Decidual Vasculopathy and Spiral Artery Remodeling Revisited III: Hypoxia and Re-oxygenation Sequence with Vascular Regeneration


Department of Pathology, New York Presbyterian—Brooklyn Methodist Hospital, Brooklyn, NY 11215, USA
Reprod. Med. 2020, 1(2), 77-90; https://0-doi-org.brum.beds.ac.uk/10.3390/reprodmed1020006
Submission received: 26 May 2020
/
Revised: 5 July 2020
/
Accepted: 9 July 2020
/
Published: 11 July 2020
(This article belongs to the Special Issue Preeclampsia: Pathogenesis, Diagnosis and Treatment)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Aim: Spiral artery remodeling at early pregnancy is characterized by two distinct mechanisms with two morphologic features, namely, trophoblastic-dependent vascular invasion with “plugging”, and trophoblastic-independent mural muscular hypertrophy/hyperplasia, both of which lead to the blocking or narrowing of the arterial lumen with the consequence of reduced maternal blood flow to the developing embryo. Methods: Review of historic literature in light of the new discovery of CD56 (NCAM) expression on endovascular trophoblasts at late gestation, in relation to placental lateral growth with vascular regeneration. Results: Reduced maternal blood flow to the embryo results in a hypoxic condition critical for trophectoderm differentiation and proliferation. Hypoxia is also important for the development of hemangioblasts of vasculogenesis, and hematopoiesis of the placental villi. Up to 13 weeks, both uteroplacental and fetoplacental circulations are established and hypoxic condition relieved for normal fetal/placenta development by ultrasonography. The persistence of trophoblastic plugging and/or mural muscular hypertrophy/hyperplasia leads to persistent reduced maternal blood flow to the placenta, resulting in persistent hypoxia and increased angiogenesis, with a constellation of pathologic features of maternal vascular malperfusion atlate gestation. Wilm’s tumor gene (WT1) expression appears to be central to steroid and peptide hormonal actions in early pregnancy, and vascular regeneration/restoration after pregnancy. Conclusions: Spiral artery remodeling at early pregnancy leads to hypoxia with vascular transformation, and the establishment of uteroplacental circulation results in relief of hypoxia. The hypoxia–re-oxygenation sequence may provide insights into the mechanism of normal fetal/placental development and associated pregnancy complications, such as preeclampsia.
1. Introduction
Decidual vasculopathy is a term used in the pathology community to depict the pathologic condition of a placenta, characterized by specific morphologic changes of the maternal spiral artery after delivery [1]. Decidual vasculopathy is classified into three morphologic variants, namely acute atherosis, fibrinoid medial necrosis and mural arteriole hypertrophy/hyperplasia [2]. Acute atherosis and fibrinoid medial necrosis are considered the two morphologic variants of the same disease spectrum, which was therefore considered “classic type vasculopathy” [1,3,4,5]. The pathogenesis of classic type vasculopathy was shown to be related to direct trophoblastic invasion of the spiral artery in early pregnancy (named “physiologic changes”) and the persistence of these changes up to late gestation [4]. Mural arterial hypertrophy/hyperplasia, however, appears to be a separate artery change, characterized by smooth muscle proliferation of the vascular wall without direct trophoblastic invasion [6,7,8,9]. Mural arterial hypertrophy can be identified in early pregnancy as “physiologic changes”, and it can also be identified as pathologic decidual vasculopathy at the late gestation after placental delivery, similar to those morphologic features described for classic type vasculopathy [7]. Decidual vasculopathy overall is historically associated with preeclampsia and the hypertensive disorders of pregnancy, and these maternal vascular changes were recently classified as maternal vascular malperfusion in the current pathology guideline, leading to a variety of placental and fetal pathologic changes [1,5].
The link between the decidual vasculopathy in late gestation and the spiral artery remodeling in the early pregnancy was found recently, due to the finding of CD56 (NCAM) expression on the “foamy cells” within the acute atherosis at late gestation [4]. CD56 (NCAM) was found to be expressed in the early embryos and trophoblasts decades ago [10,11], and the endovascular trophoblasts have also been noted within the wall of the spiral artery in decidua or myometrium in late pregnancy for half a century [12,13,14]. Our ability to link the endovascular trophoblasts in spiral artery remodeling at implantation to the “foamy cells” of acute atherosis is due to the fact that those “foamy macrophages” express the unique marker CD56 (NCAM) exclusively, which leads to a search for the cellular origin of these “foamy cells”. These “foamy cells” were found to be endovascular trophoblasts, instead of macrophages, phagocytes or inflammatory cells. To link acute atherosis at term or late pregnancy to the endovascular trophoblasts ultimately results in the link to spiral artery remodeling at early pregnancy, leading to the theory of “failure of involution” (failure to die) as a mechanism of the pathogenesis of acute atherosis. There are many historic theories and controversies related to the pathogenesis of decidual vasculopathy and preeclampsia. In the context of “failure of involution”, I will re-examine the theory of “failure of invasion” (failure to invade) with some new data and a new perspective.
