
Evaluating the Presence of Software-as-a-Medical-Device in the Australian Therapeutic Goods Register


Department of Engineering Science, University of Oxford, Oxford OX1 3PJ, UK
*
Author to whom correspondence should be addressed.
Prosthesis 2021, 3(3), 221-228; https://0-doi-org.brum.beds.ac.uk/10.3390/prosthesis3030022
Submission received: 24 June 2021
/
Revised: 18 August 2021
/
Accepted: 20 August 2021
/
Published: 24 August 2021
(This article belongs to the Special Issue Regulatory Data Science for Medical Devices)
Abstract
:In recent years, medical device regulatory bodies have recognised software-as-a-medical-device (SaMD) as a distinct subgroup of devices. The field of SaMD has been rapidly evolving and encompasses a range of different digital solutions. Many organisations have now started to look into digital healthcare, as a way to solve key global challenges. However, there remains uncertainty regarding how many of these SaMD products are entering the market and to what extent these systems achieve a desired level of general safety once they are in the market. In this study, we utilise data collected from publicly available databases. The data are evaluated for trends and a descriptive analysis is performed of the recall and adverse events associated specifically with SaMD. We find that there is a significant positive trend (p < 0.05) of SaMD registrations, although the number of SaMD registrations remains relative low compared to non-SaMD. This rise in SaMD registrations coincides with increasing levels of recalls and adverse events. More importantly, it becomes apparent that adverse events notification is not yet fit for purpose with regards to SaMD.
1. Introduction
Although the inclusion of software into medical devices is a long established practice, stand alone software offerings that provide diagnostic and therapeutic functions is relative new. Nonetheless, software applications are an increasingly fundamental feature of clinical practice and regulators around the world have responded to recognise this rise in interest within this area. Certain regulators, such as the one in Australia, have even made data available that can be used for further analysis. The use of medical devices in Australia is overseen by the Therapeutic Goods Authority (TGA). Consideration of software in medical devices has long been a concern for the TGA, which had evaluated means by which to address software through its essential principles as early as 2001 [1]. In 2020, the TGA released a study on the harms caused by software [2], outlining recalls and adverse events related to software through a literature review. The TGA report did not distinguish in its analysis between medical devices that have software and software as a stand alone medical device.
The distinction between devices that incorporate software and “software-as-a-medical-device” (SaMD) is important, as these now represent two distinct device types within regulation. SaMD are those entirely composed of software without any additional hardware. In other words, the software program itself is the medical device. Since 2013, there has been international consensus through the International Medical Device Regulators Forum (of which Australia is a member) on the definition of SaMD [3]. Although regulatory legislation now incorporates the definition, there remain unanswered questions about how the safety of such devices can be ensured. There has been work conducted related to recall and adverse events for medical devices that involve software using data from the FDA in the United States (e.g., [4,5]). However, we have been unable to find any quantitative analysis specific to SaMD in any jurisdiction after an extensive review of the available literature. Further to this, there has been no empirical treatment of reported risks associated with SaMD in Australia, or any other jurisdiction. To the best of our knowledge this work represents the first quantitative analysis of SaMD within any regulatory framework. In this work, we focus on the following three research questions; (1) Has SaMD registrations been growing in Australia and what is the origin (domestic or foreign)? (2) What types of recall events are associated with SaMD? (3) What are the types of adverse events associated with SaMD? We conclude with a discussion on results and the nature of regulatory actions for SaMD.
2. Data and Methods
2.1. Data Collection
For this study, we relied on the TGA’s publicly accessible databases relating to (a) the registration of medical devices, (b) the recall of the devices, and (c) adverse events associated with the device. We utilised these databases in order to map SaMD development in Australia, with a particular focus on risk of harms that SaMD might pose. Below is a description of databases that were used in this study:
- The Australian Register of Therapeutic Goods (ARTG), which is a publicly accessible database of medical devices that are allowed on the market in Australia. An entry in the ARTG contains the device’s description, global medical device number (GMDN), manufacturer, sponsor, classification, and date of registration [6]. A GMDN provides a broad description of a device type and consists of a unique number. The TGA’s website states that, as of October 2019, there were over 90,000 registrations on the register, which includes medicines, as well as medical devices. We extract ARTG entries resulting in 60,806 device records, comprised of 7029 unique device types (based on their GMDN), spanning a period of 18 years, from 26 November 2002 to 24 June 2020;
- The System for Australian Recall Action (SARA) is a searchable, public database containing product recall action notifications. These notifications are divided into (i) recalls; (ii) recalls for product correction; and (iii) hazard alerts. We extracted 4746 notifications, over the period between 2 July 2012 to 24 June 2020;
- The Database of Adverse Event Notifications (DAEN) records the types of harms caused by medical devices. Within this database the string ‘software’ was searched, which returned 68 events, of which there are 43 events that could be cross-referenced to the ARTG.
