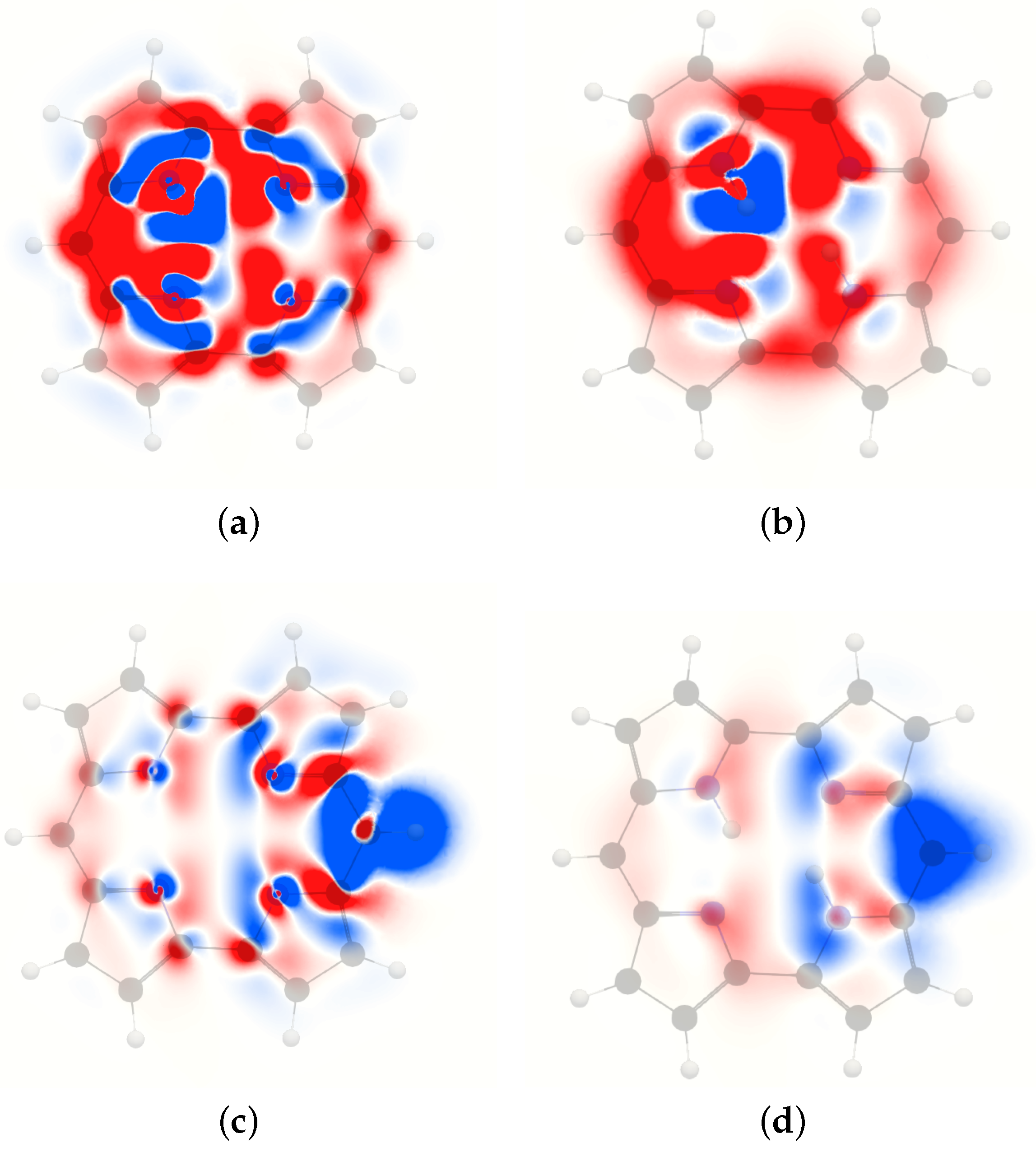
The MSD of the inner hydrogen of free-base trans-norcorrole in Figure 9 is dominated by deshielding contributions around the outer edge of the molecule that originate from the strong paratropic ring current [74,75,76]. The ring-current contribution is strong in the molecular plane as well as in a plane 1 Å away from it, where the deshielding contributions completely dominate. Diatropic contributions appear in the molecular plane between the carbons and the nitrogen atoms inside the pyrrole rings due to the local diatropic current-density flux around the nitrogen moiety, which leads to a large shielding domain near the studied inner hydrogen that extends to the plane 1 Å away from the molecular plane.