2. Spiral Artery Remodeling and Hypoxia
Spiral artery remodeling is an essential step to establishing the embryonic implantation into the hormonally primed endometrium [15]. It involves the initial exposure of the developing embryo and trophectoderm to the maternal circulation, followed by the subsequent development of intervillous spaces and intervillous circulation. The extravillous trophoblasts derived from the developing trophectoderm invade into the endometrial glands and stromal tissue, approaching the terminal segment of the spiral artery, and they subsequently replace the entire arterial wall with eosinophilic fibrinoid material and fill the entire arterial lumen by forming a trophoblastic “plug” [15]. This process was proposed as the trophoblastic-dependent remodeling of the spiral artery [16]. The endovascular trophoblasts undergo a significant phenotypic switch to express a set of cell adhesion molecules, including integrins CD59 and CD56 (NCAM), and behave similarly to endothelial cells [6,17,18,19,20]. These spiral artery changes do not induce vascular thrombosis or platelet aggregates, with no CD42b expression within the wall or the lumen [21]. The fibrinoid material of the vascular wall was shown to be a matrix-type material secreted by the trophoblasts, and not fibrin or any other coagulatory components [21]. Most of the historic literature states that spiral artery remodeling is intended to expand the capacity of maternal vasculature in order to accommodate the growing embryo’s development [15]. This is to a large degree incomplete. The critical issue of spiral artery remodeling in fetal development is timing and anatomic location, i.e., spatiotemporal vascular changes driven by genetic (intrinsic) and environmental (extrinsic) factors. When extravillous trophoblasts first invade the spiral artery at the time of implantation, plugging the lumen of the vessel, the maternal blood flow to the embryo is consequently reduced, leading to a hypoxic environment around the developing embryo and placenta [15]. These early changes of spiral artery remodeling are, in theory, not intended to expand the capacity of maternal or intervillous circulation, but to restrict the maternal flow to the intervillous spaces. Hypoxia, and fetal and placental development, have been studied and reviewed extensively, and it is reasonable to believe that hypoxia is critical for early implantation, and embryonic and trophectoderm development and differentiation [22,23,24]. It is also reasonable to believe that the change of the spiral artery at this time is the primary cause, rather than the effect, of a hypoxic environment. The driving factors of trophoblastic invasion into the spiral artery, with subsequent luminal plugging, are unknown, although the uterine natural killer cells (NK) are known to play a critical role, and the NK cell antigen CD56 (NCAM) appears to be expressed exclusively by the endovascular or intramural trophoblasts (EVT), but not by the extravillous interstitial trophoblasts outside the vessels [25,26,27]. It is reasonable to speculate that the driving factor(s) of trophoblastic invasion into the spiral artery is intrinsically (genetically) controlled, and it is not influenced by the maternal or hormonal environment, as extravillous trophoblasts in tubal pregnancy can invade the tubal arterioles in a similar manner, and the tubal epithelium and stromal tissue do not undergo decidualization nor are they affected by steroid hormones (unpublished observation). Furthermore, the extravillous trophoblasts including the endovascular trophoblasts are mostly negative for proliferation index Ki-67, indicating the low to non-existent proliferative potential of these trophoblasts [1]. However, the depth of extravillous trophoblastic invasion into the decidua and myometrium is likely influenced by the unknown maternal factor(s) (extrinsic), as there are far fewer extravillous trophoblasts within the deep myometrium than the decidua and superficial myometrium [28]. A similar argument of the unknown maternal factor(s) that influence the endovascular trophoblastic migration proximally can be made concerning the lumen of the spiral artery, as the endovascular trophoblasts are not present in the proximal uterine artery or femoral artery.
The spiral artery’s distance from the developing embryo or trophectoderm also undergoes specific morphologic changes, characterized by the smooth muscle proliferation/hypertrophy of the arterial wall, leading to significant narrowing of the arterial lumen, and consequently to reduced maternal blood flowing to the uterine cavity and endometrium [6,7]. Smooth muscle hypertrophy/hyperplasia of the muscular arterial wall can be induced in the secretory phase of the endometrium, mediated by the steroid hormone effect (progesterone) after ovulation as an essential part of endometrial decidualization. The morphologic features again likely result in a hypoxic environment for the developing embryo and placenta. This process was proposed as “trophoblastic independent remodeling” of the spiral artery. The driving factor of smooth muscle proliferation/hypertrophy is most likely endocrine factor(s), as no direct interaction of the smooth muscle cells with extravillous trophoblasts is identified, and the smooth muscle hypertrophy/hyperplasia is likely related in part to the Wilm’s tumor gene expression (WT1) [29].