All the above databases are publicly available on the internet. In order to extract the data, a webscraper was developed for the ARTG and SARA. The webscraper interfaced with the search box provided by each of these databases. We searched only for medical devices, excluding medicines and other therapies. The webscraper downloaded the returned documents in the PDF format. The data were then extracted by transforming the PDF into plain text and extracting the information from the records..
2.2. Analysis Methodology
For the first research question, we undertook a trend analysis, evaluating whether SaMD has a trend and, if so, what the nature of that trend was. In order to make this determination, we utilise the Mann–Kendall test, which determines whether a time series exhibits a monotonic upward or downward trend. In relation to second question, a visualisation of recalls in SARA was created and the nature of those recalls related to SaMD was explored. Finally, the last question was investigated by obtaining the description of the types of adverse events associated with software generally, and the presence of SaMD in the dataset was determined. We developed the webscraper and conducted our analysis using version 4.1 of the R programming language.
3. Results
3.1. Registration of Software-as-a-Medical-Device
For all devices containing any kind of software, the ARTG dataset lists 971 devices, which represents 1.6% of the entire register of therapeutic goods. Of these, 736 were identified as SaMD with the remaining 235 being devices that incorporated software to some degree. We analysed the trend in SaMD registrations by finding the values related to the monthly registrations of SaMD. Figure 1 illustrates a pronounced increase in SaMD over time when compared to software that was incorporated into medical devices. The Mann–Kendall test showed that a significant positive trend for SaMD () existed in the data. No significant trend was found for software that was incorporated into medical devices.
Of the total amount of registered devices, foreign (non-Australian) products made up 92% with the remaining products being domestic (8%). Overall devices from either the USA and the EU constitute a total of 66.6% of the Australian market. However, there is a stronger domestic representation when SaMD is considered in its own right. Australian development of SaMD has a positive trend that is significant () when tested with the Mann–Kendall test, which determines whether or not there is a linear monotonic trend, as illustrated in Figure 2.
