
Piperine Derivatives Enhance Fusion and Axonal Transport of Mitochondria by Activating Mitofusins



Abstract
:1. Introduction
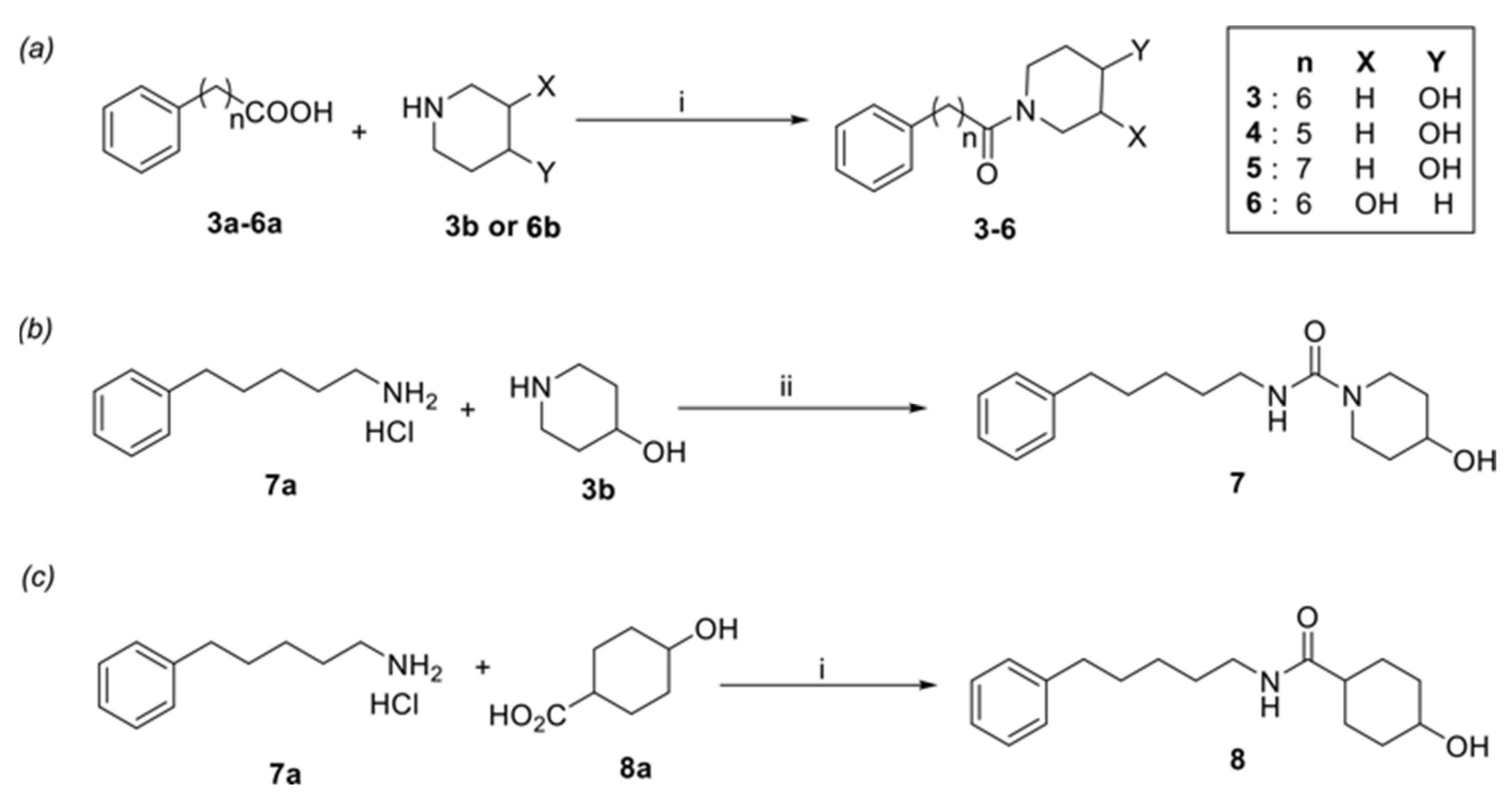
2. Materials and Methods
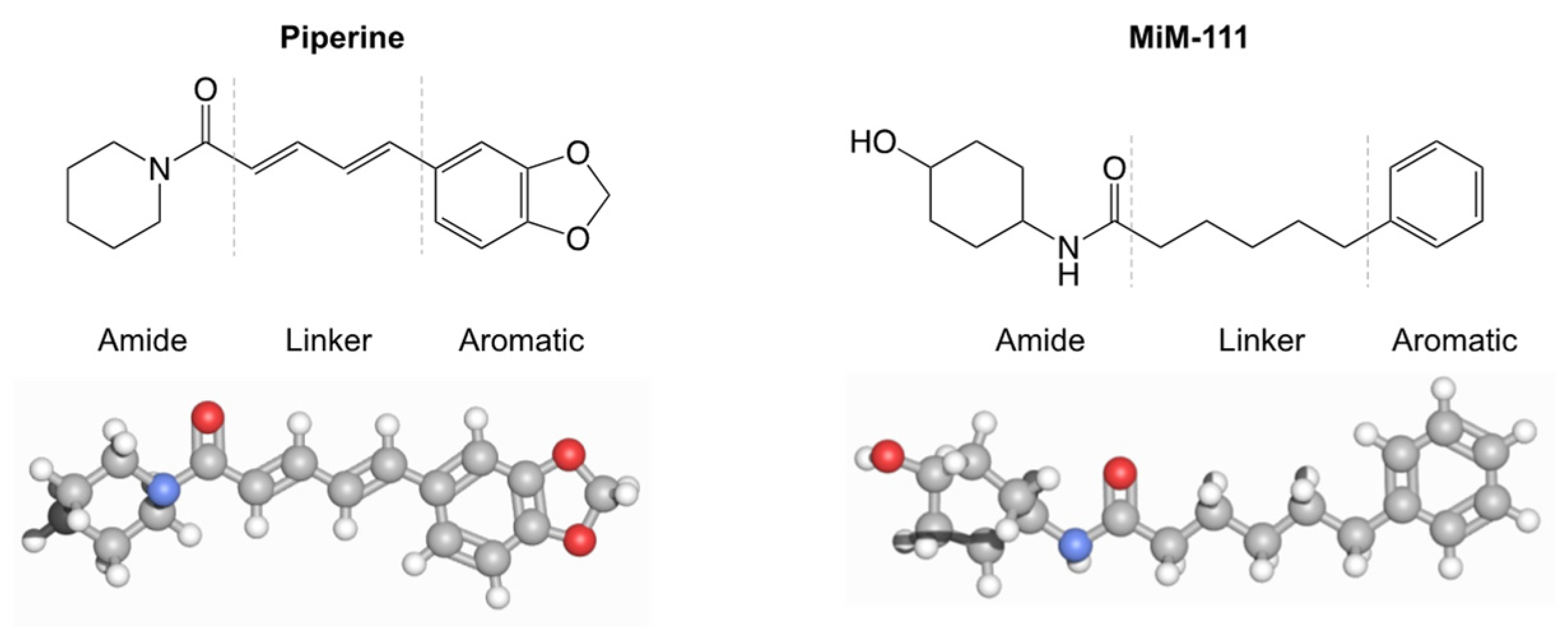
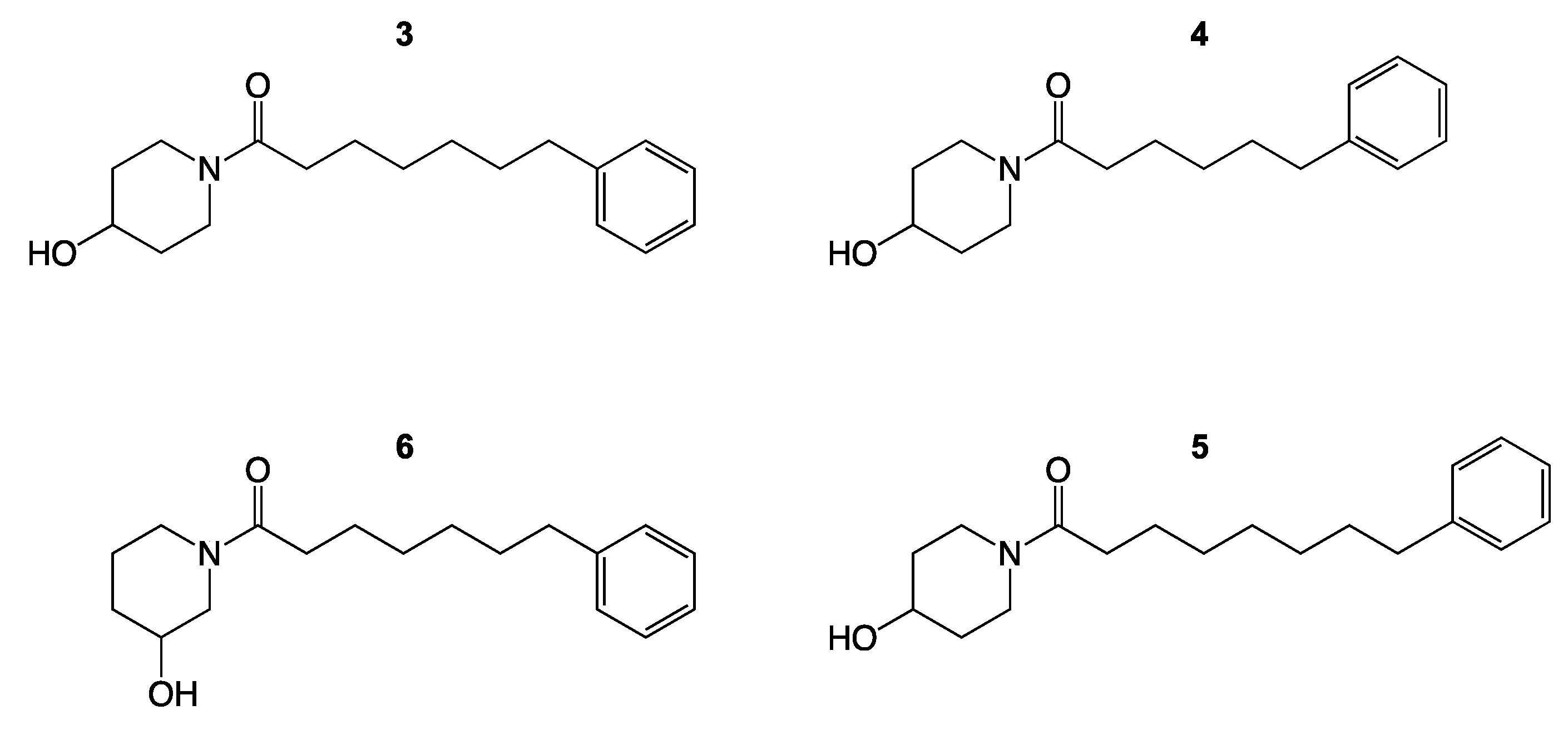
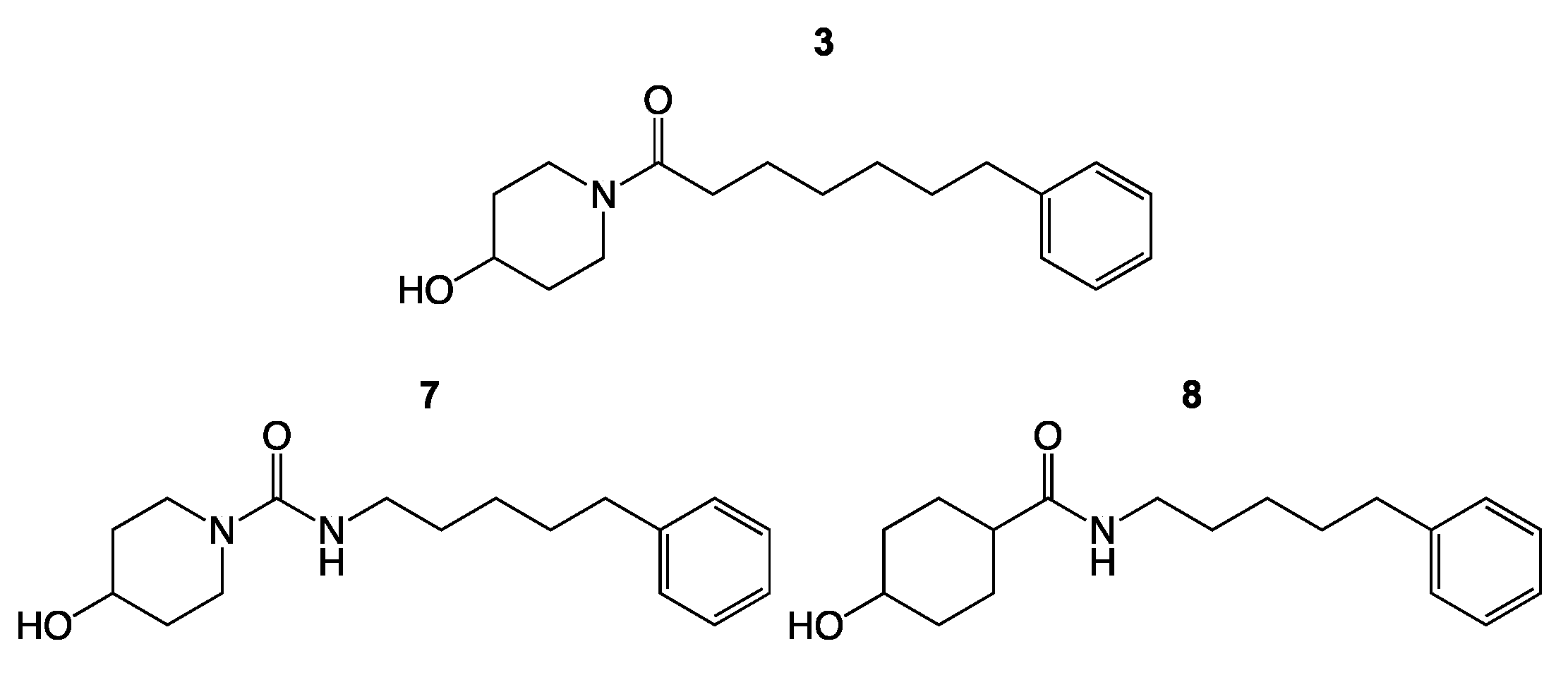
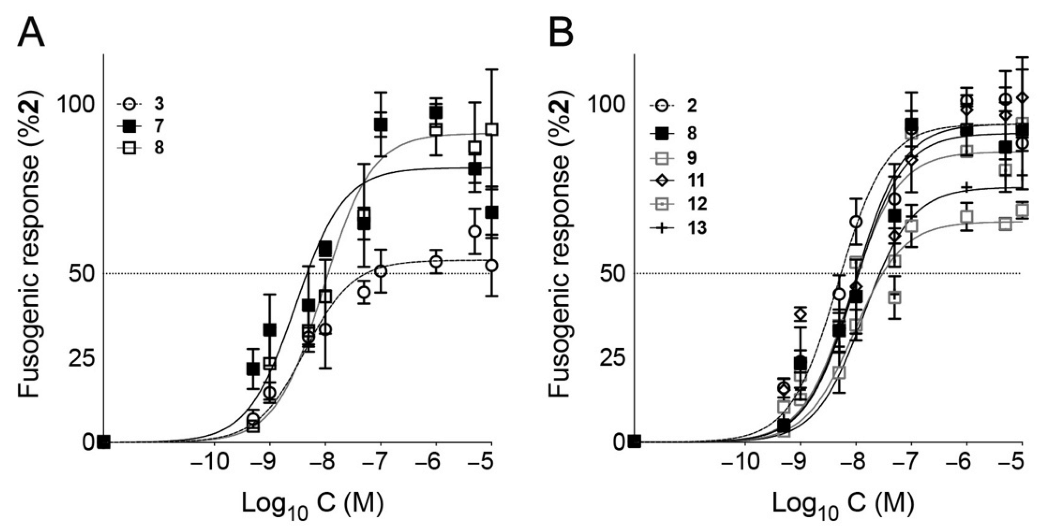
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darshan, S.; Doreswamy, R. Patented antiinflammatory plant drug development from traditional medicine. Phytother. Res. 2004, 18, 343–357. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; Sahebkar, A. Piperine and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Stojanović-Radić, Z.; Pejčić, M.; Dimitrijević, M.; Aleksić, A.; Anil Kumar, N.V.; Salehi, B.; Cho, W.C.; Sharifi-Rad, J. Piperine-A Major Principle of Black Pepper: A review of its bioactivity and studies. Appl. Sci. 2019, 9, 4270. [Google Scholar] [CrossRef] [Green Version]
- Meghwal, M.; Goswami, T.K. Piper nigrum and piperine: An update. Phytother. Res. 2013, 27, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.K.; Glaeser, H.; Becquemont, L.; Klotz, U.; Gupta, S.K.; Fromm, M.F. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002, 302, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöffmann, A.; Wimmer, L.; Goldmann, D.; Khom, S.; Hintersteiner, J.; Baburin, I.; Schwarz, T.; Hintersteininger, M.; Pakfeifer, P.; Oufir, M.; et al. Efficient modulation of γ-aminobutyric acid type A receptors by piperine derivatives. J. Med. Chem. 2014, 57, 5602–5619. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Hong, S.S.; Han, X.H.; Hwang, J.S.; Oh, G.J.; Lee, K.S.; Lee, M.K.; Hwang, B.Y.; Ro, J.S. Piperine from the fruits of Piper longum with inhibitory effect on monoamine oxidase and antidepressant-like activity. Chem. Pharm. Bull. 2005, 53, 832–835. [Google Scholar] [CrossRef] [Green Version]
- McNamara, F.N.; Randall, A.; Gunthorpe, M.J. Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). Br. J. Pharmacol. 2005, 144, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Vaibhav, K.; Shrivastava, P.; Javed, H.; Khan, A.; Ahmed, M.E.; Tabassum, R.; Khan, M.M.; Khuwaja, G.; Islam, F.; Siddiqui, M.S.; et al. Piperine suppresses cerebral ischemia-reperfusion-induced inflammation through the repression of COX-2, NOS-2, and NF-κB in middle cerebral artery occlusion rat model. Mol. Cell Biochem. 2012, 367, 73–84. [Google Scholar] [CrossRef]
- Selvendiran, K.; Thirunavukkarasu, C.; Singh, J.P.; Padmavathi, R.; Sakthisekaran, D. Chemopreventive effect of piperine on mitochondrial TCA cycle and phase-I and glutathione-metabolizing enzymes in benzo(a)pyrene induced lung carcinogenesis in Swiss albino mice. Mol. Cell Biochem. 2005, 271, 101–106. [Google Scholar] [CrossRef]
- Kim, N.; Nam, M.; Kang, M.S.; Lee, J.O.; Lee, Y.W.; Hwang, G.S.; Kim, H.S. Piperine regulates UCP1 through the AMPK pathway by generating intracellular lactate production in muscle cells. Sci. Rep. 2017, 7, 41066. [Google Scholar] [CrossRef] [PubMed]