3. Transition of Extravillous Trophoblasts to Endovascular Trophoblasts
There are dramatic morphologic changes of the spiral artery in trophoblasts-dependent spiral artery remodeling, in terms of the smooth muscle wall and endothelium, when the extravillous trophoblasts invade into the vessel. The muscular artery becomes a flaccid tubular structure walled by a poorly formed structure of eosinophilic fibrinoid material, and filled with foamy endovascular trophoblasts. The framework of the artery is somewhat maintained by an unknown structure, as the original muscular artery did not become a pool of blood or plasma filled with the random endovascular trophoblasts. Rather, it is a loosely formed lumen, although the adventitia layer of the artery cannot be readily appreciated. It is reasonably clear that the extravillous trophoblasts invade the vessel directionally from the adventitia into the lumen. The obvious question is where those smooth muscle and endothelium go, and by what mechanism, during the trophoblastic invasion. Fas and Fas–ligand systems have been shown to mediate the apoptosis of the smooth muscle cells of the spiral artery wall in in vitro models, and the cytokines and chemokines were shown to play roles in the process [30,31,32,33]. The spiral artery is continuous proximally to the radial artery, and how far the endovascular trophoblasts travel within the arterial lumen and wall remains an interesting question. The answer is likely related to the interaction between the endovascular trophoblasts and the endothelium/smooth muscle within the proximal artery. The junction of the transformed (remodeled) artery and the original muscular artery is likely a dynamic zone, as the vascular transformation changes over the course of pregnancy, and vascular restoration /regeneration/involution occurs after the delivery. In the delicate balance of endovascular trophoblasts and the endothelium in early pregnancy, the invading trophoblasts at this junction remodel the vascular wall progressively from the decidua toward myometrium. In late pregnancy, the endothelium at this junction likely serve as progenitors to re-populate the regenerating spiral artery after a certain time point, for vascular restoration. Locally, many angiocrine factors secreted by the endothelium likely play critical roles in vascular regeneration/restoration [34,35,36] (Figure 1). During the vascular transformation, the endovascular trophoblasts gain the expression of CD56 (NCAM), and the CD56 (NCAM) expression is exclusively on the endovascular trophoblasts, not on other types of trophoblasts [4]. CD56 (NCAM) expression on endovascular trophoblasts also occurs in tubal pregnancy, where the tubal artery is remodeled in a fashion similar to that of the decidua (unpublished observation). The obvious question is how the endovascular trophoblasts gain CD56 expression, i.e., through de novo synthesis by the endovascular trophoblasts, or through the transfer of CD56 (NCAM) from other cell types, such as uterine NK cells. It is known that extravillous trophoblasts, including the endovascular trophoblasts, are not proliferative with a low to non-existent Ki-67 proliferative index [24]. De novo synthesis of CD56 through transcription and translation by endovascular trophoblasts appears less likely, although such a mechanism remains a possibility. It is unclear if any of the transcription factors identified within the extravillous trophoblasts, including p63, GATA3, CDX2, ZEB2, TCF4 or hypoxia inducible factors (HIFs), regulate the CD56 gene expression during the transition into becoming endovascular trophoblasts [37]. Uterine NK cells are the likely providers of CD56 to the endovascular trophoblasts, although definitive evidence of such antigen transfer is lacking. Such a mechanism of antigen transfer between various cell types has been described for T-lymphocytes and neutrophils [38]. It is noteworthy that the smooth muscle cells of the spiral artery wall also express CD56 (NCAM)(unpublished data), and it is known that CD56 (NCAM) plays certain roles in myotube formation in smooth muscle and cardiac muscle cells [39,40]. Acquisition of CD56 expression from the smooth muscles of the spiral artery wall (kill to gain) remains an interesting, but unresolved, issue.