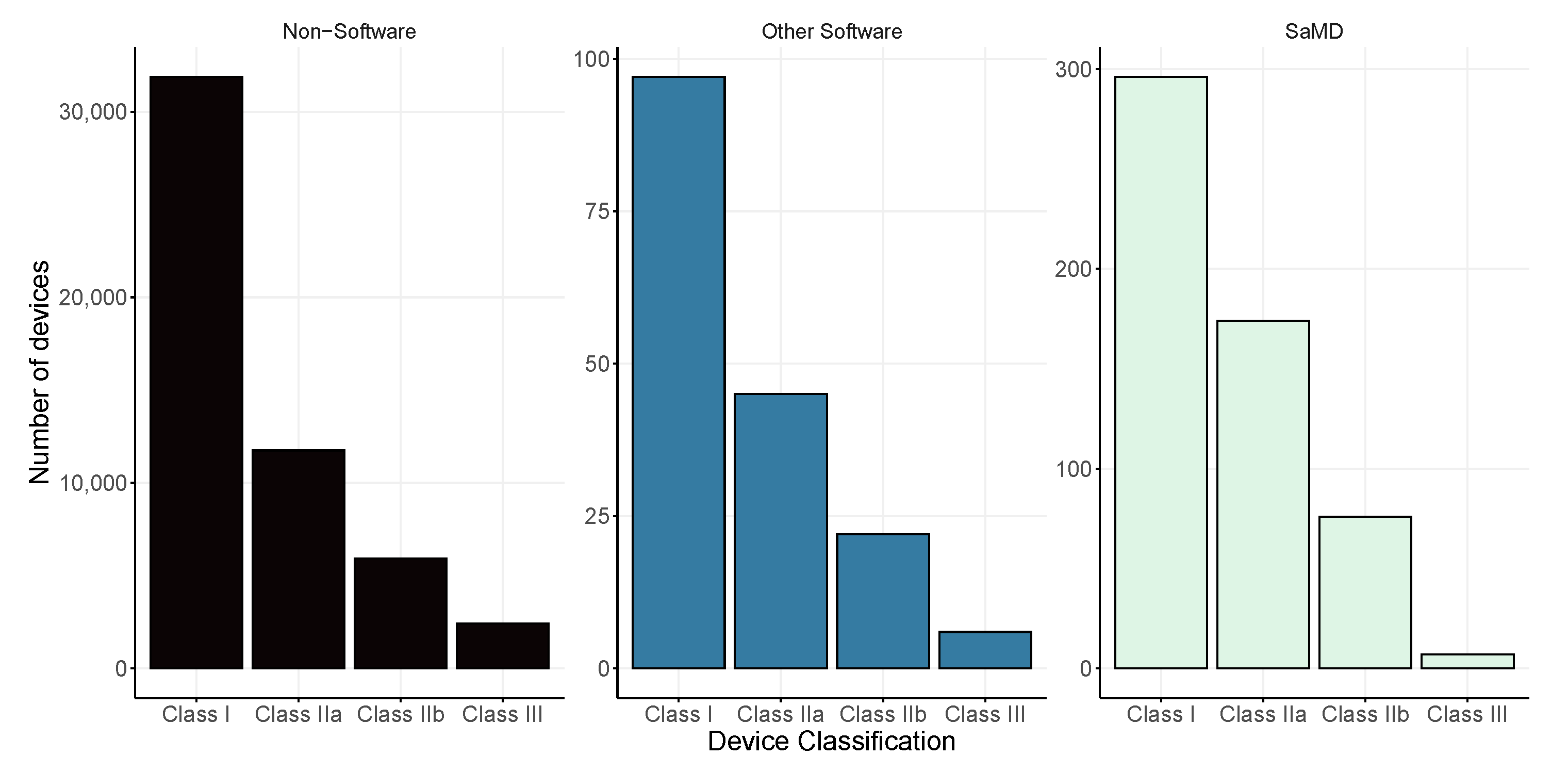
The legislation provides a system of categorisation of medical devices based on the potential risk of harm to the user. Assessment for risk consideration include: (i) the intended use of the device, (ii) location on the body during use, (iii) invasiveness into the body, as well as (iv) duration of use. The Therapeutic Goods Act 1989, along with associated subsidiary legislation, regulates the use of medical devices in Australia. This legislation provides a classification system to assess the risk that devices can pose and provides the means to categorise these devices according to risk. There are three main categories for medical devices: (i) Class I for low risk, (ii) Class II for low to medium risk, (iii) Class IIB for medium to high risk, and (iv) Class III for the highest risk devices. The largest classification group for non-software devices and SaMDs is class I, while for other non-SaMD software, the largest group consist of those from class II. In Figure 3, we illustrate the division of risk classifications based on the type of device: (i) non-software; (ii) other software, meaning that the software is part of a physical device, and (iii) SaMD. The proportions remain consistent across all device types, with Class I representing around half of the classifications (57%, 52%, and 51%, respectively). SaMD has a higher proportion of low-medium risk class devices Class IIA (32% compared to 21% for non-software devices and 28% for devices that include software) and lower proportion of high risk devices, Class III (2% compared to 9% for non-software, and 7% for software included in another device).
3.2. Recall Actions
The System for Australian Recall Action (SARA) is a searchable, public database containing product recall action notifications. These notifications are divided into (i) recalls; (ii) recalls for product correction; and (iii) hazard alerts. It is important to note that entries into SARA are commenced by the product manufacturer or product sponsor with the TGA monitoring the recall action itself. A total of 4746 notifications were extracted, from between 2 July 2012 to 24 June 2020. When cross-referencing the ARTG data set with SARA, it resulted in 3597 recall events, obtained from 1900 unique devices, as shown in Table 1.
SARA provides the “level” at which the device is recalled, which relates to hospital, retail, wholesale, or consumer. The hospital setting accounted for over 90% of the recalls actions (Table 2), which might relate to the nature of the devices.