The 3D contour of the MSD of the inner hydrogen in Figure 10 shows a completely dominating deshielding contribution originating from the paratropic ring current. The diatropic ring current along the outer perimeter of free-base trans-norcorrole results in shielding contributions that are larger in the vicinity of the inner hydrogen than on the remote side of the molecule because the magnetic vector potential declines rapidly at longer distances. The significant shielding domain appears near the studied inner hydrogen.
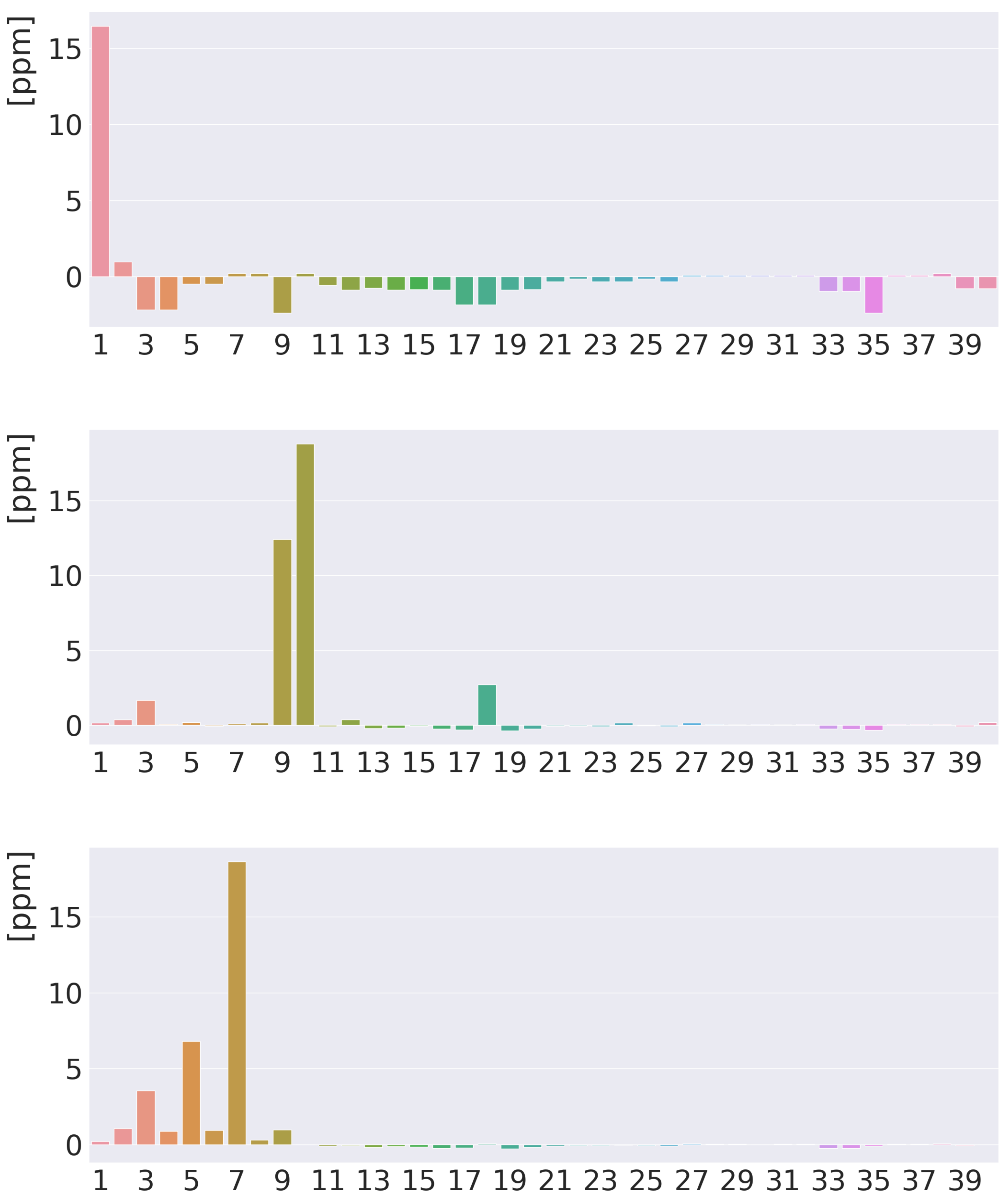
Calculations of MSD contributions from atomic domains show that many atoms contribute to of the inner hydrogen due to the short distances between it and the atoms inside norcorrole. The atomic contribution of 10.28 ppm from the studied inner hydrogen is shielding, whereas the atomic contributions from atoms in the vicinity are deshielding. The contributions from the C atoms are in the range of −1.09 to −1.99 ppm. The contribution from the nearest meso carbon is −2.04 ppm. The contributions from the adjacent and trans nitrogen atoms are −2.19 and −1.01 ppm, respectively. The contribution from the ipso nitrogen is only −0.78 ppm. The small negative contributions from many atoms leads to a negative of −8.73 ppm even though the local contribution is of about the same size but positive.