CD56 (NCAM) is a well-characterized cell adhesion molecule, expressed predominantly in the central and peripheral nervous systems and muscle system [41]. CD56 was also found in the hematopoietic systems, including NK cells, T-cells and dendritic cells. CD56 is a glycoprotein of the immunoglobulin superfamily, with multiple alternatively spliced variants of both mRNA and proteins including NCAM-120, NCAM-140 and NCAM-180 [41]. Our antibody against CD56 (mouse monoclonal 123C3) appeared to react with all three isoforms, and cannot distinguish the variants of CD56, so it is unclear which variant of CD56 is expressed on the endovascular trophoblasts during spiral artery remodeling [4]. CD56 also showed post-translational modification by polysialylation, and the polysialylated CD56 (PSA-CD56, PSA-NCAM) played important roles in trophoblast invasion in early human placenta [42,43]. The polysialylation process involves many target proteins important for early human embryonic and placental development, especially for brain development. There are two known enzymes that specifically catalyze the transfer of sialyl groups to CD56 (NCAM), St8sia2 and St8sia4, both of which are important for PSA-NCAM expression in embryonic and adult tissues [44]. The extracellular domains of CD56 (immunoglobulin-like domains) have been shown to play various roles in signal transduction between fibronectin and the fibroblast grow factor receptor (FGFR) in cell–cell adhesion, cell–matrix adhesion and neuronal cell migration [45,46]. The role of CD56 with regards to endovascular trophoblasts in spiral artery remodeling, and how PSA-NCAM involves the phenotypic switch of endovascular trophoblasts, is yet to be defined in cell or animal models [43].
4. Hypoxia and Placental/Fetal Development in First Trimester
A hypoxic environment during early pregnancy, resulting from the spiral artery remodeling, was further demonstrated by blood flow analysis via two-dimensional color ultrasonography [47]. Numerous studies demonstrated that no meaningful maternal blood flow to intervillous spaces is detected until 13 weeks into the gestational age [48,49,50,51,52]. From week 3 after fertilization to week 13, the endovascular trophoblasts only allow small amounts of maternal blood to flow through the modified spiral arteries to the intervillous spaces (Figure 2). Examining the photomicrographs of Hertig’s work (embryo project) and Hamilton/Boyd’s work revealed the presence of a small number of red blood cells within the intervillous spaces lined by syncytiotrophoblasts during the early weeks of gestations, indicating the endovascular “plugging” of the spiral artery is not the absolute blocking of the entire lumen [53,54,55]. Therefore, the resulting environment for embryonic and placental development is hypoxic, but not anoxic. This view is shared by other investigators of recent decades [47]. It is unknown, however, how the placenta extends the lateral growth and remodels the corresponding spiral artery in a sequential and spatial fashion (spatiotemporal), as the centrally located spiral artery will be in close contact with the developing embryo and trophoblasts first, and there is a time sequence between the center and the periphery of the developing placenta. Establishment of intervillous circulation seems to be a gradual process, and the endovascular trophoblastic plugs within the spiral artery started to dislodge at week 7, although the molecular mechanism of endovascular cell death is still controversial [48]. The disappearance of endovascular trophoblasts results in maternal blood flow into the intervillous spaces and relief of hypoxia, and the process of cell death of the endovascular trophoblasts is likely associated with the regeneration/restoration of the endothelium and smooth muscle cells within the spiral artery wall, although experimental data of vascular regeneration is lacking at the early stages of pregnancy. Our own data showed a significant difference in frequencies of decidual vasculopathy in the central region and peripheral region of the placenta at late gestation [56]. It seems reasonable to believe that between week 7 and week 13 of gestation, the centrally located spiral artery is undergoing luminal recovery with associated endovascular cell death, and the laterally located spiral artery is undergoing trophoblastic-dependent remodeling via the formation of the trophoblastic plugging, until meaningful blood flow can be visualized and intervillous circulation is established. In other words, the spiral artery remodeling in the central and peripheral regions is not synchronous, and neither is the vascular luminal regeneration in the two different regions during placental lateral growth.
Significant advances have been made in the understanding of oxygen sensing by hypoxia-inducible factor genes (HIFs) and the mechanism of molecular signaling, and HIFs can induce a host of target genes in both villous trophoblasts, and villous vasculogenesis and hematopoiesis [23,51,57,58]. Hypoxia and mammalian gene expression in general has been reviewed extensively elsewhere, and is beyond the scope of this writing. However, spiral artery remodeling in the first trimester, in trophoblasts-dependent and -independent manners, results in hypoxic conditions, and the subsequent relief of the hypoxic environment in the second and third trimesters (re-oxygenation) may represent a pathway of the pathogenic mechanism of a variety of clinical conditions, if the re-oxygenation process becomes abnormal (Figure 3).
