
Effect of Components and Operating Conditions on the Performance of PEM Electrolyzers: A Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Catalysts
2.1. Oxygen Evolution Reaction Catalysts
2.2. Nano Structuring of Ir
2.3. Alloying Ir with Hetero Metal (Oxide)
2.4. Depositing Ir on Supporting Substrates
3. Gas Diffusion Layers
3.1. Gas Diffusion Layers Modification by Microporous Layer (MPL)
3.2. Novel Thin Tunable Gas Diffusion Layer
3.3. Gas Diffusion Layers with Electrodeposited Catalyst
4. Bipolar Plates
4.1. Titanium-Based Bipolar Plates
4.2. Stainless Steel Bipolar Plates
5. Performance of PEMWE According to Operating Conditions
5.1. Operating Temperature
5.2. Flow Rate
5.3. H2 Pressurized Operation
6. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chi, J.; Yu, H. Water electrolysis based on renewable energy for hydrogen production. Chin. J. Catal. 2018, 39, 390–394. [Google Scholar] [CrossRef]
- LeValley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies–a review. Int. J. Hydrogen Energy 2014, 39, 16983–17000. [Google Scholar] [CrossRef]
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Midilli, A.; Dincer, I. Hydrogen as a renewable and sustainable solution in reducing global fossil fuel consumption. Int. J. Hydrogen Energy 2008, 33, 4209–4222. [Google Scholar] [CrossRef]
- Barbir, F. PEM electrolysis for production of hydrogen from renewable energy sources. Sol. Energy 2005, 78, 661–669. [Google Scholar] [CrossRef]
- Aricò, A.; Siracusano, S.; Briguglio, N.; Baglio, V.; Di Blasi, A.; Antonucci, V. Polymer electrolyte membrane water electrolysis: Status of technologies and potential applications in combination with renewable power sources. J. Appl. Electrochem. 2013, 43, 107–118. [Google Scholar] [CrossRef]
- Mo, J.; Dehoff, R.R.; Peter, W.H.; Toops, T.J.; Green, J.B., Jr.; Zhang, F. Additive manufacturing of liquid/gas diffusion layers for low-cost and high-efficiency hydrogen production. Int. J. Hydrogen Energy 2016, 41, 3128–3135. [Google Scholar] [CrossRef]
- Chisholm, G.; Kitson, P.J.; Kirkaldy, N.D.; Bloor, L.G.; Cronin, L. 3D printed flow plates for the electrolysis of water: An economic and adaptable approach to device manufacture. Energy Environ. Sci. 2014, 7, 3026–3032. [Google Scholar] [CrossRef]
- Song, S.; Zhang, H.; Ma, X.; Shao, Z.; Baker, R.T.; Yi, B. Electrochemical investigation of electrocatalysts for the oxygen evolution reaction in PEM water electrolyzers. Int. J. Hydrogen Energy 2008, 33, 4955–4961. [Google Scholar] [CrossRef]
- Han, B.; Steen, S.M., III; Mo, J.; Zhang, F. Electrochemical performance modeling of a proton exchange membrane electrolyzer cell for hydrogen energy. Int. J. Hydrogen Energy 2015, 40, 7006–7016. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, W.; Li, X.; Ouyang, T.; Liu, Z. Strong hydrophilicity NiS2/Fe7S8 heterojunctions encapsulated in N-doped carbon nanotubes for enhanced oxygen evolution reaction. Chem. Commun. 2020, 56, 1489–1492. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, M.; Zhou, X.; Huang, Z.; Xia, Z.; Chang, C.; Ma, Y.; Qu, Y. Mechanistic insights on ternary Ni2−xCoxP for hydrogen evolution and their hybrids with graphene as highly efficient and robust catalysts for overall water splitting. Adv. Funct. Mater. 2016, 26, 6785–6796. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, T.; Wang, L.; Zhong, J.; Liu, Z. Surface Reorganization on Electrochemically-Induced Zn–Ni–Co Spinel Oxides for Enhanced Oxygen Electrocatalysis. Angew. Chem. 2020, 132, 6554–6561. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef]
- Zhao, D.; Zhuang, Z.; Cao, X.; Zhang, C.; Peng, Q.; Chen, C.; Li, Y. Atomic site electrocatalysts for water splitting, oxygen reduction and selective oxidation. Chem. Soc. Rev. 2020, 49, 2215–2264. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Kirshenbaum, M.J.; Richter, M.H.; Dasog, M. Electrochemical Water Oxidation in Acidic Solution Using Titanium Diboride (TiB2) Catalyst. ChemCatChem 2019, 11, 3877–3881. [Google Scholar] [CrossRef]
- Gatto, I.; Stassi, A.; Baglio, V.; Carbone, A.; Passalacqua, E.; Aricò, A.; Schuster, M.; Bauer, B. Optimization of perfluorosulphonic ionomer amount in gas diffusion electrodes for PEMFC operation under automotive conditions. Electrochim. Acta 2015, 165, 450–455. [Google Scholar] [CrossRef]
- Mo, J.; Steen, S.; Kang, Z.; Yang, G.; Taylor, D.A.; Li, Y.; Toops, T.J.; Brady, M.P.; Retterer, S.T.; Cullen, D.A. Study on corrosion migrations within catalyst-coated membranes of proton exchange membrane electrolyzer cells. Int. J. Hydrogen Energy 2017, 42, 27343–27349. [Google Scholar] [CrossRef]
- Steen, S.M., III; Mo, J.; Kang, Z.; Yang, G.; Zhang, F. Investigation of titanium liquid/gas diffusion layers in proton exchange membrane electrolyzer cells. Int. J. Green Energy 2017, 14, 162–170. [Google Scholar] [CrossRef]
- Kang, Z.; Yu, S.; Yang, G.; Li, Y.; Bender, G.; Pivovar, B.S.; Green, J.B., Jr.; Zhang, F. Performance improvement of proton exchange membrane electrolyzer cells by introducing in-plane transport enhancement layers. Electrochim. Acta 2019, 316, 43–51. [Google Scholar] [CrossRef]
- Kang, Z.; Mo, J.; Yang, G.; Li, Y.; Talley, D.A.; Retterer, S.T.; Cullen, D.A.; Toops, T.J.; Brady, M.P.; Bender, G. Thin film surface modifications of thin/tunable liquid/gas diffusion layers for high-efficiency proton exchange membrane electrolyzer cells. Appl. Energy 2017, 206, 983–990. [Google Scholar] [CrossRef]