The MSD of the meso hydrogen consists of alternating shielding and deshielding contributions in the molecular plane, as well as in the plane 1 Å away from it. The main shielding areas appear in the vicinity of the studied meso hydrogen originating from the paratropic ring current inside it and the diatropic current density outside the meso hydrogen. The current density around the nitrogen moieties leads to alternating shielding and deshielding contributions, whereas the typical ring-current contributions from other parts of the molecule are not very pronounced. The main atomic contributions to the meso hydrogen are 18.56, 8.93, and 1.06 ppm from the meso hydrogen, C and one of the nearest C atoms, respectively. The atomic shielding contributions are localized to the nearest atoms because other atoms are very far away from the meso hydrogen, implying that the amplitude of the vector potential of its nuclear magnetic moment vanishes at atoms on the other side of the porphyrinoid ring.
The magnetic shieldings of the hydrogen atoms are in the range of 26.89 to 28.16 ppm. Since the differences are small, we only discuss one type of hydrogen atoms. The value of atom 21, which is one of the hydrogen atoms of a pyrrole ring without an inner hydrogen, has a local contribution of 18.24 ppm. The largest atomic contributions from adjacent atoms are 3.73, 2.23, 1.25, and 1.16 ppm from C, nearest C, nearest C that is not C and the nitrogen of the same pyrrole ring, respectively. Only the nearest atoms contribute to the of the hydrogen atoms due to the long distances to many of the other atoms.
Contributions from each atom to the of the hydrogen atoms are summarized in Figure 11. The atomic contributions to the values from all atoms are reported in the ESI. The 3D pictures of the MSD of the meso and hydrogen atoms are reported in the ESI.
5. Summary and Conclusions
In the present studies on free-base trans-porphyrin, free-base isophlorin, and free-base trans-norcorrole, we have employed a method to calculate and visualize nuclear magnetic shielding densities (MSD) that was recently implemented in the Gimic program. The MSD of the isotropic H NMR shielding constants has been visualized in the molecular plane and in a plane 1 Å away from it. We also presented 3D pictures of the MSD showing the spatial origin of magnetic shielding and deshielding contributions. We have divided the molecules into atomic domains and determined atomic contributions to the isotropic H NMR shielding constants by numerically integrating each domain separately.
Visualization of the MSD in the two planes shows that the main shielding contributions to the isotropic H NMR shielding constant of the inner hydrogen of free-base trans-porphyrin originate from the diatropic ring current along the inner pathway, whereas the contributions from the outer one is smaller due to the longer distance between the inner hydrogen and the atoms along the edge of the molecule. The MSD contribution fades out as one goes away from the molecular plane. The current density in the vicinity of the studied inner hydrogen also contributes significantly to its shielding constant. The MSD of the meso hydrogen of free-base trans-porphyrin has shielding contributions from the ring current along the inner pathway and on the outside of the meso hydrogen. The diatropic ring current inside the meso hydrogen deshields its nuclear magnetic moment. Many atomic domains contribute to isotropic H NMR shielding constant of the inner hydrogen, whereas the contributions to the shielding constants of the meso and hydrogen atoms atoms are dominated by contributions from the local atomic domains.