5. Re-Oxygenation (2nd Trimester and 3rd Trimester)
The endovascular trophoblastic plugs starting to dislodge allows the maternal blood to flow into the intervillous spaces, establishing the intervillous circulation and relieving the hypoxic condition induced by the early spiral artery remodeling (Figure 3). The experimental data indicated that the dislodging of the trophoblastic plug started from week 7, becoming continuous until week 13, when meaningful maternal blood flow to the intervillous spaces can be detected. The exact cell death process of endovascular trophoblasts is still controversial, and it is likely the key to understanding the establishment of maternal flow to the intervillous spaces. The molecular signals influencing the cell death process of endovascular trophoblasts within the spiral artery are likely within the maternal circulation, and/or within the arterial wall, including the remaining intact smooth muscle and endothelium. The function of the endothelium within the spiral artery on the endovascular trophoblasts is unclear, but the mutually exclusive relationship between the endovascular trophoblasts (fetal) and the endothelium (maternal) within the lumen suggests certain cellular signals are required to maintain the survival of one or the other between the two cell types in close proximity. Whether the disappearance of the endovascular trophoblastic plugs represents the end of spiral artery remodeling and the beginning of the restoration/regeneration of the maternal vascular structure, and how long this sequence of the disappearance/restoration/regeneration of trophoblastic plugs and the maternal vessels will take, are yet to be convincingly established experimentally. It is uncommon to see endovascular trophoblastic plugs in the placentas delivered for chorioamnionitis (otherwise normal pregnancy) at the end of the second trimester or in the early third trimester. Our own experience is that vascular regeneration/restoration with the endothelium and smooth muscle cells can be frequently identified at the end of the second trimester. Similar observations were also made by other investigators many decades ago [59]. Naturally, the presence of endovascular trophoblasts within the lumen or wall of the vessels in the third trimester, regardless of clinical manifestations, raised the question of the abnormal cell death process of endovascular trophoblasts (failure to die) [3]. A large spectrum of the “failure to die” manifestations of endovascular trophoblasts can be correlated with a large spectrum of clinically important pregnancy complications, including preeclampsia, and various degrees of maternal vascular malperfusion (ischemia). Placental ischemic changes lead to a cascade of molecular mechanisms, including hypoxic signaling, placental tissue damage and increased angiogenesis. It is true that severe maternal vascular malperfusion can result from classic type vasculopathy, such as acute atherosis and fibrinoid medial necrosis, and the presence of endovascular trophoblasts within the lumen or wall of the spiral artery may interfere with the maternal blood flow, leading to reduced maternal blood flow to the intervillous spaces, and poor development of the placenta, with the consequence of ischemic placental lesions like infarcts and intervillous thrombosis [1]. The presence of fewer endovascular trophoblasts within the lumen will have a reduced effect on the maternal blood flow, due to mechanics alone, with a significantly reduced ischemic consequence within the placenta and intervillous circulation. The question then comes to concern the quantitative cell death process of endovascular trophoblasts. This particular view of the quantitative cell death process of endovascular trophoblasts in the pathogenesis of pregnancy complications has not been investigated vigorously, and it requires significantly more additional studies, both on biomolecular signaling and gene expressions.
Pijnenborg showed that the extravillous trophoblasts invade and migrate to the spiral artery in two separate waves, and the second wave of trophoblastic invasion and migration started between weeks 14 and 16 [60]. It was believed that the failure of the second wave of trophoblastic invasion and migration potentially links to the etiology of preeclampsia (failure to invade theory) [61,62]. Whether the two separate waves of trophoblastic invasion and migration represent the placental lateral growth and asynchronous spiral artery remodeling remains unknown. Placental lateral growth is a poorly understood area of research, but it is required to fully understand how placental developments in normal pregnancies, and complications, occur (discussed below). The two wave theory is also in direct contrast with the quantitative cell death process described above. To resolve these issues requires additional studies using both in vivo and in vitro cellular and animal models.