- Ito, H.; Maeda, T.; Nakano, A.; Kato, A.; Yoshida, T. Influence of pore structural properties of current collectors on the performance of proton exchange membrane electrolyzer. Electrochim. Acta 2013, 100, 242–248. [Google Scholar] [CrossRef]
- Hwang, C.M.; Ishida, M.; Ito, H.; Maeda, T.; Nakano, A.; Kato, A.; Yoshida, T. Effect of titanium powder loading in gas diffusion layer of a polymer electrolyte unitized reversible fuel cell. J. Power Sources 2012, 202, 108–113. [Google Scholar] [CrossRef]
- Liu, F.; Lu, G.; Wang, C. Low crossover of methanol and water through thin membranes in direct methanol fuel cells. J. Electrochem. Soc. 2006, 153, A543. [Google Scholar] [CrossRef]
- Nam, J.H.; Lee, K.; Hwang, G.; Kim, C.; Kaviany, M. Microporous layer for water morphology control in PEMFC. Int. J. Heat Mass Transfer 2009, 52, 2779–2791. [Google Scholar] [CrossRef]
- Minke, C.; Hickmann, T.; dos Santos, A.R.; Kunz, U.; Turek, T. Cost and performance prospects for composite bipolar plates in fuel cells and redox flow batteries. J. Power Sources 2016, 305, 182–190. [Google Scholar] [CrossRef]
- Bertuccioli, L.; Chan, A.; Hart, D.; Lehner, F.; Madden, B.; Standen, E. Study on Development of Water Electrolysis in the EU; Fuel Cells and Hydrogen Joint Undertaking: Lausanne, Switzerland, 2014; pp. 1–160. [Google Scholar]
- Yang, G.; Yu, S.; Mo, J.; Kang, Z.; Dohrmann, Y.; List, F.A., III; Green, J.B., Jr.; Babu, S.S.; Zhang, F. Bipolar plate development with additive manufacturing and protective coating for durable and high-efficiency hydrogen production. J. Power Sources 2018, 396, 590–598. [Google Scholar] [CrossRef]
- Wang, H.; Brady, M.P.; Teeter, G.; Turner, J. Thermally nitrided stainless steels for polymer electrolyte membrane fuel cell bipolar plates: Part 1: Model Ni–50Cr and austenitic 349™ alloys. J. Power Sources 2004, 138, 86–93. [Google Scholar] [CrossRef]
- Scotti, G.; Matilainen, V.; Kanninen, P.; Piili, H.; Salminen, A.; Kallio, T.; Franssila, S. Laser additive manufacturing of stainless steel micro fuel cells. J. Power Sources 2014, 272, 356–361. [Google Scholar] [CrossRef]
- Gago, A.; Ansar, S.; Saruhan, B.; Schulz, U.; Lettenmeier, P.; Cañas, N.A.; Gazdzicki, P.; Morawietz, T.; Hiesgen, R.; Arnold, J. Protective coatings on stainless steel bipolar plates for proton exchange membrane (PEM) electrolysers. J. Power Sources 2016, 307, 815–825. [Google Scholar] [CrossRef]
- Langemann, M.; Fritz, D.L.; Müller, M.; Stolten, D. Validation and characterization of suitable materials for bipolar plates in PEM water electrolysis. Int. J. Hydrogen Energy 2015, 40, 11385–11391. [Google Scholar] [CrossRef]
- Sun, X.; Xu, K.; Fleischer, C.; Liu, X.; Grandcolas, M.; Strandbakke, R.; Bjørheim, T.S.; Norby, T.; Chatzitakis, A. Earth-abundant electrocatalysts in proton exchange membrane electrolyzers. Catalysts 2018, 8, 657. [Google Scholar] [CrossRef]
- Jang, M.J.; Yang, J.; Lee, J.; Park, Y.S.; Jeong, J.; Park, S.M.; Jeong, J.; Yin, Y.; Seo, M.; Choi, S.M. Superior performance and stability of anion exchange membrane water electrolysis: PH-controlled copper cobalt oxide nanoparticles for the oxygen evolution reaction. J. Mater. Chem. A 2020, 8, 4290–4299. [Google Scholar] [CrossRef]
- Suen, N.; Hung, S.; Quan, Q.; Zhang, N.; Xu, Y.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, X.; Zhang, J.; Wan, S.; Guo, S.; Lu, G.; Yao, J.; Huang, X. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Jin, H.; Ruqia, B.; Park, Y.; Kim, H.J.; Oh, H.; Choi, S.; Lee, K. Nanocatalyst Design for Long-Term Operation of Proton/Anion Exchange Membrane Water Electrolysis. Adv. Energy Mater. 2021, 11, 2003188. [Google Scholar] [CrossRef]
- Song, F.; Bai, L.; Moysiadou, A.; Lee, S.; Hu, C.; Liardet, L.; Hu, X. Transition metal oxides as electrocatalysts for the oxygen evolution reaction in alkaline solutions: An application-inspired renaissance. J. Am. Chem. Soc. 2018, 140, 7748–7759. [Google Scholar] [CrossRef]
- Li, L.; Wang, P.; Shao, Q.; Huang, X. Recent progress in advanced electrocatalyst design for acidic oxygen evolution reaction. Adv. Mater. 2021, 33, 2004243. [Google Scholar] [CrossRef]
- McCrory, C.C.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, L.; Pan, L.; Yan, T.; He, Z.; Li, Y.; Shi, C.; Huang, Z.; Zhang, X.; Zou, J. Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv. Energy Mater. 2022, 12, 2103670. [Google Scholar] [CrossRef]
- Jang, H.; Lee, J. Iridium oxide fabrication and application: A review. J. Energy Chem. 2020, 46, 152–172. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Xu, J.; Liu, G.; Li, J.; Wang, X. The electrocatalytic properties of an IrO2/SnO2 catalyst using SnO2 as a support and an assisting reagent for the oxygen evolution reaction. Electrochim. Acta 2012, 59, 105–112. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Van Dijk, N.; Merlo, L.; Aricò, A.S. Enhanced performance and durability of low catalyst loading PEM water electrolyser based on a short-side chain perfluorosulfonic ionomer. Appl. Energy 2017, 192, 477–489. [Google Scholar] [CrossRef]