The MSD of the inner hydrogen of free-base isophlorin has large deshielding contributions from the paratropic ring current and the shielding domain appears mainly in the vicinity of the studied inner hydrogen. The contributions to MSD are stronger in the molecular plane than in the plane 1 Å away from it. The 3D picture of the MSD shows that the contribution from the inner pathway is larger than for the inner one at the remote pyrrole ring, whereas at the adjacent pyrrole rings the deshielding contribution mainly appears along the outer pathway. The meso hydrogen has deshielding contributions from the remote part of the paratropic ring current, whereas its contribution is shielding near the meso hydrogen. The current density on the outside of the meso hydrogen also shields its nuclear magnetic moment. Many atomic domains deshield the nuclear magnetic moment of the inner hydrogen, whereas the local contribution is shielding. The shielding contributions to the shielding constants of the meso and hydrogen atoms are dominated by contributions from the atomic domains near the studied nucleus, whereas the deshielding contributions are, in comparison, very small.
Free-base trans-norcorrole sustains a paratropic ring current that leads to strong deshielding contributions to the MSD of the inner hydrogen. The deshielding contributions are strong in the molecular plane and in a plane 1 Å away from it. The local diatropic current density around the nitrogen moieties leads to shielding contributions that are largest at the studied inner hydrogen. The 3D picture of the MSD of the inner hydrogen shows that the molecule is dominated by deshielding contributions. Shielding contributions appear at the outer edge of the molecular ring and near the studied inner hydrogen. Integration of atomic domains shows that the largest shielding contribution appear locally, whereas the deshielding contributions from many atomic domains are almost twice as large, yielding a net deshielding contribution to the shielding constant of the inner hydrogen. The MSD of the meso hydrogen is dominated by local shielding contributions, whereas the remote part of the paratropic ring current deshields it. The local diatropic current density around the nitrogen moieties yields alternating shielding and deshielding contributions depending on the relative direction of the current-density flux with respect to the nucleus of the meso hydrogen. The shielding contributions from the atomic domains near the meso and hydrogen atoms dominate their MSDs, whereas they have very few and small deshielding atomic contributions.
The present study shows the power of the present approach that can be used for determining spatial contributions to H NMR magnetic shieldings. The shielding constants can be assigned to atoms by numerically integrating the MSD in atomic domains. The calculations yield the magnetically induced ring current contributions to the shielding constants of the the inner and outer hydrogen atoms of typical aromatic and anti-aromatic porphyrinoids. Here, we have focused on H NMR shielding constants. However, a similar analysis can also be performed for C NMR and N NMR shielding constants.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/chemistry3030072/s1, electronic supporting information (ESI) available: the optimized molecular structures for free-base trans-porphyrin, free-base isophlorin and free-base trans-norcorrole. The atomic contributions to the isotropic H NMR shielding constants and 3D pictures of the are magnetic shielding densities of the isotropic H NMR shielding constants are also reported.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
Funded by the Academy of Finland, the Finnish Cultural Foundation, the Swedish Cultural Foundation in Finland, and Magnus Ehrnrooth Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The work has been supported by the Academy of Finland through project number 314821, by the Finnish Cultural Foundation, the Swedish Cultural Foundation in Finland, and by Magnus Ehrnrooth Foundation. We acknowledge computational resources from CSC—IT Center for Science, Finland and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lazzeretti, P.; Zanasi, R. Inconsistency of the ring-current model for the cyclopropenyl cation. Chem. Phys. Lett. 1981, 80, 533–536. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Malagoli, M.; Zanasi, R. Computational Approach to Molecular Magnetic Properties by Continuous Transformation of the Origin of the Current Density. Chem. Phys. Lett. 1994, 220, 299–304. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Malagoli, M.; Zanasi, R. SYSMO Package; Technical Report on Project ”Sistemi Informatici e Calcolo, Parallelo”; Research Report, 1/67; Additional routines for the evaluation and plotting of current densities by E. Steiner, P.W. Fowler, R.W.A. Havenith, A. Soncini; University of Exeter: Exeter, UK; University of Modena: Modena, Italy; University of Salerno: Rome, Italy, 1991. [Google Scholar]