6. Decidual Vasculopathy and Failure of Trophoblastic Invasion
A review of Robertson, Brosens and Pijnenborgs’ early work showed essentially similar or identical observations to the decidual vasculopathy in the current pathology guideline, with the exception that those findings in their original descriptions were in the inner layer of the myometrium, or the myometrial/decidual junction [12,13,60,63]. These investigators proposed the term “physiological changes” in normal pregnancy and the theory of “failure of invasion” to depict the lack of trophoblastic remnants within the arterial walls and within the inner layers of myometrium, and defined the lack of trophoblastic invasion of the vascular wall as the cardinal feature of the preeclampsia or hypertensive disorders of pregnancy, as well as of other obstetric disorders [64]. The morphologic changes of the spiral artery in the decidua in preeclampsia have been described by many other early investigators, and it seems that all the investigators agree that these vascular changes are associated with preeclampsia [5,65]. The issue appears to be the interpretation of the same, or a similar, morphologic vascular transformation in a spatiotemporal manner among the early investigators. Hertig described these vascular changes in preeclampsia as “acute degenerative arteriolitis”, with the “collection of foamy fat laden mononuclear leukocytes or phagocytes” [5]. These vascular changes observed by Hertig appeared to be within the decidua, and not in the myometrium, and these morphologic changes were considered the basis of pathologic findings associated with the preeclampsia and hypertensive disorders of pregnancy following placental examination. There is a large body of literature that followed the initial description of Hertig in the pathology texts [1]. The vascular changes within the myometrium, described early by Robertson, Brosens and Pijnenborg, can be classified today as acute atherosis and mural arterial hypertrophy/hyperplasia, but these changes were found in the myometrium and not in the decidua, raising the question of their equivalency to decidual vasculopathy today [2]. Some of the vessels in Brosens and Pijnenborgs’ work showed various degrees of arterial restoration, with early endothelial and smooth muscle regeneration, and these regenerative changes were described as a “cushion” with or without mural thrombosis, which was found to be mutually exclusive with endovascular trophoblasts or trophoblasts within the fibrinoid vascular walls [12,13]. The concept of vascular restoration/regeneration after “physiologic changes”, or decidual vasculopathy, is only appropriate when the vascular changes are considered in a dynamic and spatiotemporal manner. There was only a brief mention of repair change in the work of Brosens and Pijnenborg, in either decidua or myometrium, without data or systemic study of “vascular regeneration/restoration”. Much of the placental bed biopsies revealed results similar to those described in the myometrium, as these biopsies were taken after the placentas were delivered [66]. The theory of “failure to invade” acknowledges the presence of the vascular changes, such as acute atherosis and fibrinoid medial necrosis, in the location of decidua as “pathological”, but defines the changes within the myometrium as “physiological”. It is possible that the “physiologic changes” described by these investigators were located in the central portion of the placenta, and the asynchronous spiral artery remodeling associated with the placental lateral growth pattern is described below. The “physiologic changes” within the myometrium could be an extension and continuation of the same morphologic features of decidual vasculopathy into the myometrial segment of the spiral artery, and these “physiologic changes” could represent various degrees of vascular restoration/regeneration after the initial vascular transformation in early pregnancy. From an anatomical viewpoint, the decidual segment of the spiral artery will undergo vascular transformation first during early pregnancy, extending into the myometrial segment of the spiral artery, whereas the myometrial segment of the spiral artery will recover (regenerate/restore) first, if the endothelial cells within the proximal artery serve as progenitor cells for both endothelial cells and smooth muscle cells in the vascular lumen. Many angiocrine factors have been described for tissue-specific endothelium in vascular regeneration, and these angiocrine factors are likely the keys to understanding the role of endothelium as progenitor cells during and after pregnancy [34,35,36,67]. We examined one hysterectomy specimen, with the placenta in the central region, with regards to decidua and the myometrial segments of the spiral artery. The presence of CD56-positive endovascular and intramural trophoblasts within the decidua supports the view of decidual vasculopathy, and these vascular changes were connected to the myometrial segments of the spiral artery without CD56-positive endovascular or intramural trophoblasts, but with WT1-positive endothelium and SMYOHC-positive smooth muscle cells [29,56]. The key morphologic features of “failure to invade” in the early work are those of smooth muscle mural hypertrophy/hyperplasia, as defined in the current placental pathology guideline [2]. These particular morphologic features were shown to be more associated with essential hypertension in the early work [13,68]. It is important to note again that current guidelines on placental examination concern placentas after delivery, whereas the “failure to invade” theory was based on the vascular changes within the myometrium. Our own experience of the placentas after delivery is that mural hypertrophy/hyperplasia is commonly seen in small caliber arteries, and more commonly seen in the decidua capsularis (vera). Mural hypertrophy/hyperplasia alone, similar to classic type vasculopathy alone, does not significantly affect the placental growth and weight [4]. However, mural hypertrophy/hyperplasia associated with classic vasculopathy in the same placenta significantly affects the placental weight and growth at the late gestation stage [4]. As suggested previously, mural hypertrophy/hyperplasia appears to be affected by endocrine or circulating factors, without direct interaction with trophoblasts, and the presence of the mixed type of vasculopathy (mural hypertrophy/hyperplasia and classic type vasculopathy) reflects abnormal interactions both locally, between the endothelium and trophoblasts, and globally, within the maternal circulation [6]. While mural hypertrophy/hyperplasia is a part of the disease process in late gestation, as a cardinal feature of “failure to invade” theory, mural hypertrophy/hyperplasia represents a limited aspect of the pathogenesis of the preeclampsia or hypertensive disorders of pregnancy, as well as other pregnancy-related complications.