- Piyachaturawat, P.; Glinsukon, T.; Toskulkao, C. Acute and subacute toxicity of piperine in mice, rats and hamsters. Toxicol. Lett. 1983, 16, 351–359. [Google Scholar] [CrossRef]
- Allameh, A.; Saxena, M.; Biswas, G.; Raj, H.G.; Singh, J.; Srivastava, N. Piperine, a plant alkaloid of the piper species, enhances the bioavailability of aflatoxin B1 in rat tissues. Cancer Lett. 1992, 61, 195–199. [Google Scholar] [CrossRef]
- Rao, P.J.; Kolla, S.D.; Elshaari, F.; Elshaari, F.; Awamy, H.E.; Elfrady, M.; Singh, R.; Belkhier, A.; Srikumar, S.; Said, A.R.; et al. Effect of piperine on liver function of CF-1 albino mice. Infect. Disord. Drug Targets 2015, 15, 131–134. [Google Scholar] [CrossRef]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, G.W., II. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051. [Google Scholar] [CrossRef]
- Dang, X.; Williams, S.B.; Devanathan, S.; Franco, A.; Fu, L.; Bernstein, P.R.; Walters, D.; Dorn, G.W., II. Pharmacophore-Based Design of Phenyl-[hydroxycyclohexyl] Cycloalkyl-Carboxamide Mitofusin Activators with Improved Neuronal Activity. J. Med. Chem. 2021, 64, 12506–12524. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782. [Google Scholar] [CrossRef]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 2020, 9, e61119. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipis, M.; Feely, S.M.E.; Polke, J.M.; Skorupinska, M.; Perez, L.; Shy, R.R.; Laura, M.; Morrow, J.M.; Moroni, I.; Pisciotta, C.; et al. Natural history of Charcot-Marie-Tooth disease type 2A: A large international multicentre study. Brain 2020, 143, 3589–3602. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics fusion, fission, movement, and mitophagy in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012, 32, 4145–4155. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.P.; Choudhary, A. Piperine and Derivatives: Trends in Structure-Activity Relationships. Curr. Top. Med. Chem. 2015, 15, 1722–1734. [Google Scholar] [CrossRef]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W., II. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., II; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Dang, X.; Walton, E.K.; Zablocka, B.; Baloh, R.H.; Shy, M.E.; Dorn, G.W., II. Mitochondrial Phenotypes in Genetically Diverse Neurodegenerative Diseases and Their Response to Mitofusin Activation. Cells 2022, 11, 1053. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Miret-Casals, L.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, G.W., II. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. Mitofusins as mitochondrial anchors and tethers. J. Mol. Cell Cardiol. 2020, 142, 146–153. [Google Scholar] [CrossRef]
- Li, J.; Dang, X.; Franco, A.; Dorn, G.W., II. Reciprocal Regulation of Mitofusin 2-Mediated Mitophagy and Mitochondrial Fusion by Different PINK1 Phosphorylation Events. Front. Cell Dev. Biol. 2022, 10, 868465. [Google Scholar] [CrossRef]
- Wang, L.; Gao, J.; Liu, J.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Huntley, M.L.; Jiang, Y.; Ji, H.; Yan, T.; et al. Mitofusin 2 Regulates Axonal Transport of Calpastatin to Prevent Neuromuscular Synaptic Elimination in Skeletal Muscles. Cell Metab. 