There are 455 recall actions in SARA that mention software as the reason for the reported error, comprising 9.6% of all the reported recall events. A total of 72 software recalls were related to SaMD, constituting 16% of the software reported recalls and 2.3% of total recalls. All of these SaMD recalls come from 29 unique SaMD registrations, making up 3.9% of all registered SaMD that are in the ARTG. The connection between recall action, level and recall class is shown in Figure 4 using an alluvial plot to map them out for both SaMD and all others. The figure is set to a logarithmic scale in order to highlight the differences between SaMD and all other medical devices.
3.3. Adverse Events
The Database of Adverse Event Notifications (DAEN) records the types of harms caused by medical devices. We searched for the string ‘software’ in DAEN, which returned 68 events, of which there are 43 events that could be cross-referenced with data from the ARTG. This resulted in 17 devices, of which 7 were SaMD. Due to the nature of DAEN, we were unable to retrieve all reported events for all device types.
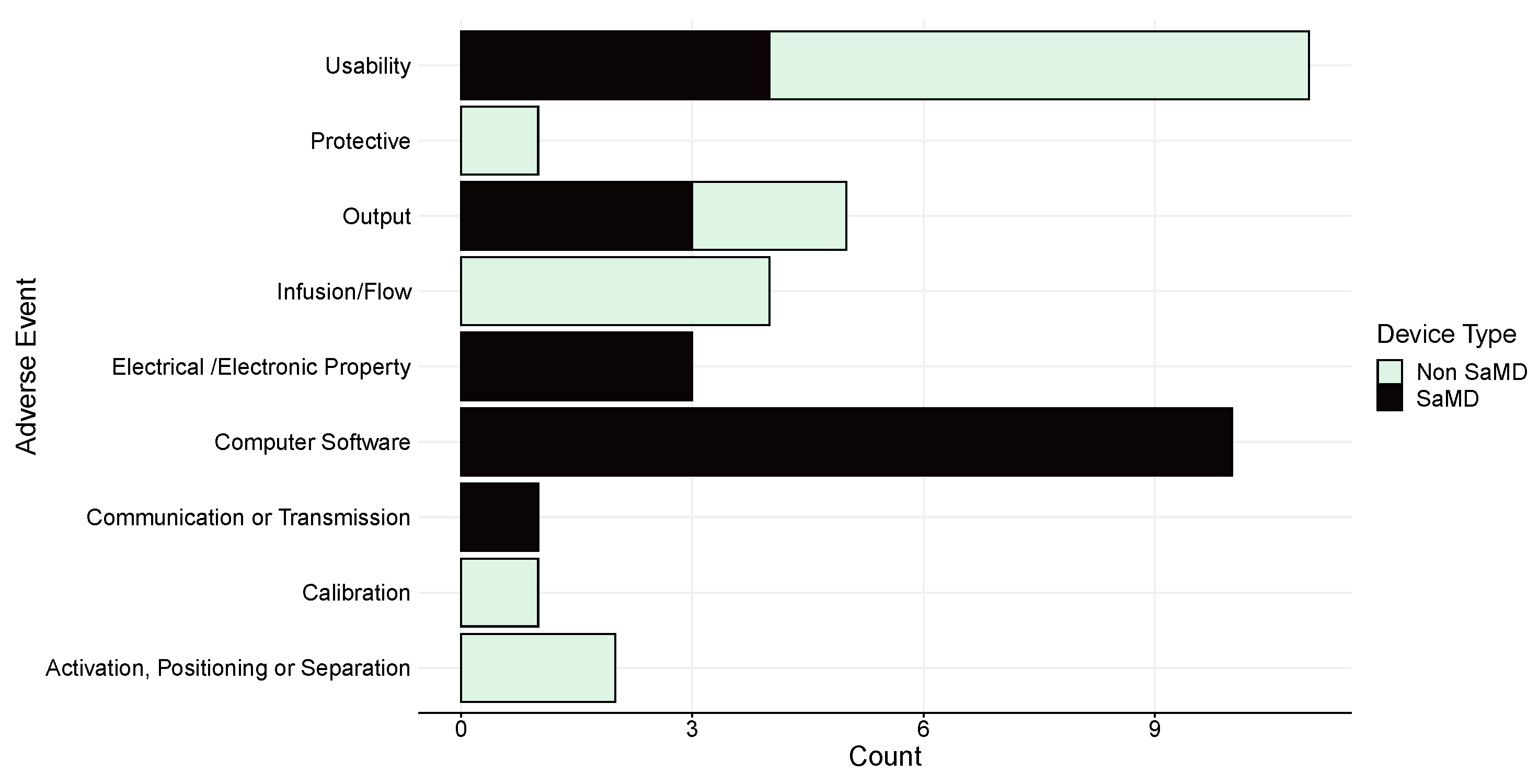
The event types in DAEN are categories according to the standard on medical device adverse event reporting [7]. The standard lists 20 different event types, 9 of which have been reported for software. According to the standard, all software errors fall under the broad event label of problems related to ‘Computer Software’, rather than providing specific software issue labels. As such, Figure 5 shows that the second-most frequent of the events in DAEN for software devices fall under this heading (n = 10, 26.3%) after usability issues (n = 11, 28.9%). Non-SaMD include a label describing software issues related to hardware control as well.