7. Spiral Artery Remodeling and Placental Lateral Growth
Placental growth is continuous throughout the entire pregnancy, and the placental growth primarily takes place through the lateral growth pattern, due in part to the mechanical limit of the endometrial cavity. The size and the cell number of the mature placenta in late gestation are determined by intrinsic and extrinsic factors, as proposed for all mammalian animals [69,70]. From early implantation, the trophoblasts-dependent spiral artery remodeling occurs at the center of the placental trophectoderm, expanding laterally, involving more spiral arteries of the decidua with the formation of more cotyledons, in a spatiotemporal manner. This spatiotemporal remodeling of the spiral artery raises the question of the timing sequence of the trophoblastic invasion into the vessel in the central portion versus in the peripheral portion of the placenta. Formation of the cotyledons is continuous throughout the formation of the anchoring villi and trophoblastic cell column, where the trophoblastic proliferation occurs. The Ki-67 index was only detected in the trophoblasts in the anchoring villi and trophoblastic cell column, and the extravillous trophoblasts destined to remodel the spiral arteries are derived from the proliferating trophoblastic cell columns [71]. Theoretically, the central portion of the placenta will undergo spiral artery remodeling early, and the peripheral portion of the placenta later, with a lag of time of uncertain duration. Consequently, the vascular restoration/regeneration after remodeling will not be synchronous between the central and peripheral portions of the placenta. We collected 105 placentas at term with decidual vasculopathy, and examined the presence of vasculopathy in the central and peripheral regions using the method of Khong (en face horizontal sectioning of decidua surface), with respect to central and peripheral recovery after the vasculopathy (restoration/regeneration) [72]. There is a statistically significant difference between the frequencies of decidual vasculopathy in the central region and that in the peripheral region of the placentas [56]. There is also a significant difference in vascular recovery (regeneration/restoration) between the central and peripheral regions. These data suggest that the placental lateral growth occurs in a spatiotemporal manner, with asynchronous vascular recovery taking place after the initial vascular transformation [56]. We have noticed the vascular recovery/regeneration at the end of the second trimester, with markedly diminished endovascular and intramural trophoblasts, similar to those identified by the early investigators [12,59], and we felt that the abnormal vascular recovery/regeneration and failure of death of the endovascular trophoblasts (failure to die) both play a central role in the pathogenesis of decidual vasculopathy in late gestation [3].
8. Hormonal Actions and WT1 Gene Expression
Pregnancy is characterized by significant hormonal changes, and these hormonal changes are mediated by the hormone receptors. Steroid hormone receptors, including estrogen (ER) and progesterone receptors (PR), play profound roles in menstrual cycles and throughout pregnancy. ER and PR are both nuclear receptors and transcription factors, regulating a large set of target genes involved in fetal and placental development, as evidenced by the profound phenotypic changes taking place in the gene deletion mouse models. Wilm’s tumor gene (WT1) is also a versatile transcription regulator of many cellular processes, which functions in a fashion similar to that of the steroid hormone receptors. The WT1 gene is a tumor suppressor discovered in patients with a Wilm’s tumor, a high grade pediatric malignancy [73,74]. The WT1 gene was found to be involved in many aspects of developmental and cellular functions, in addition to tumor suppression [29]. WT1 gene mutations are found in patients with Denys-Drash syndrome and Frasier syndrome, and the deletion of the short arm of chromosome 11, containing the WT1 gene, is associated with Wilm’s tumor, anirida, genitourinary anomalies, and mental retardation (WAGR) syndrome [75]. WT1 is expressed abundantly at the basal level in the endometrial stromal cells, and it appears that WT1 expression is increased in response to progesterone in both endometrial stromal cells and smooth muscle cells within the spiral artery, during menstrual cycle and early pregnancy. WT1 is a known regulator of the progenitor cells of the endothelium, the mesothelium, fat, and smooth muscle. WT1 is also a key regulator of steroidogenic factor 1 (SF1) expression during the development of gonads and the urogenital system, and SF1 is the key regulator of many enzyme genes for steroid biosynthesis (P450s steroidogenic enzymes) (see diagram) [76,77]. Our interest in WT1 gene expression in spiral artery remodeling and regeneration relies on the fact that WT1 gene expression was shown to be critical for coronary artery development, and the smooth muscle wall of the coronary artery appeared to be derived from the endothelial progenitor (stem) cells in which WT1 expression is abundant [78]. WT1 was also shown to be critical for abdominal fat tissue development, and the fat cells appeared to be derived from the mesothelial progenitor cells with WT1 gene expression [79,80]. The function of the WT1 gene within the progenitor cells of fat (mesothelium) and the vascular smooth muscle (endothelium) makes WT1 a candidate in regulating the restoration/regeneration of the spiral artery after the initial trophoblasts-dependent remodeling. A study of the upstream factors influencing WT1 gene expression in the endothelium and spiral artery will likely yield information useful in understanding the mechanisms of the cell death process of the endovascular trophoblasts and endothelial regeneration. This information will likely be useful in understanding the pathogenesis of pregnancy complications, including preeclampsia. It is true that there are multiple WT1 gene deletion and transgenic mice models available for study, but none of the reproductive/pregnancy aspects of these mouse models are currently published. Searching for maternal factors, present in the circulation, affecting the WT1 gene’s expression in the endothelial cells and the smooth muscle cells of the vascular wall will be critical in the study of the molecular mechanism of preeclampsia. The design of a quantitative gene expression model is likely important in pathogenesis, as a dramatic phenotype of the binary gene expression model is rare in common diseases such as preeclampsia.