- Monaco, G.; Summa, F.F.; Zanasi, R. Program Package for the Calculation of Origin-Independent Electron Current Density and Derived Magnetic Properties in Molecular Systems. J. Chem. Inf. Model. 2021, 61, 270–283. [Google Scholar] [CrossRef]
- Jinger, R.K.; Fliegl, H.; Bast, R.; Dimitrova, M.; Lehtola, S.; Sundholm, D. Spatial Contributions to Nuclear Magnetic Shieldings. J. Phys. Chem. A 2021, 125, 1778–1786. [Google Scholar] [CrossRef]
- Jusélius, J.; Sundholm, D. The aromatic pathways of porphins, chlorins and bacteriochlorins. Phys. Chem. Chem. Phys. 2000, 2, 2145–2151. [Google Scholar] [CrossRef] [Green Version]
- Fliegl, H.; Pichierri, F.; Sundholm, D. Antiaromatic Character of 16 π Electron Octaethylporphyrins: Magnetically Induced Ring Currents from DFT-GIMIC Calculations. J. Phys. Chem. A 2015, 119, 2344–2350. [Google Scholar] [CrossRef]
- Valiev, R.R.; Fliegl, H.; Sundholm, D. The aromatic character of thienopyrrole-modified 20π-electronporphyrinoids. Phys. Chem. Chem. Phys. 2014, 16, 11010–11016. [Google Scholar] [CrossRef]
- Pople, J.A. Molecular orbital theory of aromatic ring currents. Mol. Phys. 1958, 1, 175–180. [Google Scholar] [CrossRef]
- McWeeny, R. Ring currents and proton magnetic resonance in aromatic molecules. Mol. Phys. 1958, 1, 311–321. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Klod, S.; Kleinpeter, E. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes-application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2001, 2, 1893–1898. [Google Scholar]
- Merino, G.; Heine, T.; Seifert, G. The Induced Magnetic Field in Cyclic Molecules. Chem. Eur. J. 2004, 10, 4367–4371. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.M.; Pitzer, R.M.; Lipscomb, W.N. Perturbed Hartree-Fock Calculations. I. Magnetic Susceptibility and Shielding in the LiH Molecule. J. Chem. Phys. 1963, 38, 550–560. [Google Scholar] [CrossRef]
- Woodward, R.B. Totalsynthese des chlorophylls. Angew. Chem. Int. Ed. 1960, 72, 651–662. [Google Scholar] [CrossRef]
- Reddy, B.K.; Basavarajappa, A.; Ambhore, M.D.; Anand, V.G. Isophlorinoids: The Antiaromatic Congeners of Porphyrinoids. Chem. Rev. 2017, 117, 3420–3443. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Wasbotten, I.H.; Davis, W.; Swarts, J.C. Norcorrole and Dihydronorcorrole: A Predictive Quantum Chemical Study. Eur. J. Inorg. Chem. 2005, 2005, 4479–4485. [Google Scholar] [CrossRef]
- Bröring, M.; Köhler, S.; Kleeberg, C. Norcorrole: Observation of the Smallest Porphyrin Variant with a N4 Core. Angew. Chem. Int. Ed. 2008, 47, 5658–5660. [Google Scholar] [CrossRef]
- Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.Y.; Kobayashi, N.; Shinokubo, H. Gram-Scale Synthesis of Nickel(II) Norcorrole: The Smallest Antiaromatic Porphyrinoid. Angew. Chem. Int. Ed. 2012, 51, 8542–8545. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Helgaker, T.; Jaszuński, M.; Ruud, K. Ab initio Methods for the Calculation of NMR Shielding and Indirect Spin-Spin Coupling Constants. Chem. Rev. 1999, 99, 293–352. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F. Electron-correlated approaches for the calculation of NMR chemical shifts. Adv. Chem. Phys. 2002, 123, 355–422. [Google Scholar]
- Facelli, J.C. Calculations of chemical shieldings: Theory and applications. Concepts Magn. Reson. Part A 2004, 20A, 42–69. [Google Scholar] [CrossRef]
- Helgaker, T.; Coriani, S.; Jørgensen, P.; Kristensen, K.; Olsen, J.; Ruud, K. Recent Advances in Wave Function-Based Methods of Molecular-Property Calculations. Chem. Rev. 2012, 112, 543–631. [Google Scholar] [CrossRef] [PubMed]
- Jameson, C.J.; Buckingham, A.D. Nuclear magnetic shielding density. J. Phys. Chem. 1979, 83, 3366–3371. [Google Scholar] [CrossRef]
- Jameson, C.J.; Buckingham, A.D. Molecular electronic property density functions: The nuclear magnetic shielding density. J. Chem. Phys. 1980, 73, 5684–5692. [Google Scholar] [CrossRef]
- Bieger, W.; Seifert, G.; Eschrig, H.; Grossmann, G. LCAO Xα calculations of nuclear magnetic shielding in molecules. Chem. Phys. Lett. 1985, 115, 275–280. [Google Scholar] [CrossRef]
- Mohr, P.J.; Newell, D.B.; Taylor, B.N. CODATA Recommended Values of the Fundamental Physical Constants: 2014. J. Phys. Chem. Ref. Data 2016, 45, 043102. [Google Scholar] [CrossRef] [Green Version]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of Current Densities using Gauge-Including Atomic Orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef] [PubMed]
- Taubert, S.; Sundholm, D.; Jusélius, J. Calculation of spin-current densities using gauge-including atomic orbitals. J. Chem. Phys. 2011, 134, 054123. [Google Scholar] [CrossRef]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. WIREs Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Dimitrova, M.; Sundholm, D. Current density, current-density pathways and molecular aromaticity. In Aromaticity: Modern Computational Methods and Applications; Fernández López, I., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; Chapter 5; pp. 155–194. [Google Scholar]
- GIMIC, Version 2.0, a Current Density Program. December 2020. Available online: https://github.com/qmcurrents/gimic (accessed on 3 September 2021).