The DAEN database provides injury outcomes of the reported events. Figure 6 shows that SaMD are associated with low levels of injury, but it should be noted that the ”unknown” category is also high. This suggests that it is difficult to ascertain the exact level of injury from SaMD events.
4. Discussion
The results demonstrate that SaMD (and software more generally) are growing facets of the medical device landscape, as shown based on the Australian data. Caution needs to be taken in case of generalising too much, as there might regional differences. Nonetheless, the suggestion seems to be in-line with the popularity of machine learning and artificial intelligence for healthcare within the research arena.
The outcomes also highlight that there are some challenges in evaluating the risks associated with SaMD, as the limited regulatory vocabulary for software defects prevents further granular analysis. The blunt categorisation of the ISO code ”Computer Software Problem” in DAEN makes it hard to identify clear areas of improvement for the SaMD. It acts as an obstacle for the regulator, and those assessing regulatory data, as it prevents a better understanding of the types of events that occur for SaMD and reduces the potential for better risk management. Within the recall data, there are no fields specifying the type of problem that prompted the recall. This is not because a lack of vocabulary to describe software defects. This does exist, in, for example, the IEEE Standard for Classification of Software Anomalies [8]. The inclusion of more detailed software-specific recall reports and adverse events may facilitate the policy prioritisation for the regulator, which, in turn, will ensure that devices on the market reach an appropriate level of safety and performance.
The issue of applying appropriate standards for clinical safety are more acute for software than for non-software. McHugh et al. [9] already suggested that the rapid, iterative approach to software development may be in tension with the regulatory requirements for medical devices. Non-software device manufactures require specific fabrication infrastructure. This is in stark contrast to what happens in software testing. The costs associated with changing software are different compared to those associated with hardware manufacturing. For example, a software developer could quickly push an update to customers upon the discovery of a problem, whilst this is impossible for a (hardware) manufacture to do at scale. This rapid response option is not available to non-software devices, which must collect all defective items and fix the issue physically. Increasing the regulatory support offered to software developers during the early phases of research and development, as well as integrating this kind of support into, e.g., computer science courses, will potentially create more robust solutions for these regulatory issues in the future [10].
The difference in how risks need to be addressed by the manufacturers of products also relates to the origin of the product itself. The Australian market for medical devices relies heavily on foreign suppliers of medical devices. Herpin et al. [11] reports that the funding opportunities within the Australian biomedical industry may motivate innovators within the country to seek to establish manufacturing operations in larger markets for profit maximisation as the distance to these markets has an impact on costs. These economic dynamics seem to affect SaMD development less as there is no need for a factory to manufacture these devices, just access to skilled software developers. The lower initial cost for a more scalable product might account for the higher levels of Australian representation in SaMD.
In their work on safety and security of products in supply chain management, Marucheck et al. [12] found that recalls and adverse events of products, rather than regulation, may limit innovation in medical device development. Although Thirumalai and Sinha [13] showed that within the medical device industry, recalls costs are expected and they normally do not influence the market too much. As mentioned software might be better suited for ongoing improvements and this is also reflected in the fact that the for SaMD corrections are taking place if defects are detected.
We note that this study is limited by the amount of available data. The categorisation of ‘SaMD’ in this study was derived through the provided descriptions and the GMDN of the medical devices that were reported in the ARTG. There could be a potential for misclassification because of this. The results presented do not represent definitive evidence of trends, only an illustration of patterns available based on the data collected by the TGA. With the popularity of SaMD continuing to grow, along with the concerns that the TGA has with the regulation of these devices, it may be useful for records to indicate which devices are solely composed of software.