- Grigoriev, S.; Mamat, M.; Dzhus, K.; Walker, G.; Millet, P. Platinum and palladium nano-particles supported by graphitic nano-fibers as catalysts for PEM water electrolysis. Int. J. Hydrogen Energy 2011, 36, 4143–4147. [Google Scholar] [CrossRef]
- Yu, H.; Bonville, L.; Jankovic, J.; Maric, R. Microscopic insights on the degradation of a PEM water electrolyzer with ultra-low catalyst loading. Appl. Catal. B Environ. 2020, 260, 118194. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Briguglio, N.; Brunaccini, G.; Di Blasi, A.; Stassi, A.; Ornelas, R.; Trifoni, E.; Antonucci, V.; Aricò, A. An electrochemical study of a PEM stack for water electrolysis. Int. J. Hydrogen Energy 2012, 37, 1939–1946. [Google Scholar] [CrossRef]
- Su, H.; Bladergroen, B.J.; Linkov, V.; Pasupathi, S.; Ji, S. Study of catalyst sprayed membrane under irradiation method to prepare high performance membrane electrode assemblies for solid polymer electrolyte water electrolysis. Int. J. Hydrogen Energy 2011, 36, 15081–15088. [Google Scholar] [CrossRef]
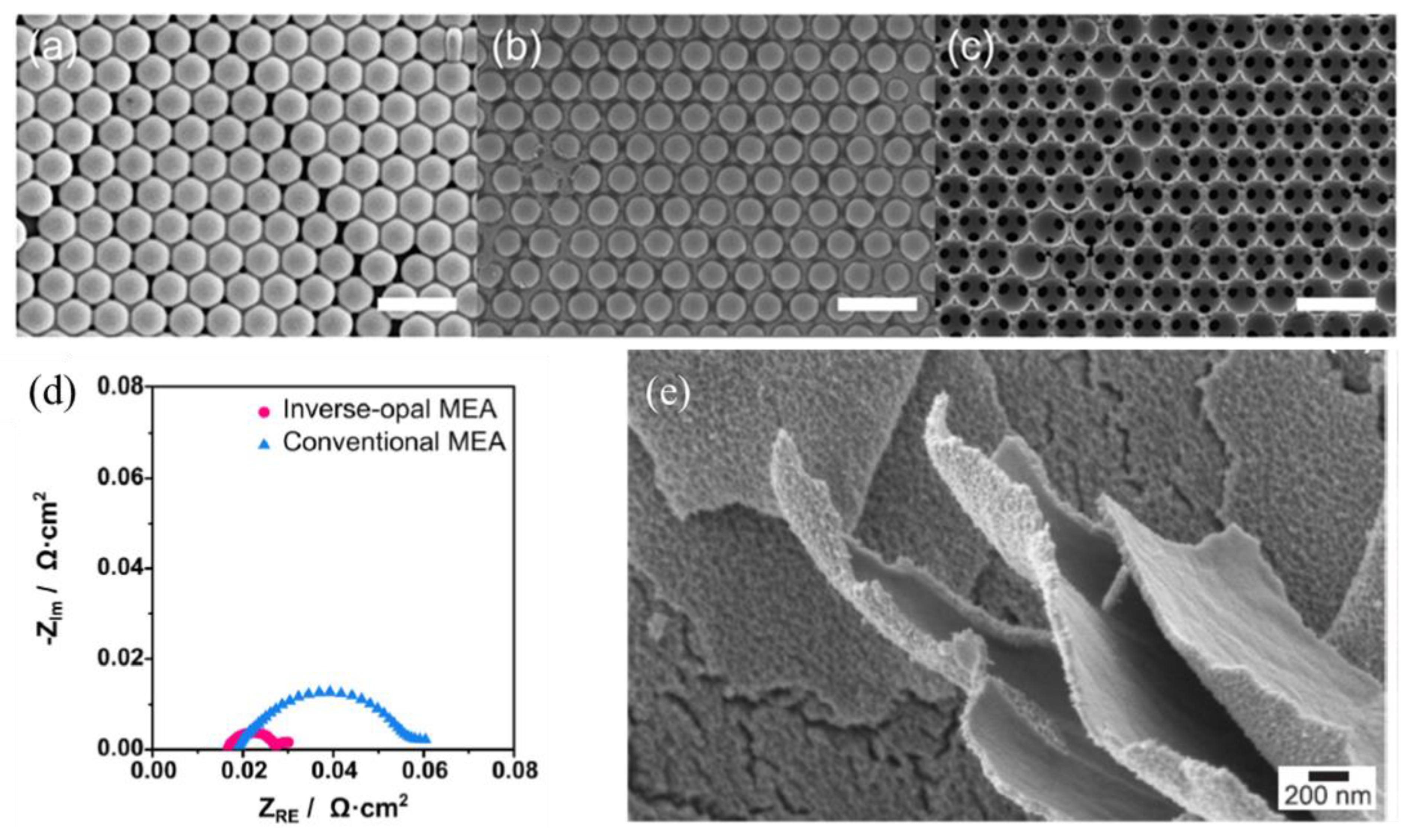
- Park, J.E.; Kim, S.; Kim, O.; Ahn, C.; Kim, M.; Kang, S.Y.; Jeon, T.I.; Shim, J.; Lee, D.W.; Lee, J.H. Ultra-low loading of IrO2 with an inverse-opal structure in a polymer-exchange membrane water electrolysis. Nano Energy 2019, 58, 158–166. [Google Scholar] [CrossRef]
- Oh, J.H.; Han, G.H.; Kim, H.; Jang, H.W.; Park, H.S.; Kim, S.Y.; Ahn, S.H. Activity and stability of Ir-based gas diffusion electrode for proton exchange membrane water electrolyzer. Chem Eng J. 2021, 420, 127696. [Google Scholar] [CrossRef]
- Lim, J.; Kang, G.; Lee, J.W.; Jeon, S.S.; Jeon, H.; Kang, P.W.; Lee, H. Amorphous Ir atomic clusters anchored on crystalline IrO2 nanoneedles for proton exchange membrane water oxidation. J. Power Sources 2022, 524, 231069. [Google Scholar] [CrossRef]
- Lee, S.W.; Baik, C.; Kim, D.; Pak, C. Control of Ir oxidation states to overcome the trade-off between activity and stability for the oxygen evolution reaction. J. Power Sources 2021, 493, 229689. [Google Scholar] [CrossRef]
- Lee, B.; Ahn, S.H.; Park, H.; Choi, I.; Yoo, S.J.; Kim, H.; Henkensmeier, D.; Kim, J.Y.; Park, S.; Nam, S.W. Development of electrodeposited IrO2 electrodes as anodes in polymer electrolyte membrane water electrolysis. Appl. Catal. B Environ. 2015, 179, 285–291. [Google Scholar] [CrossRef]
- Chatterjee, S.; Peng, X.; Intikhab, S.; Zeng, G.; Kariuki, N.N.; Myers, D.J.; Danilovic, N.; Snyder, J. Nanoporous Iridium Nanosheets for Polymer Electrolyte Membrane Electrolysis. Adv. Energy Mater. 2021, 11, 2101438. [Google Scholar] [CrossRef]
- Ghadge, S.D.; Velikokhatnyi, O.I.; Datta, M.K.; Shanthi, P.M.; Tan, S.; Damodaran, K.; Kumta, P.N. Experimental and theoretical validation of high efficiency and robust electrocatalytic response of one-dimensional (1D)(Mn, Ir) O2: 10F nanorods for the oxygen evolution reaction in PEM-based water electrolysis. ACS Catal. 2019, 9, 2134–2157. [Google Scholar] [CrossRef]
- Jiang, G.; Yu, H.; Hao, J.; Chi, J.; Fan, Z.; Yao, D.; Qin, B.; Shao, Z. An effective oxygen electrode based on Ir0.6Sn0.4O2 for PEM water electrolyzers. J. Energy Chem. 2019, 39, 23–28. [Google Scholar] [CrossRef]
- Lebedev, D.; Povia, M.; Waltar, K.; Abdala, P.M.; Castelli, I.E.; Fabbri, E.; Blanco, M.V.; Fedorov, A.; Copéret, C.; Marzari, N. Highly active and stable iridium pyrochlores for oxygen evolution reaction. Chem. Mater. 2017, 29, 5182–5191. [Google Scholar] [CrossRef]
- Lv, F.; Zhang, W.; Yang, W.; Feng, J.; Wang, K.; Zhou, J.; Zhou, P.; Guo, S. Ir-based alloy nanoflowers with optimized hydrogen binding energy as bifunctional electrocatalysts for overall water splitting. Small Methods 2020, 4, 1900129. [Google Scholar] [CrossRef]
- Ai, L.; Su, J.; Wang, M.; Jiang, J. Bamboo-structured nitrogen-doped carbon nanotube coencapsulating cobalt and molybdenum carbide nanoparticles: An efficient bifunctional electrocatalyst for overall water splitting. ACS Sustain. Chem. Eng. 2018, 6, 9912–9920. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, F.; Wang, G.; Lai, D.; Zou, L.; Cheng, Q.; Li, J.; Zou, Z.; Yang, H. CO induced phase-segregation to construct robust and efficient IrRux@ Ir core-shell electrocatalyst towards acidic oxygen evolution. J. Power Sources 2022, 528, 231189. [Google Scholar] [CrossRef]
- Saveleva, V.A.; Wang, L.; Kasian, O.; Batuk, M.; Hadermann, J.; Gallet, J.; Bournel, F.; Alonso-Vante, N.; Ozouf, G.; Beauger, C. Insight into the mechanisms of high activity and stability of iridium supported on antimony-doped tin oxide aerogel for anodes of proton exchange membrane water electrolyzers. ACS Catal. 2020, 10, 2508–2516. [Google Scholar] [CrossRef]
- Kúš, P.; Ostroverkh, A.; Ševčíková, K.; Khalakhan, I.; Fiala, R.; Skála, T.; Tsud, N.; Matolin, V. Magnetron sputtered Ir thin film on TiC-based support sublayer as low-loading anode catalyst for proton exchange membrane water electrolysis. Int. J. Hydrogen Energy 2016, 41, 15124–15132. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.; Kim, J.; Han, G.H.; Guo, W.; Hong, S.; Park, H.S.; Jang, H.W.; Kim, S.Y.; Ahn, S.H. Dendritic gold-supported iridium/iridium oxide ultra-low loading electrodes for high-performance proton exchange membrane water electrolyzer. Appl. Catal. B Environ. 2021, 283, 119596. [Google Scholar] [CrossRef]