2018, 28, 400–414.e408. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Bobylev, I.; Joshi, A.R.; Barham, M.; Neiss, W.F.; Lehmann, H.C. Depletion of Mitofusin-2 Causes Mitochondrial Damage in Cisplatin-Induced Neuropathy. Mol. Neurobiol. 2018, 55, 1227–1235. [Google Scholar] [CrossRef]
- Yamashita, Y.; Irie, K.; Kochi, A.; Kimura, N.; Hayashi, T.; Matsuo, K.; Myose, T.; Sano, K.; Nakano, T.; Takase, Y.; et al. Involvement of Charcot-Marie-Tooth disease gene mitofusin 2 expression in paclitaxel-induced mechanical allodynia in rats. Neurosci. Lett. 2017, 653, 337–340. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W., II; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Daniele, T.; Hurbain, I.; Vago, R.; Casari, G.; Raposo, G.; Tacchetti, C.; Schiaffino, M.V. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol. 2014, 24, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanwar, J.; Saurav, S.; Basu, R.; Singh, J.B.; Priya, A.; Dutta, M.; Santhanam, U.; Joshi, M.; Madison, S.; Singh, A.; et al. Mitofusin-2 Negatively Regulates Melanogenesis by Modulating Mitochondrial ROS Generation. Cells 2022, 11, 701. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Piperine Target | Action | EC50/IC50 (nM) | Reference |
|---|---|---|---|
| Mitofusin | Activator | 8 | current paper |
| GABAAR | Agonist | 52,400 | Schoffman J Med Chem |
| Vanilloid R (TRPV1) | Agonist | 37,900 | McNamara Brit J Pharm |
| P-gp/MDR1 | Inhibitor | 15,500; 74,100 | Bhardwaj JPET |
| MAO-A | Inhibitor | 20,900 | Lee Chem Pharm Bull |