5. Conclusions
SaMD is a growing trend within medical device innovation. In this work, we provide the first empirical analysis of SaMD. Within Australia, which relies heavily on importation of medical devices, SaMD shows a greater domestic production than other types of medical devices.
The increased registrations of SaMD has coincided with an increased number of recall events. However, this might be explained by the fact that software can be more readily updated compared to hardware-based devices. Thus, recalls are easier to deal with for SaMD than for a traditional (non-software) medical device. Further research is required to determine the most efficient way that this difference can be reflected in the regulations. With regards to adverse events, it is hard to establish the type of adverse event, as the largest category of events is based on issues originating from the ‘Computer Software’ itself. This does not help to determine the precise type of event. To this end, adverse event reporting for SaMD might be improved with a greater software-specific granularity in order to better inform regulatory authorities, as well as clinical professionals and patients. More detailed data on this will provide an improved understand of the risks associated with SaMD. It will also allow the regulator to develop further insights into ways to better control these devices.
Author Contributions
Conceptualization, A.C. and J.B.; methodology, A.C. and J.B.; software, A.C.; formal analysis, A.C.; investigation, A.C. and J.B.; resources, A.C.; data curation, A.C. and J.B.; writing—original draft preparation, A.C. and J.B.; writing—review and editing, A.C. and J.B.; visualization, A.C.; project administration, J.B.; funding acquisition, J.B. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Institute of Innovation and Technology (EIT) Health and supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC).
Data Availability Statement
All data were obtained from public databases and references are provided in the manuscript whenever they are discussed.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jamieson, J. Regulation of medical devices involving software in Australia: An overview. In Proceedings of the Sixth Australian Workshop on Safety Critical Systems and Software, Brisbane, Australia, 1 July 2001; Australian Computer Society, Inc.: Darlinghurst, Australia, 2001; Volume 3, pp. 7–12. [Google Scholar]
- Therapeutic Goods Administration. Actual and Potential Harm Caused by Medical Software. July 2020. Available online: https://www.tga.gov.au/resource/actual-and-potential-harm-caused-medical-software (accessed on 6 February 2021).
- IMDRF SaMDWorking Group. Software as a Medical Device (SaMD): Key Definitions; Tech. Rep.; International Medical Device Regulators Forum; December 2013; Available online: http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-samd-key-definitions-140901.pdf (accessed on 6 February 2021).
- Fu, Z.; Guo, C.; Zhang, Z.; Ren, S.; Jiang, Y.; Sha, L. Study of software-related causes in the fda medical device recalls. In Proceedings of the 2017 22nd IEEE International Conference on Engineering of Complex Computer Systems (ICECCS), Fukuoka, Japan, 6–8 November 2017; pp. 60–69. [Google Scholar]
- Ronquillo, J.G.; Zuckerman, D.M. Software-related recalls of health information technology and other medical devices: Implications for fda regulation of digital health. Milbank Q. 2017, 95, 535–553. [Google Scholar] [CrossRef] [PubMed]
- Therapeutic Goods Authority. Therapeutic Goods Authority Website. 2021. Available online: https://www.tga.gov.au/ (accessed on 25 June 2021).
- International Organization for Standardization. ISO/TS 19218-1:2011Medical Devices—Hierarchical Coding Structure for Adverse Events—Part 1: Event-Type Codes. Available online: https://www.iso.org/standard/55589.html (accessed on 2 April 2021).
- IEEE Standards Association. IEEE 1044-2009—IEEE Standard Classification for Software Anomalies. Available online: https://0-standards-ieee-org.brum.beds.ac.uk/standard/1044-2009.html (accessed on 2 April 2021).
- McHugh, M.; McCaffery, F.; Casey, V. Barriers to adopting agile practices when developing medical device software. In Software Process Improvement and Capability Determination; Mas, A., Mesquida, A., Rout, T., O’Connor, R.V., Dorling, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 141–147. [Google Scholar]
- Hendricusdottir, R.; Hussain, A.; Milnthorpe, W.; Bergmann, J.H.M. Lack of Support in Medical Device Regulation within Academia. Prosthesis 2021, 3, 1–8. [Google Scholar] [CrossRef]
- Herpin, T.F.; Karuso, H.; Foley, J.E. Australian biotech companies: Navigating the maze. J. Commer. Biotechnol. 2005, 11, 111–120. [Google Scholar] [CrossRef]
- Marucheck, A.; Greis, N.; Mena, C.; Cai, L. Product safety and security in the global supply chain: Issues, challenges and research opportunities. J. Oper. Manag. 2011, 29, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Thirumalai, S.; Sinha, K.K. Product recalls in the medical device industry: An empirical exploration of the sources and financial consequences. Manag. Sci. 2011, 57, 376–392. [Google Scholar] [CrossRef]
Figure 1.