- Kim, E.; Shin, J.; Bak, J.; Lee, S.J.; hyun Kim, K.; Song, D.; Roh, J.; Lee, Y.; Kim, H.; Lee, K. Stabilizing role of Mo in TiO2-MoOx supported Ir catalyst toward oxygen evolution reaction. Appl. Catal. B Environ. 2021, 280, 119433. [Google Scholar] [CrossRef]
- Jiang, G.; Yu, H.; Li, Y.; Yao, D.; Chi, J.; Sun, S.; Shao, Z. Low-Loading and Highly Stable Membrane Electrode Based on an Ir@ WO x NR Ordered Array for PEM Water Electrolysis. ACS Appl. Mater. Interfaces 2021, 13, 15073–15082. [Google Scholar] [CrossRef]
- Islam, J.; Kim, S.; Thien, P.T.; Kim, M.; Cho, H.; Cho, W.; Kim, C.; Lee, C.; Lee, J.H. Enhancing the activity and durability of iridium electrocatalyst supported on boron carbide by tuning the chemical state of iridium for oxygen evolution reaction. J. Power Sources 2021, 512, 230506. [Google Scholar] [CrossRef]
- Hartig-Weiss, A.; Miller, M.; Beyer, H.; Schmitt, A.; Siebel, A.; Freiberg, A.T.; Gasteiger, H.A.; El-Sayed, H.A. Iridium oxide catalyst supported on antimony-doped tin oxide for high oxygen evolution reaction activity in acidic media. ACS Appl. Nano Mater. 2020, 3, 2185–2196. [Google Scholar] [CrossRef]
- Genova-Koleva, R.V.; Alcaide, F.; Álvarez, G.; Cabot, P.L.; Grande, H.; Martínez-Huerta, M.V.; Miguel, O. Supporting IrO2 and IrRuOx nanoparticles on TiO2 and Nb-doped TiO2 nanotubes as electrocatalysts for the oxygen evolution reaction. J. Energy Chem. 2019, 34, 227–239. [Google Scholar] [CrossRef]
- Steegstra, P.; Ahlberg, E. Influence of oxidation state on the pH dependence of hydrous iridium oxide films. Electrochim. Acta 2012, 76, 26–33. [Google Scholar] [CrossRef]
- Yamanaka, K. Anodically electrodeposited iridium oxide films (AEIROF) from alkaline solutions for electrochromic display devices. Jpn. J. Appl. Phys. 1989, 28, 632. [Google Scholar] [CrossRef]
- Terashima, C.; Rao, T.N.; Sarada, B.V.; Spataru, N.; Fujishima, A. Electrodeposition of hydrous iridium oxide on conductive diamond electrodes for catalytic sensor applications. J. Electroanal. Chem. 2003, 544, 65–74. [Google Scholar] [CrossRef]
- Ges, I.A.; Ivanov, B.L.; Werdich, A.A.; Baudenbacher, F.J. Differential pH measurements of metabolic cellular activity in nl culture volumes using microfabricated iridium oxide electrodes. Biosens. Bioelectron. 2007, 22, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lv, F.; Zhang, W.; Li, P.; Wang, K.; Yang, C.; Wang, B.; Yang, Y.; Zhou, J.; Lin, F. Iridium-based multimetallic porous hollow nanocrystals for efficient overall-water-splitting catalysis. Adv. Mater. 2017, 29, 1703798. [Google Scholar] [CrossRef] [PubMed]
- Nong, H.N.; Oh, H.; Reier, T.; Willinger, E.; Willinger, M.; Petkov, V.; Teschner, D.; Strasser, P. Oxide-Supported IrNiOx Core–Shell Particles as Efficient, Cost-Effective, and Stable Catalysts for Electrochemical Water Splitting. Angew. Chem. Int. Ed. 2015, 54, 2975–2979. [Google Scholar] [CrossRef]
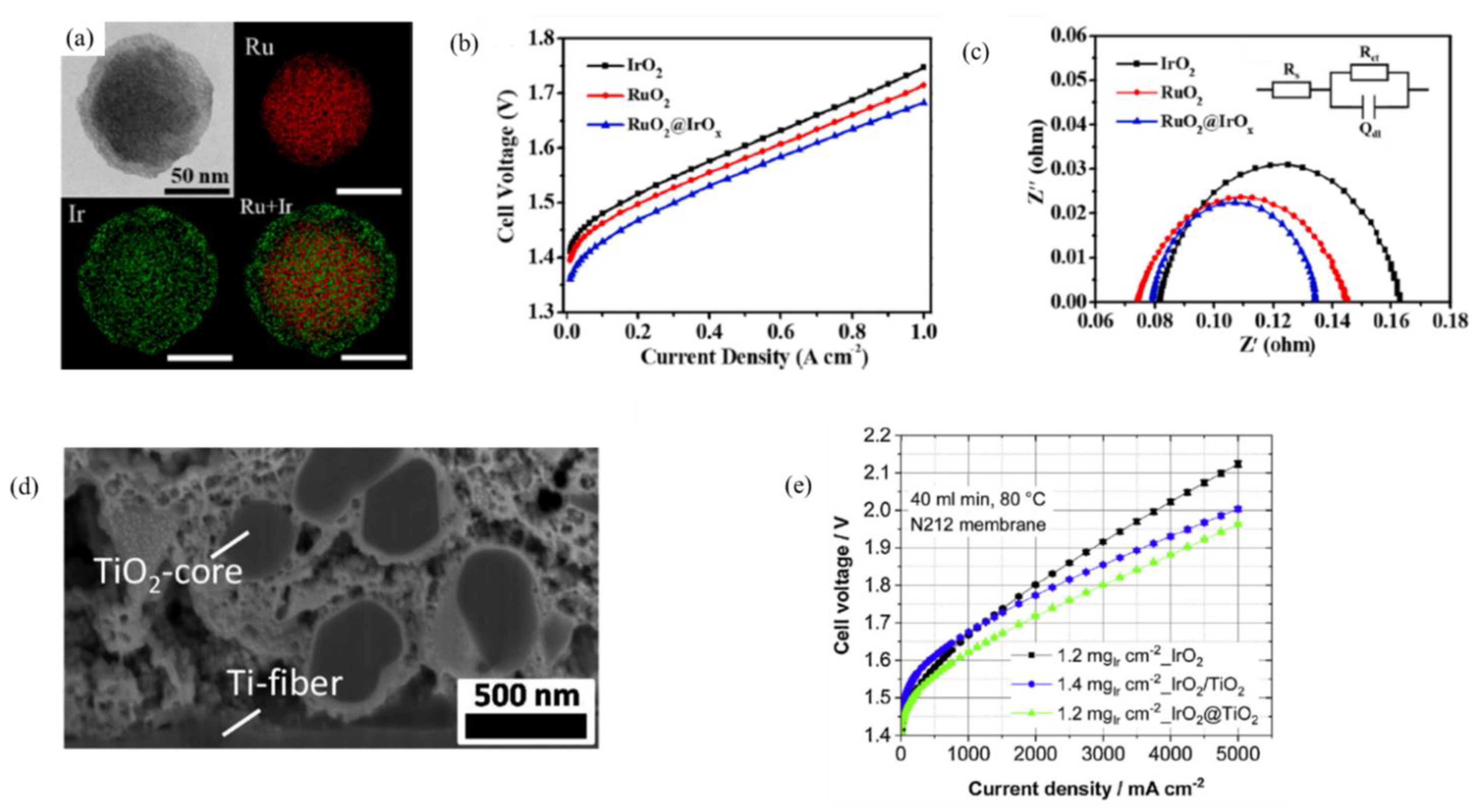
- Ma, Z.; Zhang, Y.; Liu, S.; Xu, W.; Wu, L.; Hsieh, Y.; Liu, P.; Zhu, Y.; Sasaki, K.; Renner, J.N. Reaction mechanism for oxygen evolution on RuO2, IrO2, and RuO2@ IrO2 core-shell nanocatalysts. J. Electroanal. Chem. 2018, 819, 296–305. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Pedersen, A.F.; Paoli, E.A.; Frydendal, R.; Friebel, D.; Malacrida, P.; Rossmeisl, J.; Stephens, I.E.; Chorkendorff, I. Importance of Surface IrO x in Stabilizing RuO2 for Oxygen Evolution. J. Phys. Chem. B 2018, 122, 947–955. [Google Scholar] [CrossRef]
- Lv, H.; Wang, S.; Li, J.; Shao, C.; Zhou, W.; Shen, X.; Xue, M.; Zhang, C. Self-assembled RuO2@ IrOx core-shell nanocomposite as high efficient anode catalyst for PEM water electrolyzer. Appl. Surf. Sci 2020, 514, 145943. [Google Scholar] [CrossRef]
- Van Pham, C.; Bühler, M.; Knöppel, J.; Bierling, M.; Seeberger, D.; Escalera-López, D.; Mayrhofer, K.J.; Cherevko, S.; Thiele, S. IrO2 coated TiO2 core-shell microparticles advance performance of low loading proton exchange membrane water electrolyzers. Appl. Catal. B Environ. 2020, 269, 118762. [Google Scholar] [CrossRef]
- Faustini, M.; Giraud, M.; Jones, D.; Rozière, J.; Dupont, M.; Porter, T.R.; Nowak, S.; Bahri, M.; Ersen, O.; Sanchez, C. Hierarchically structured ultraporous iridium-based materials: A novel catalyst architecture for proton exchange membrane water electrolyzers. Adv. Energy Mater. 2019, 9, 1802136. [Google Scholar] [CrossRef]
- Siracusano, S.; Hodnik, N.; Jovanovic, P.; Ruiz-Zepeda, F.; Šala, M.; Baglio, V.; Aricò, A.S. New insights into the stability of a high performance nanostructured catalyst for sustainable water electrolysis. Nano Energy 2017, 40, 618–632. [Google Scholar] [CrossRef]
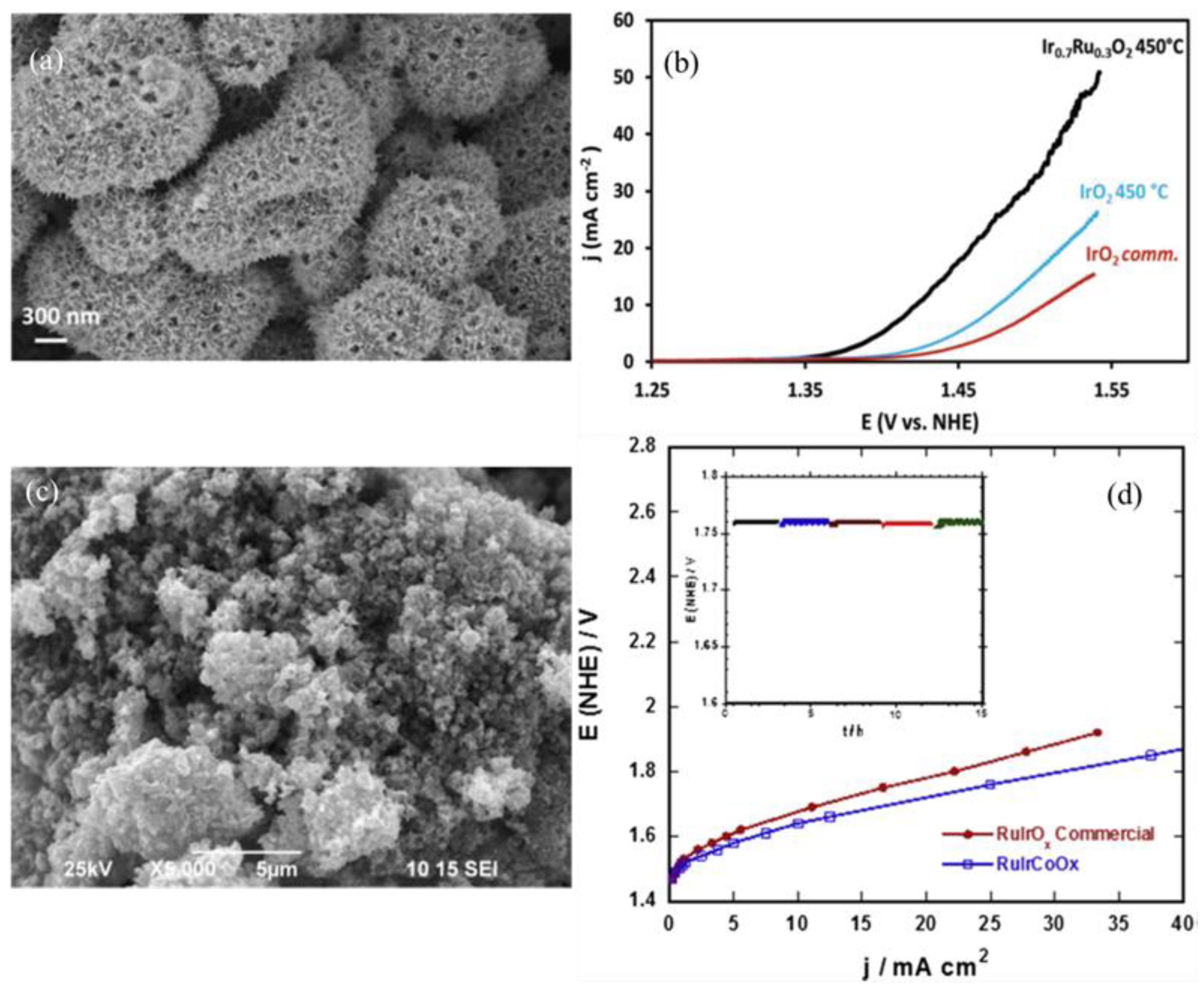
- Corona-Guinto, J.; Cardeño-García, L.; Martínez-Casillas, D.; Sandoval-Pineda, J.M.; Tamayo-Meza, P.; Silva-Casarin, R.; González-Huerta, R. Performance of a PEM electrolyzer using RuIrCoOx electrocatalysts for the oxygen evolution electrode. Int. J. Hydrogen Energy 2013, 38, 12667–12673. [Google Scholar] [CrossRef]
- Bernt, M.; Gasteiger, H.A. Influence of ionomer content in IrO2/TiO2 electrodes on PEM water electrolyzer performance. J. Electrochem. Soc. 2016, 163, F3179. [Google Scholar] [CrossRef]
- Marshall, A.T.; Sunde, S.; Tsypkin, M.; Tunold, R. Performance of a PEM water electrolysis cell using IrxRuyTazO2 electrocatalysts for the oxygen evolution electrode. Int. J. Hydrogen Energy 2007, 32, 2320–2324. [Google Scholar] [CrossRef]