| MAO-B | Inhibitor | 7000 | “ |
| Compound | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| MW | 289.4 | 289.41 | 275.39 | 303.44 | 289.41 |
| EC50 mito elongation mean (95% CI); nM | 8.4 (6.1−11.4) | 53.6 (22.4−ND) | >10,000 | 13.9 (8.5−21.8) | >10,000 |
| Emax (% of 2) mean ± SEM | 95.6 ± 2.9 | 54.0 ± 2.7 | n/a | 81.0 ± 3.7 | 37.0 ± 2.4 |
| Plasma Protein Binding | % Bound | % Bound | % Bound | % Bound | % Bound |
| Human | 91 | 95.1 | 83.7 | 98.6 | 96.6 |
| Mouse | 96.3 | 95.7 | 91.2 | 98.9 | 95.9 |
| Liver Microsomes | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) |
| Human | >145 | 103.3 | >145 | 64.5 | 60.6 |
| Mouse | 92.4 | 52.5 | 82.4 | 26 | 22.5 |
| PAMPA (Pe) | nm/s | nm/s | nm/s | nm/s | nm/s |
| 26.277 | 145.127 | 96.106 | 142.782 | 180.027 |
| Compound | 3 | 7 | 8 |
|---|---|---|---|
| MW | 289.41 | 290.4 | 289.41 |
| EC50 mito elongation mean (95% CI); nM | 53.5 (22.4−ND) | 4.7 (2.6−7.9) | 13.5 (6.5−25.4) |
| Emax (% of 2) | 54.0 ± 2.7 | 81.4 ± 4.0 | 69.4 ± 4.3 |
| Plasma Protein Binding | % Bound | % Bound | % Bound |
| Human | 95.1 | 91.7 | 89.3 |
| Mouse | 95.7 | 91.6 | 92.9 |
| Liver Microsomes | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) |
| Human | 103.3 | 133.6 | >145 |
| Mouse | 52.5 | 94.8 | 102.1 |
| PAMPA (Pe) | nm/s | nm/s | nm/s |
| 145.127 | 14.585 | 30.249 |
| Compound | 2 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|
| MW | 289.4 | 289.41 | 291.18 | 301.2 | 303.18 | 289.17 | 317.2 |
| EC50 mito elongation mean (95% CI); nM | 5.4 (4.1−7.1) | 11.2 (6.3−19.2) | 30.3 (18.9−47.2) | >10,000 | 10.6 (6.3−17.2) | 10.8 (6.2−18.0) | 27.0 (17.1−40.6) |