Times series comparison of software-as-a-medical-device (SaMD) and “software that incorporated into a medical device” (non-SaMD) based on the registrations in the ARTG over a period ranging from 2014–2019 (n = 558). The dotted red line represent the lowess regression smoothing of all the time series data.
Figure 1.
Times series comparison of software-as-a-medical-device (SaMD) and “software that incorporated into a medical device” (non-SaMD) based on the registrations in the ARTG over a period ranging from 2014–2019 (n = 558). The dotted red line represent the lowess regression smoothing of all the time series data.

Figure 2.
Time series data of software-as-a-medical-device (SaMD) for registrations, based on the region of origin as reported in the ARTG. The data covers a period between 2014 and 2019. Dotted red lines represent the lowess regression for each of the time series.
Figure 2.
Time series data of software-as-a-medical-device (SaMD) for registrations, based on the region of origin as reported in the ARTG. The data covers a period between 2014 and 2019. Dotted red lines represent the lowess regression for each of the time series.

Figure 3.
Comparison of device classification between software-as-a-medical-device (SaMD), other software and non-software. Please note the difference in scales for each of the figures.
Figure 3.
Comparison of device classification between software-as-a-medical-device (SaMD), other software and non-software. Please note the difference in scales for each of the figures.

Figure 4.
Alluvial diagram of recalls types, levels, and recall class in the SARA dataset. Data are plotted for SaMD and all other medical devices (Non-SaMD). The scale is logarithmic.
Figure 4.
Alluvial diagram of recalls types, levels, and recall class in the SARA dataset. Data are plotted for SaMD and all other medical devices (Non-SaMD). The scale is logarithmic.

Figure 5.
Number and type of adverse events for both SaMD and non-SaMD software.

Figure 6.
Outcomes of adverse event related to software in DAEN for both SaMD and non-SaMD software.
Figure 6.
Outcomes of adverse event related to software in DAEN for both SaMD and non-SaMD software.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Recall actions obtained from the System for Australian Recall Action (SARA) database for the period from July 2012 to June 2020.
Table 1.
Recall actions obtained from the System for Australian Recall Action (SARA) database for the period from July 2012 to June 2020.
| Action | Count | Percentage |
|---|---|---|
| Recall for Product Correction | 1396 | 38.81 |
| Recall | 1193 | 33.17 |
| Product Defect Correction | 891 | 24.77 |
| Hazard Alert | 104 | 2.89 |
| Product Defect Alert | 13 | 0.36 |
Table 2.
The level at which recalls are made for all medical devices in System for Australian Recall Action (SARA).
Table 2.
The level at which recalls are made for all medical devices in System for Australian Recall Action (SARA).
| Action Level | Count | Percentage |
|---|---|---|
| Hospital | 3286 | 91.35 |
| Retail | 178 | 4.95 |
| Consumer | 118 | 3.28 |
| Wholesale | 15 | 0.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ceross, A.; Bergmann, J. Evaluating the Presence of Software-as-a-Medical-Device in the Australian Therapeutic Goods Register. Prosthesis 2021, 3, 221-228. https://0-doi-org.brum.beds.ac.uk/10.3390/prosthesis3030022
AMA Style
Ceross A, Bergmann J. Evaluating the Presence of Software-as-a-Medical-Device in the Australian Therapeutic Goods Register. Prosthesis. 2021; 3(3):221-228. https://0-doi-org.brum.beds.ac.uk/10.3390/prosthesis3030022
Chicago/Turabian StyleCeross, Aaron, and Jeroen Bergmann. 2021. "Evaluating the Presence of Software-as-a-Medical-Device in the Australian Therapeutic Goods Register" Prosthesis 3, no. 3: 221-228. https://0-doi-org.brum.beds.ac.uk/10.3390/prosthesis3030022