- Li, G.; Li, K.; Yang, L.; Chang, J.; Ma, R.; Wu, Z.; Ge, J.; Liu, C.; Xing, W. Boosted performance of Ir species by employing TiN as the support toward oxygen evolution reaction. ACS Appl. Mater. Interfaces 2018, 10, 38117–38124. [Google Scholar] [CrossRef] [PubMed]
- Rozain, C.; Mayousse, E.; Guillet, N.; Millet, P. Influence of iridium oxide loadings on the performance of PEM water electrolysis cells: Part I–Pure IrO2-based anodes. Appl. Catal. B Environ. 2016, 182, 153–160. [Google Scholar] [CrossRef]
- Liu, G.; Xu, J.; Wang, Y.; Wang, X. An oxygen evolution catalyst on an antimony doped tin oxide nanowire structured support for proton exchange membrane liquid water electrolysis. J. Mater. Chem. A 2015, 3, 20791–20800. [Google Scholar] [CrossRef]
- Geiger, S.; Kasian, O.; Mingers, A.M.; Mayrhofer, K.J.; Cherevko, S. Stability limits of tin-based electrocatalyst supports. Sci. Rep. 2017, 7, 4595. [Google Scholar] [CrossRef]
- Polonský, J.; Mazúr, P.; Paidar, M.; Christensen, E.; Bouzek, K. Performance of a PEM water electrolyser using a TaC-supported iridium oxide electrocatalyst. Int. J. Hydrogen Energy 2014, 39, 3072–3078. [Google Scholar] [CrossRef]
- Oh, H.; Nong, H.N.; Reier, T.; Bergmann, A.; Gliech, M.; Ferreira de Arauújo, J.; Willinger, E.; Schloögl, R.; Teschner, D.; Strasser, P. Electrochemical catalyst–support effects and their stabilizing role for IrO x nanoparticle catalysts during the oxygen evolution reaction. J. Am. Chem. Soc. 2016, 138, 12552–12563. [Google Scholar] [CrossRef]
- Mazúr, P.; Polonský, J.; Paidar, M.; Bouzek, K. Non-conductive TiO2 as the anode catalyst support for PEM water electrolysis. Int. J. Hydrogen Energy 2012, 37, 12081–12088. [Google Scholar] [CrossRef]
- Karimi, F.; Peppley, B.A. Metal carbide and oxide supports for iridium-based oxygen evolution reaction electrocatalysts for polymer-electrolyte-membrane water electrolysis. Electrochim. Acta 2017, 246, 654–670. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, G.; Hao, C.; Mi, C.; Zhou, W.; Yang, D.; Li, B.; Zhang, C. Activity of IrO2 supported on tantalum-doped TiO2 electrocatalyst for solid polymer electrolyte water electrolyzer. Rsc Adv. 2017, 7, 40427–40436. [Google Scholar] [CrossRef]
- Tariq, M.; Zaman, W.Q.; Wu, Y.; Nabi, A.; Abbas, Z.; Iqbal, W.; Sun, W.; Hao, Z.; Zhou, Z.H.; Cao, L. Facile synthesis of IrO2 nanoparticles decorated@ WO3 as mixed oxide composite for outperformed oxygen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 31082–31093. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Di Blasi, A.; Briguglio, N.; Stassi, A.; Ornelas, R.; Trifoni, E.; Antonucci, V.; Arico, A. Electrochemical characterization of single cell and short stack PEM electrolyzers based on a nanosized IrO2 anode electrocatalyst. Int. J. Hydrogen Energy 2010, 35, 5558–5568. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Stassi, A.; Ornelas, R.; Antonucci, V.; Aricò, A. Investigation of IrO2 electrocatalysts prepared by a sulfite-couplex route for the O2 evolution reaction in solid polymer electrolyte water electrolyzers. Int. J. Hydrogen Energy 2011, 36, 7822–7831. [Google Scholar] [CrossRef]
- Seitz, L.C.; Dickens, C.F.; Nishio, K.; Hikita, Y.; Montoya, J.; Doyle, A.; Kirk, C.; Vojvodic, A.; Hwang, H.Y.; Norskov, J.K.; et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 2016, 353, 1011–1014. [Google Scholar] [CrossRef]
- Puthiyapura, V.K.; Pasupathi, S.; Su, H.; Liu, X.; Pollet, B.; Scott, K. Investigation of supported IrO2 as electrocatalyst for the oxygen evolution reaction in proton exchange membrane water electrolyser. Int. J. Hydrogen Energy 2014, 39, 1905–1913. [Google Scholar] [CrossRef]
- Park, S.; Lee, J.; Popov, B.N. A review of gas diffusion layer in PEM fuel cells: Materials and designs. Int. J. Hydrogen Energy 2012, 37, 5850–5865. [Google Scholar] [CrossRef]
- Park, J.; Oh, H.; Lee, Y.I.; Min, K.; Lee, E.; Jyoung, J. Effect of the pore size variation in the substrate of the gas diffusion layer on water management and fuel cell performance. Appl. Energy 2016, 171, 200–212. [Google Scholar] [CrossRef]
- Mamaca, N.; Mayousse, E.; Arrii-Clacens, S.; Napporn, T.; Servat, K.; Guillet, N.; Kokoh, K. Electrochemical activity of ruthenium and iridium based catalysts for oxygen evolution reaction. Appl. Catal. B Environ. 2012, 111, 376–380. [Google Scholar] [CrossRef]
- Manzo-Robledo, A.; Boucher, A.; Pastor, E.; Alonso-Vante, N. Electro-oxidation of Carbon Monoxide and Methanol on Carbon-Supported Pt–Sn Nanoparticles: A DEMS Study. Fuel Cells 2002, 2, 109–116. [Google Scholar] [CrossRef]
- Ito, H.; Maeda, T.; Nakano, A.; Hwang, C.M.; Ishida, M.; Kato, A.; Yoshida, T. Experimental study on porous current collectors of PEM electrolyzers. Int. J. Hydrogen Energy 2012, 37, 7418–7428. [Google Scholar] [CrossRef]
- Lettenmeier, P.; Kolb, S.; Burggraf, F.; Gago, A.; Friedrich, K.A. Towards developing a backing layer for proton exchange membrane electrolyzers. J. Power Sources 2016, 311, 153–158. [Google Scholar] [CrossRef]
- Polonský, J.; Kodým, R.; Vágner, P.; Paidar, M.; Bensmann, B.; Bouzek, K. Anodic microporous layer for polymer electrolyte membrane water electrolysers. J. Appl. Electrochem. 2017, 47, 1137–1146. [Google Scholar] [CrossRef]