- Sambe, H. Properties of induced electron current density of a molecule under a static uniform magnetic field. J. Chem. Phys. 1973, 59, 555. [Google Scholar] [CrossRef]
- Lazzeretti, P. Current density tensors. J. Chem. Phys. 2018, 148, 134109. [Google Scholar] [CrossRef] [PubMed]
- Lazzeretti, P. Ring currents. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 36, 1–88. [Google Scholar] [CrossRef]
- Pelloni, S.; Lazzeretti, P. On the existence of a natural common gauge-origin for the calculation of magnetic properties of atoms and molecules via gaugeless basis sets. J. Chem. Phys. 2012, 136, 164110. [Google Scholar] [CrossRef] [PubMed]
- Lazzeretti, P.; Malagoli, M.; Zanasi, R. Electronic Current-Density Induced by Nuclear Magnetic Dipoles. J. Mol. Struct. THEOCHEM 1994, 119, 299–304. [Google Scholar] [CrossRef]
- Komorovsky, S.; Jakubowska, K.; Świder, P.; Repisky, M.; Jaszuński, M. NMR Spin-Spin Coupling Constants Derived from Relativistic Four-Component DFT Theory-Analysis and Visualization. J. Phys. Chem. A 2020, 124, 5157–5169. [Google Scholar] [CrossRef]
- Lehtola, S.; Dimitrova, M.; Fliegl, H.; Sundholm, D. Benchmarking Magnetizabilities with Recent Density Functionals. J. Chem. Theory Comput. 2021, 17, 1457–1468. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of Magnetic Response Properties Using a Continuous Set of Gauge Transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Patterns of Ring Currents in Conjugated Molecules: A Few-Electron Model Based on Orbital Contributions. J. Phys. Chem. A 2001, 105, 9553–9562. [Google Scholar] [CrossRef]
- Soncini, A.; Fowler, P.; Lazzeretti, P.; Zanasi, R. Ring-current signatures in shielding-density maps. Chem. Phys. Lett. 2005, 401, 164–169. [Google Scholar] [CrossRef]
- Pelloni, S.; Ligabue, A.; Lazzeretti, P. Ring-Current Models from the Differential Biot-Savart Law. Org. Lett. 2004, 6, 4451–4454. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Lazzeretti, P.; Viglione, R.; Zanasi, R. Understanding proton magnetic shielding in the benzene molecule. Chem. Phys. Lett. 2004, 390, 268–271. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. On the orbital analysis of magnetic properties. Phys. Chem. Chem. Phys. 2004, 6, 261–272. [Google Scholar] [CrossRef]
- Ferraro, M.B.; Faglioni, F.; Ligabue, A.; Pelloni, S.; Lazzeretti, P. Ring current effects on nuclear magnetic shielding of carbon in the benzene molecule. Magn. Res. Chem. 2005, 43, 316–320. [Google Scholar] [CrossRef]
- Acke, G.; Van Damme, S.; Havenith, R.W.A.; Bultinck, P. Interpreting the behavior of the NICSzz by resolving in orbitals, sign, and positions. J. Comp. Chem. 2018, 39, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Acke, G.; Van Damme, S.; Havenith, R.W.A.; Bultinck, P. Quantifying the conceptual problems associated with the isotropic NICS through analyses of its underlying density. Phys. Chem. Chem. Phys. 2019, 21, 3145–3153. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chim. Acta 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-Consistent Perturbation-Theory Of Diamagnetism 1. Gauge-Invariant LCAO Method For NMR Chemical-Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Kollwitz, M.; Häser, M.; Gauss, J. Non-Abelian point group symmetry in direct second-order many-body perturbation theory calculations of NMR chemical shifts. J. Chem. Phys. 1998, 108, 8295–8301. [Google Scholar] [CrossRef] [Green Version]