| Emax (% of 2) | 94.3 ± 2.4 | 91.5 ± 5.1 | 65.4 ± 2.7 | n/a | 94.3 ± 4.7 | 75.6 ± 4.5 | 86.1 ± 3.3 |
| Plasma Protein Binding | % Bound | % Bound | % Bound | % Bound | % Bound | % Bound | % Bound |
| Human | 91 | 89.3 | 68.51 | 89.13 | 46.48 | 37.36 | 62.95 |
| Mouse | 96.3 | 92.9 | unstable | 95.34 | 72.14 | 63.93 | 77.92 |
| Liver Microsomes | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) | T 1/2 (min) |
| Human | >145 | >145 | 10.3 | >145 | >145 | >145 | >145 |
| Mouse | 92.4 | 102.1 | 3.8 | 77.8 | >145 | >145 | >145 |
| PAMPA (Pe) | nm/s | nm/s | nm/s | nm/s | nm/s | nm/s | nm/s |
| 26.277 | 30.249 | 10.1 | 50.9 | 5.43 | 2.28 | 10.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Dang, X.; Franco, A.; Zhao, H.; Dorn, G.W., II. Piperine Derivatives Enhance Fusion and Axonal Transport of Mitochondria by Activating Mitofusins. Chemistry 2022, 4, 655-668. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry4030047
Zhang L, Dang X, Franco A, Zhao H, Dorn GW II. Piperine Derivatives Enhance Fusion and Axonal Transport of Mitochondria by Activating Mitofusins. Chemistry. 2022; 4(3):655-668. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry4030047
Chicago/Turabian StyleZhang, Lihong, Xiawei Dang, Antonietta Franco, Haiyang Zhao, and Gerald W. Dorn, II. 2022. "Piperine Derivatives Enhance Fusion and Axonal Transport of Mitochondria by Activating Mitofusins" Chemistry 4, no. 3: 655-668. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry4030047