- Bystron, T.; Vesely, M.; Paidar, M.; Papakonstantinou, G.; Sundmacher, K.; Bensmann, B.; Hanke-Rauschenbach, R.; Bouzek, K. Enhancing PEM water electrolysis efficiency by reducing the extent of Ti gas diffusion layer passivation. J. Appl. Electrochem. 2018, 48, 713–723. [Google Scholar] [CrossRef]
- Wang, H.; Turner, J. Reviewing metallic PEMFC bipolar plates. Fuel Cells 2010, 10, 510–519. [Google Scholar] [CrossRef]
- Jung, H.; Huang, S.; Ganesan, P.; Popov, B.N. Performance of gold-coated titanium bipolar plates in unitized regenerative fuel cell operation. J. Power Sources 2009, 194, 972–975. [Google Scholar] [CrossRef]
- Choe, S.; Lee, B.; Cho, M.K.; Kim, H.; Henkensmeier, D.; Yoo, S.J.; Kim, J.Y.; Lee, S.Y.; Park, H.S.; Jang, J.H. Electrodeposited IrO2/Ti electrodes as durable and cost-effective anodes in high-temperature polymer-membrane-electrolyte water electrolyzers. Appl. Catal. B Environ. 2018, 226, 289–294. [Google Scholar] [CrossRef]
- Kang, Z.; Yang, G.; Mo, J.; Yu, S.; Cullen, D.A.; Retterer, S.T.; Toops, T.J.; Brady, M.P.; Bender, G.; Pivovar, B.S. Developing titanium micro/nano porous layers on planar thin/tunable LGDLs for high-efficiency hydrogen production. Int. J. Hydrogen Energy 2018, 43, 14618–14628. [Google Scholar] [CrossRef]
- Su, H.; Xu, Q.; Chong, J.; Li, H.; Sita, C.; Pasupathi, S. Eliminating micro-porous layer from gas diffusion electrode for use in high temperature polymer electrolyte membrane fuel cell. J. Power Sources 2017, 341, 302–308. [Google Scholar] [CrossRef]
- Hu, W.; Chen, S.; Xia, Q. IrO2/Nb–TiO2 electrocatalyst for oxygen evolution reaction in acidic medium. Int. J. Hydrogen Energy 2014, 39, 6967–6976. [Google Scholar] [CrossRef]
- Puthiyapura, V.K.; Mamlouk, M.; Pasupathi, S.; Pollet, B.G.; Scott, K. Physical and electrochemical evaluation of ATO supported IrO2 catalyst for proton exchange membrane water electrolyser. J. Power Sources 2014, 269, 451–460. [Google Scholar] [CrossRef]
- Grigoriev, S.; Millet, P.; Volobuev, S.; Fateev, V. Optimization of porous current collectors for PEM water electrolysers. Int. J. Hydrogen Energy 2009, 34, 4968–4973. [Google Scholar] [CrossRef]
- Schuler, T.; Ciccone, J.M.; Krentscher, B.; Marone, F.; Peter, C.; Schmidt, T.J.; Büchi, F.N. Hierarchically structured porous transport layers for polymer electrolyte water electrolysis. Adv. Energy Mater. 2020, 10, 1903216. [Google Scholar] [CrossRef]
- Sung, C.; Liu, C. A novel micro protective layer applied on a simplified PEM water electrolyser. Int. J. Hydrogen Energy 2013, 38, 10063–10067. [Google Scholar] [CrossRef]
- Liu, C.; Hu, L.; Sung, C. Micro-protective layer for lifetime extension of solid polymer electrolyte water electrolysis. J. Power Sources 2012, 207, 81–85. [Google Scholar] [CrossRef]
- Kang, Z.; Mo, J.; Yang, G.; Retterer, S.T.; Cullen, D.A.; Toops, T.J.; Green, J.B., Jr.; Mench, M.M.; Zhang, F. Investigation of thin/well-tunable liquid/gas diffusion layers exhibiting superior multifunctional performance in low-temperature electrolytic water splitting. Energy Environ. Sci. 2017, 10, 166–175. [Google Scholar] [CrossRef]
- Lettenmeier, P.; Kolb, S.; Sata, N.; Fallisch, A.; Zielke, L.; Thiele, S.; Gago, A.; Friedrich, K.A. Comprehensive investigation of novel pore-graded gas diffusion layers for high-performance and cost-effective proton exchange membrane electrolyzers. Energy Environ. Sci. 2017, 10, 2521–2533. [Google Scholar] [CrossRef]
- Kim, H.; Choe, S.; Park, H.; Jang, J.H.; Ahn, S.H.; Kim, S. An extremely low Pt loading cathode for a highly efficient proton exchange membrane water electrolyzer. Nanoscale 2017, 9, 19045–19049. [Google Scholar] [CrossRef]
- Egetenmeyer, A.; Radev, I.; Durneata, D.; Baumgärtner, M.; Peinecke, V.; Natter, H.; Hempelmann, R. Pulse electrodeposited cathode catalyst layers for PEM fuel cells. Int. J. Hydrogen Energy 2017, 42, 13649–13660. [Google Scholar] [CrossRef]
- Han, Y.J.; Zhang, X.; Leach, G.W. Shape control of electrodeposited copper films and nanostructures through additive effects. Langmuir 2014, 30, 3589–3598. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wippermann, K.; Rasinski, M.; Suo, Y.; Shviro, M.; Carmo, M.; Lehnert, W. Constructing a Multifunctional Interface between Membrane and Porous Transport Layer for Water Electrolyzers. ACS Appl. Mater. Interfaces 2021, 13, 16182–16196. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, J.; Park, Y.S.; Yang, J.; Jang, M.J.; Jeong, J.; Choe, S.; Lee, J.W.; Kwon, J.; Choi, S.M. Electrodeposition of high-surface-area IrO2 films on Ti felt as an efficient catalyst for the oxygen evolution reaction. Front. Chem. 2020, 8, 593272. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Z.; Gong, X.; Guo, Z. The intensification technologies to water electrolysis for hydrogen production—A review. Renew. Sustain. Energy Rev. 2014, 29, 573–588. [Google Scholar] [CrossRef]
- Chalker, P.; Bull, S.; Rickerby, D. A review of the methods for the evaluation of coating-substrate adhesion. Mater. Sci. Eng. A 1991, 140, 583–592. [Google Scholar] [CrossRef]
- Lee, B.; Park, H.; Choi, I.; Cho, M.K.; Kim, H.; Yoo, S.J.; Henkensmeier, D.; Kim, J.Y.; Nam, S.W.; Park, S. Polarization characteristics of a low catalyst loading PEM water electrolyzer operating at elevated temperature. J. Power Sources 2016, 309, 127–134. [Google Scholar] [CrossRef]