- Reiter, K.; Mack, F.; Weigend, F. Calculation of Magnetic Shielding Constants with meta-GGA Functionals Employing the Multipole-Accelerated Resolution of the Identity: Implementation and Assessment of Accuracy and Efficiency. J. Chem. Theory Comput. 2018, 14, 191–197. [Google Scholar] [CrossRef]
- Jensen, F. Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Pecul, M.; Ruud, K. The ab initio calculation of optical rotation and electronic circular dichroism. Adv. Quantum Chem. 2005, 50, 185–212. [Google Scholar]
- Fliegl, H.; Sundholm, D.; Taubert, S.; Jusélius, J.; Klopper, W. Magnetically Induced Current Densities in Aromatic, Antiaromatic, Homoaromatic, and Nonaromatic Hydrocarbons. J. Phys. Chem. A 2009, 113, 8668–8676. [Google Scholar] [CrossRef] [PubMed]
- Bast, R. April 2020. Numgrid v1.1.2: Numerical Integration Grid for Molecules. Available online: https://zenodo.org/record/4815722#.YThHNty-s2w (accessed on 3 September 2021).
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Lebedev, V.I. A quadrature formula for the sphere of 59th algebraic order of accuracy. Russ. Acad. Sci. Dokl. Math. 1995, 50, 283–286. [Google Scholar]
- Lindh, R.; Malmqvist, P.Å.; Gagliardi, L. Molecular integrals by numerical quadrature. I. Radial integration. Theor. Chem. Acc. 2001, 106, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Fliegl, H.; Sundholm, D. Aromatic Pathways of Porphins, Chlorins and Bacteriochlorins. J. Org. Chem. 2012, 77, 3408–3414. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, R.; Kim, J.; Oh, J.; Lamping, A.; Wang, Y.; Shimizu, S.; Hisaki, I.; Kowalczyk, T.; Fliegl, H.; Kim, D.; et al. Three-dimensional aromaticity in an antiaromatic cyclophane. Nat. Commun. 2019, 10, 3576. [Google Scholar] [CrossRef]
- Sundholm, D.; Fliegl, H. Aromatic Pathways in Porphyrinoids by Magnetically Induced Ring Currents. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2021; Volume 46, in press. [Google Scholar]
- Kawashima, H.; Ukai, S.; Nozawa, R.; Fukui, N.; Fitzsimmons, G.; Kowalczyk, T.; Fliegl, H.; Shinokubo, H. Determinant Factors of Three-Dimensional Aromaticity in Antiaromatic Cyclophanes. J. Am. Chem. Soc. 2021, 143, 10676–10685. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of the induced magnetic field (black arrows) when the aromatic benzene is exposed to an external magnetic field (yellow) perpendicular to the molecular ring. The magnetically induced current density circling in the classical direction is called diatropic (blue) and the one flowing in the opposite direction is paratropic (red). The induced magnetic field in the center of the ring points in the opposite direction relative to the applied field, while it deshields the nuclear magnetic moment of the hydrogen atoms outside the ring.
Figure 1.
Illustration of the induced magnetic field (black arrows) when the aromatic benzene is exposed to an external magnetic field (yellow) perpendicular to the molecular ring. The magnetically induced current density circling in the classical direction is called diatropic (blue) and the one flowing in the opposite direction is paratropic (red). The induced magnetic field in the center of the ring points in the opposite direction relative to the applied field, while it deshields the nuclear magnetic moment of the hydrogen atoms outside the ring.

Figure 2.
The molecular structure of (a) free-base trans-porphyrin, (b) free-base isophlorin, and (c) free-base trans-norcorrole.
Figure 2.
The molecular structure of (a) free-base trans-porphyrin, (b) free-base isophlorin, and (c) free-base trans-norcorrole.