- Antonucci, V.; Di Blasi, A.; Baglio, V.; Ornelas, R.; Matteucci, F.; Ledesma-Garcia, J.; Arriaga, L.; Aricò, A. High temperature operation of a composite membrane-based solid polymer electrolyte water electrolyser. Electrochim. Acta 2008, 53, 7350–7356. [Google Scholar] [CrossRef]
- Baglio, V.; Ornelas, R.; Matteucci, F.; Martina, F.; Ciccarella, G.; Zama, I.; Arriaga, L.; Antonucci, V.; Aricò, A. Solid Polymer Electrolyte Water Electrolyser Based on Nafion-TiO2 Composite Membrane for High Temperature Operation. Fuel Cells 2009, 9, 247–252. [Google Scholar] [CrossRef]
- Xu, W.; Scott, K.; Basu, S. Performance of a high temperature polymer electrolyte membrane water electrolyser. J. Power Sources 2011, 196, 8918–8924. [Google Scholar] [CrossRef]
- Kim, J.Y.; Choi, J.; Kim, H.Y.; Hwang, E.; Kim, H.; Ahn, S.H.; Kim, S. Activity and stability of the oxygen evolution reaction on electrodeposited Ru and its thermal oxides. Appl. Surf. Sci 2015, 359, 227–235. [Google Scholar] [CrossRef]
- Lee, B.; Park, H.; Cho, M.K.; Jung, J.W.; Kim, H.; Henkensmeier, D.; Yoo, S.J.; Kim, J.Y.; Park, S.; Lee, K. Development of porous Pt/IrO2/carbon paper electrocatalysts with enhanced mass transport as oxygen electrodes in unitized regenerative fuel cells. Electrochem. Commun. 2016, 64, 14–17. [Google Scholar] [CrossRef]
- Lim, A.; Kim, J.; Lee, H.J.; Kim, H.; Yoo, S.J.; Jang, J.H.; Park, H.Y.; Sung, Y.; Park, H.S. Low-loading IrO2 supported on Pt for catalysis of PEM water electrolysis and regenerative fuel cells. Appl. Catal. B Environ. 2020, 272, 118955. [Google Scholar] [CrossRef]
- Liu, C.; Shviro, M.; Gago, A.S.; Zaccarine, S.F.; Bender, G.; Gazdzicki, P.; Morawietz, T.; Biswas, I.; Rasinski, M.; Everwand, A. Exploring the Interface of Skin-Layered Titanium Fibers for Electrochemical Water Splitting. Adv. Energy Mater. 2021, 11, 2002926. [Google Scholar] [CrossRef]
- Li, X.; Sabir, I. Review of bipolar plates in PEM fuel cells: Flow-field designs. Int. J. Hydrogen Energy 2005, 30, 359–371. [Google Scholar] [CrossRef]
- Park, J.; Li, X. Effect of flow and temperature distribution on the performance of a PEM fuel cell stack. J. Power Sources 2006, 162, 444–459. [Google Scholar] [CrossRef]
- Lædre, S.; Kongstein, O.E.; Oedegaard, A.; Karoliussen, H.; Seland, F. Materials for Proton Exchange Membrane water electrolyzer bipolar plates. Int. J. Hydrogen Energy 2017, 42, 2713–2723. [Google Scholar] [CrossRef]
- Lettenmeier, P.; Wang, R.; Abouatallah, R.; Saruhan, B.; Freitag, O.; Gazdzicki, P.; Morawietz, T.; Hiesgen, R.; Gago, A.; Friedrich, K. Low-cost and durable bipolar plates for proton exchange membrane electrolyzers. Sci. Rep. 2017, 7, 44035. [Google Scholar] [CrossRef] [PubMed]
- Bessarabov, D.; Wang, H.; Li, H.; Zhao, N. PEM Electrolysis for Hydrogen Production: Principles and Applications; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
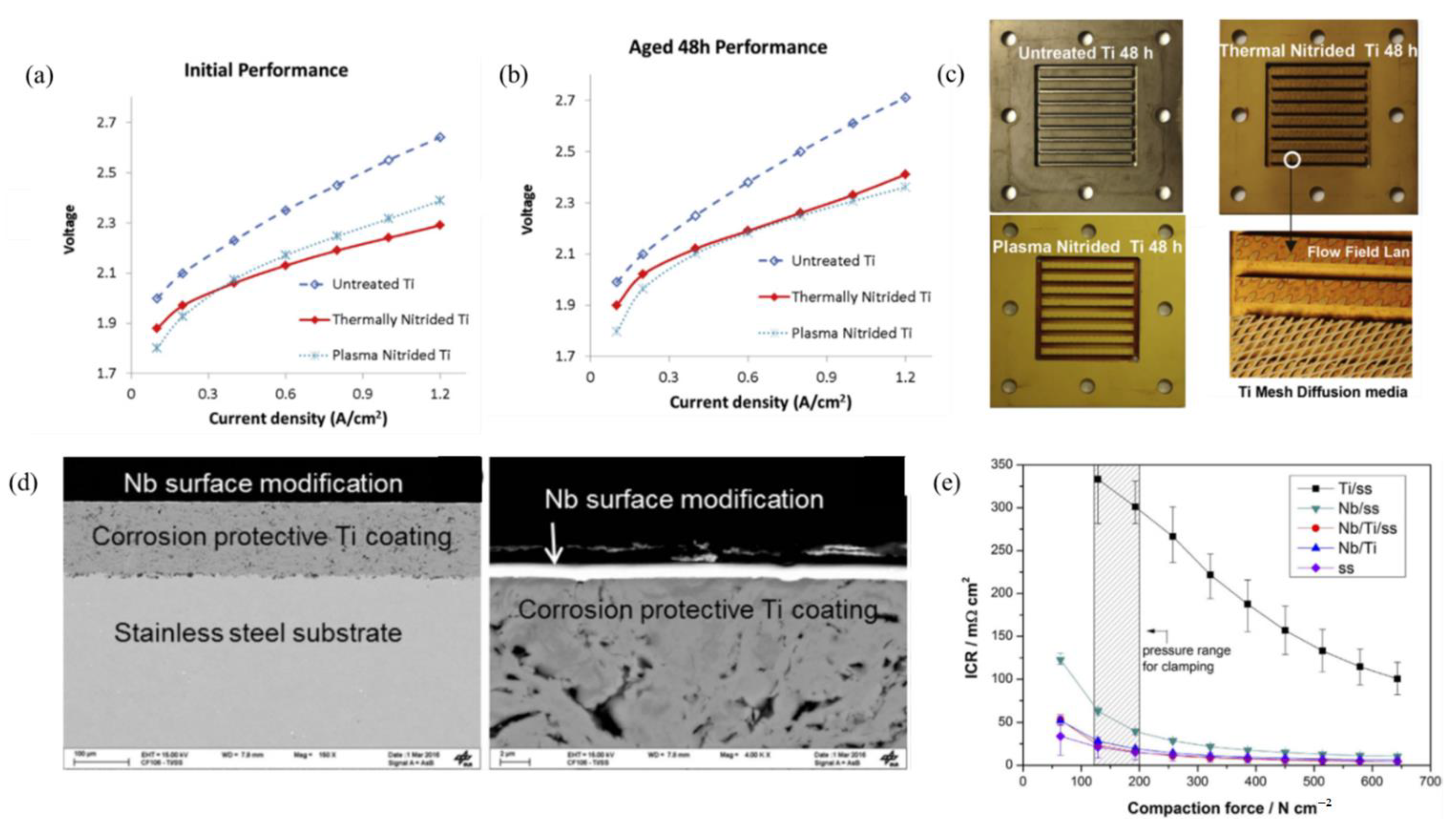
- Toops, T.J.; Brady, M.P.; Zhang, F.; Meyer, H.M., III; Ayers, K.; Roemer, A.; Dalton, L. Evaluation of nitrided titanium separator plates for proton exchange membrane electrolyzer cells. J. Power Sources 2014, 272, 954–960. [Google Scholar] [CrossRef]
- Jung, H.; Huang, S.; Popov, B.N. High-durability titanium bipolar plate modified by electrochemical deposition of platinum for unitized regenerative fuel cell (URFC). J. Power Sources 2010, 195, 1950–1956. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Wang, C.; Mao, Z. Corrosion behavior of three bipolar plate materials in simulated SPE water electrolysis environment. Int. J. Hydrogen Energy 2012, 37, 12069–12073. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, L.; Lin, G.; Shao, Z. Honeycomb-like nanocomposite Ti-Ag-N films prepared by pulsed bias arc ion plating on titanium as bipolar plates for unitized regenerative fuel cells. J. Power Sources 2012, 198, 196–202. [Google Scholar] [CrossRef]
- Lin, M.; Wan, C.; Wu, W. Comparison of corrosion behaviors between SS304 and Ti substrate coated with (Ti, Zr) N thin films as Metal bipolar plate for unitized regenerative fuel cell. Thin Solid Films 2013, 544, 162–169. [Google Scholar] [CrossRef]
- Nikiforov, A.; Petrushina, I.; Christensen, E.; Tomás-García, A.; Bjerrum, N. Corrosion behaviour of construction materials for high temperature steam electrolysers. Int. J. Hydrogen Energy 2011, 36, 111–119. [Google Scholar] [CrossRef]
- Lettenmeier, P.; Wang, R.; Abouatallah, R.; Burggraf, F.; Gago, A.; Friedrich, K. Coated stainless steel bipolar plates for proton exchange membrane electrolyzers. J. Electrochem. Soc. 2016, 163, F3119. [Google Scholar] [CrossRef]
- Wu, J.; Yuan, X.Z.; Martin, J.J.; Wang, H.; Zhang, J.; Shen, J.; Wu, S.; Merida, W. A review of PEM fuel cell durability: Degradation mechanisms and mitigation strategies. J. Power Sources 2008, 184, 104–119. [Google Scholar] [CrossRef]
- Sun, S.; Shao, Z.; Yu, H.; Li, G.; Yi, B. Investigations on degradation of the long-term proton exchange membrane water electrolysis stack. J. Power Sources 2014, 267, 515–520. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Li, G.; Zhang, G.; Shao, Z.; Yi, B. The influence of Ferric ion contamination on the solid polymer electrolyte water electrolysis performance. Electrochim. Acta 2015, 158, 253–257. [Google Scholar] [CrossRef]