Figure 3.
The isotropic H NMR shielding density of the inner hydrogen of trans-porphyrin in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the trans-porphyrin plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .
Figure 3.
The isotropic H NMR shielding density of the inner hydrogen of trans-porphyrin in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the trans-porphyrin plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .

Figure 4.
The isotropic H NMR shielding density of the inner hydrogen of trans-porphyrin. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of has been used.
Figure 4.
The isotropic H NMR shielding density of the inner hydrogen of trans-porphyrin. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of has been used.

Figure 5.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base trans-porphyrin. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring with an inner hydrogen, and the hydrogen atoms of the pyrrole ring without an inner hydrogen. The atoms are numbered along the x axis in the same order as in the ESI.
Figure 5.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base trans-porphyrin. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring with an inner hydrogen, and the hydrogen atoms of the pyrrole ring without an inner hydrogen. The atoms are numbered along the x axis in the same order as in the ESI.

Figure 6.
The isotropic H NMR shielding density of the inner hydrogen of free-base isophlorin in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the free-base isophlorin plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .
Figure 6.
The isotropic H NMR shielding density of the inner hydrogen of free-base isophlorin in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the free-base isophlorin plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .

Figure 7.
The isotropic H NMR shielding density of the inner hydrogen of free-base isophlorin. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of ±0.04 has been used. The corresponding pictures of the H NMR shielding densities of the hydrogen atoms are given in the ESI.
Figure 7.
The isotropic H NMR shielding density of the inner hydrogen of free-base isophlorin. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of ±0.04 has been used. The corresponding pictures of the H NMR shielding densities of the hydrogen atoms are given in the ESI.

Figure 8.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base isophlorin. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring. The atoms are numbered along the x axis in the same order as in ESI.
Figure 8.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base isophlorin. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring. The atoms are numbered along the x axis in the same order as in ESI.

Figure 9.
The isotropic H NMR shielding density of the inner hydrogen of trans-norcorrole in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the trans-norcorrole plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .
Figure 9.
The isotropic H NMR shielding density of the inner hydrogen of trans-norcorrole in (a) the molecular plane and (b) 1 Å above the plane. The H NMR shielding density of the meso hydrogen in (c) the molecular plane and (d) 1 Å above the trans-norcorrole plane. The shielding contributions are shown in blue and the deshielding contributions in red. The employed range is .

Figure 10.
The isotropic H NMR shielding density of the inner hydrogen of -norcorrole. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of ±0.04 has been used. The corresponding pictures of the H NMR shielding densities of the meso and hydrogen atoms are given in the ESI.
Figure 10.
The isotropic H NMR shielding density of the inner hydrogen of -norcorrole. The shielding contributions are shown in blue and the deshielding contributions in red. An isovalue of ±0.04 has been used. The corresponding pictures of the H NMR shielding densities of the meso and hydrogen atoms are given in the ESI.

Figure 11.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base trans-norcorrole. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring with an inner hydrogen, and the hydrogen atoms of the pyrrole ring without an inner hydrogen. The atoms are numbered along the x axis in the same order as in ESI.
Figure 11.
Atomic contributions (in ppm) to the isotropic H NMR shielding constants of free-base trans-norcorrole. From above: the inner hydrogen, the meso hydrogen, the hydrogen atoms of the pyrrole ring with an inner hydrogen, and the hydrogen atoms of the pyrrole ring without an inner hydrogen. The atoms are numbered along the x axis in the same order as in ESI.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fliegl, H.; Dimitrova, M.; Berger, R.J.F.; Sundholm, D. Spatial Contributions to 1H NMR Chemical Shifts of Free-Base Porphyrinoids. Chemistry 2021, 3, 1005-1021. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030072
AMA Style
Fliegl H, Dimitrova M, Berger RJF, Sundholm D. Spatial Contributions to 1H NMR Chemical Shifts of Free-Base Porphyrinoids. Chemistry. 2021; 3(3):1005-1021. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030072
Chicago/Turabian StyleFliegl, Heike, Maria Dimitrova, Raphael J. F. Berger, and Dage Sundholm. 2021. "Spatial Contributions to 1H NMR Chemical Shifts of Free-Base Porphyrinoids" Chemistry 3, no. 3: 1005-1021. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry3030072