- Gago, A.S.; Ansar, A.S.; Gazdzicki, P.; Wagner, N.; Arnold, J.; Friedrich, K.A. Low cost bipolar plates for large scale PEM electrolyzers. ECS Trans. 2014, 64, 1039. [Google Scholar] [CrossRef]
- Gago, A.; Ansar, A.; Wagner, N.; Arnold, J.; Friedrich, K.A. Titanium coatings deposited by thermal spraying for bipolar plates of PEM electrolysers. In Proceedings of the 4th European PEFC and H2 Forum, Lucerne, Switzerland, 2–5 July 2013. [Google Scholar]
- Kim, K.M.; Kim, J.H.; Lee, Y.Y.; Kim, K.Y. Electrodeposition of ruthenium oxide on ferritic stainless steel bipolar plate for polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 2012, 37, 1653–1660. [Google Scholar] [CrossRef]
- Kumar, A.; Ricketts, M.; Hirano, S. Ex situ evaluation of nanometer range gold coating on stainless steel substrate for automotive polymer electrolyte membrane fuel cell bipolar plate. J. Power Sources 2010, 195, 1401–1407. [Google Scholar] [CrossRef]
- Rojas, N.; Sánchez-Molina, M.; Sevilla, G.; Amores, E.; Almandoz, E.; Esparza, J.; Vivas, M.R.C.; Colominas, C. Coated stainless steels evaluation for bipolar plates in PEM water electrolysis conditions. Int. J. Hydrogen Energy 2021, 46, 25929–25943. [Google Scholar] [CrossRef]
- Lee, S.; Woo, S.; Kakati, N.; Lee, Y.; Yoon, Y. Corrosion and electrical properties of carbon/ceramic multilayer coated on stainless steel bipolar plates. Surf. Coat. Technol. 2016, 303, 162–169. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, W.; Na, Y. Effect of Gravity and Various Operating Conditions on Proton Exchange Membrane Water Electrolysis Cell Performance. Membranes 2021, 11, 822. [Google Scholar] [CrossRef] [PubMed]
- Kai, J.; Saito, R.; Terabaru, K.; Li, H.; Nakajima, H.; Ito, K. Effect of temperature on the performance of polymer electrolyte membrane water electrolysis: Numerical analysis of electrolysis voltage considering gas/liquid two-phase flow. J. Electrochem. Soc. 2019, 166, F246. [Google Scholar] [CrossRef]
- Chandesris, M.; Médeau, V.; Guillet, N.; Chelghoum, S.; Thoby, D.; Fouda-Onana, F. Membrane degradation in PEM water electrolyzer: Numerical modeling and experimental evidence of the influence of temperature and current density. Int. J. Hydrogen Energy 2015, 40, 1353–1366. [Google Scholar] [CrossRef]
- Inaba, M.; Kinumoto, T.; Kiriake, M.; Umebayashi, R.; Tasaka, A.; Ogumi, Z. Gas crossover and membrane degradation in polymer electrolyte fuel cells. Electrochim. Acta 2006, 51, 5746–5753. [Google Scholar] [CrossRef]
- Yoshitake, M.; Tamura, M.; Yoshida, N.; Ishisaki, T. Studies of perfluorinated ion exchange membranes for polymer electrolyte fuel cells. Denki Kagaku Oyobi Kogyo Butsuri Kagaku 1996, 64, 727–736. [Google Scholar] [CrossRef]
- Kaya, M.F.; Demir, N.; Rees, N.V.; El-Kharouf, A. Improving PEM water electrolyser’s performance by magnetic field application. Appl. Energy 2020, 264, 114721. [Google Scholar] [CrossRef]
- Olesen, A.C.; Rømer, C.; Kær, S.K. A numerical study of the gas-liquid, two-phase flow maldistribution in the anode of a high pressure PEM water electrolysis cell. Int. J. Hydrogen Energy 2016, 41, 52–68. [Google Scholar] [CrossRef] [Green Version]
- Hoeh, M.A.; Arlt, T.; Manke, I.; Banhart, J.; Fritz, D.L.; Maier, W.; Lehnert, W. In operando synchrotron X-ray radiography studies of polymer electrolyte membrane water electrolyzers. Electrochem. Commun. 2015, 55, 55–59. [Google Scholar] [CrossRef]
- Lopata, J.; Kang, Z.; Young, J.; Bender, G.; Weidner, J.; Shimpalee, S. Effects of the transport/catalyst layer interface and catalyst loading on mass and charge transport phenomena in polymer electrolyte membrane water electrolysis devices. J. Electrochem. Soc. 2020, 167, 064507. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Yang, W.; Liu, W.; Min, F.; Mao, S.S.; Xie, J. Catalyst-coated proton exchange membrane for hydrogen production with high pressure water electrolysis. Appl. Phys. Lett 2021, 119, 123903. [Google Scholar] [CrossRef]
- Stiber, S.; Balzer, H.; Wierhake, A.; Wirkert, F.J.; Roth, J.; Rost, U.; Brodmann, M.; Lee, J.K.; Bazylak, A.; Waiblinger, W. Porous Transport Layers for Proton Exchange Membrane Electrolysis Under Extreme Conditions of Current Density, Temperature, and Pressure. Adv. Energy Mater. 2021, 11, 2100630. [Google Scholar] [CrossRef]
- Bernt, M.; Schröter, J.; Möckl, M.; Gasteiger, H. Analysis of gas permeation phenomena in a PEM water electrolyzer operated at high pressure and high current density. J. Electrochem. Soc. 2020, 167, 124502. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, K.W.; Jang, M.J.; Park, M.G.; Chen, Z.; Fowler, M. Effect of Components and Operating Conditions on the Performance of PEM Electrolyzers: A Review. Electrochem 2022, 3, 581-612. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem3040040
Ahmed KW, Jang MJ, Park MG, Chen Z, Fowler M. Effect of Components and Operating Conditions on the Performance of PEM Electrolyzers: A Review. Electrochem. 2022; 3(4):581-612. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem3040040
Chicago/Turabian StyleAhmed, Khaja Wahab, Myeong Je Jang, Moon Gyu Park, Zhongwei Chen, and Michael Fowler. 2022. "Effect of Components and Operating Conditions on the Performance of PEM Electrolyzers: A Review" Electrochem 3, no. 4: 581-612. https://0-doi-org.brum.beds.ac.uk/10.3390/electrochem3040040