9. Conclusions
Decidual vasculopathy represents a large spectrum of maternal vascular changes, involving a wide spectrum of clinical manifestations, in addition to preeclampsia and hypertensive disorders. Understanding the mechanism of the pathogenesis of decidual vasculopathy will ultimately help in understanding the pathogenesis of preeclampsia.
Funding
There is no external funding to the work in this article.
Conflicts of Interest
The author declares no financial conflict of interest.
References
- Benirschke, K.; Burton, G.J.; Baergen, R.N. Pathology of the Human Placenta, 6th ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Khong, T.Y.; Mooney, E.E.; Ariel, I.; Balmus, N.C.M.; Boyd, T.K.; Brundelr, M.-A.; Derricott, H.; Evans, M.J.; Faye-Petersen, O.M.; Gillan, J.E.; et al. Sampling and definitions of placental lesions: Amsterdam placental workshop group consensus statement. Arch. Pathol. Lab. Med. 2016, 140, 698–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P. Decidual vasculopathy in preeclampsia and spiral artery remodeling revisited: Shallow invasion versus failure of involution. AJP Rep. 2018, 8, e241–e246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P. Phenotypic switch of endovascular trophoblasts in decidual vasculopathy with implication for preeclampsia and other pregnancy complications. Fetal. Pediatr. Pathol. 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hertig, A. Vascular pathology in the hypertensive albuminuric toxemias of pregnancy. Clinics 1945, 4, 602–614. [Google Scholar]
- Zhang, P. Decidual vasculopathy and spiral artery remodeling revisited II: Relations to trophoblastic dependent and independent vascular transformation. J. Matern. Fetal. Neonatal. Med. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Craven, C.M.; Morgan, T.; Ward, K. Decidual spiral artery remodelling begins before cellular interaction with cytotrophoblasts. Placenta 1998, 19, 241–252. [Google Scholar] [CrossRef]
- Hecht, J.L.; Zsengeller, Z.K.; Spiel, M.; Karumachi, S.A.; Rosen, S. Revisiting decidual vasculopathy. Placenta 2016, 42, 37–43. [Google Scholar] [CrossRef]
- Chan, J.S.; Heller, D.S.; Baergen, R.N. Decidual vasculopathy: Placental location and association with ischemic lesions. Pediatr. Dev. Pathol. 2017, 20, 44–48. [Google Scholar] [CrossRef]
- Pröll, J.; Blaschitz, A.; Hartmann, M.; Thalhamer, J.; Dohr, G. Human first-trimester placenta intra-arterial trophoblast cells express the neural cell adhesion molecule. Early Pregnancy 1996, 2, 271–275. [Google Scholar]
- Kam, E.P.; Gardner, L.; Loke, Y.W.; King, A. The role of trophoblast in the physiological change in decidual spiral arteries. Hum. Reprod. 1999, 14, 2131–2138. [Google Scholar] [CrossRef]
- Brosens, I.; Robertson, W.B.; Dixon, H.G. The physiological response of the vessels of the placental bed to normal pregnancy. J. Pathol. Bacteriol. 1967, 93, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, H.G. The pathological response of the vessels of the placental bed to hypertensive pregnancy. J. Pathol. Bacteriol. 1967, 93, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, R.; Kracke, R. The probable role of the hypercholesteremia of pregnancy in producing vascular changes in the placenta, predisposing to placental infarction and eclampsia. Am. J. Obstet. Gynecol. 1936, 31, 549–562. [Google Scholar] [CrossRef]
- Cunningham, F.G. Williams Obstetrics, 25th ed.; McGraw-Hill: New York, NY, USA, 2018. [Google Scholar]
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The uterine spiral arteries in human pregnancy: Facts and controversies. Placenta 2006, 27, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Damsky, C.H.; Librach, C.; Lim, K.H.; Fitzgerald, M.L.; McMaster, M.T.; Janatpour, M.; Zhou, Y.; Logan, S.K.; Fisher, S.J. Integrin switching regulates normal trophoblast invasion. Development 1994, 120, 3657–3666. [Google Scholar]
- Damsky, C.; Librach, C.; Lim, K.H.; Fitzgerald, M.L.; McMaster, M.T.; Janatpour, M.; Zhou, Y.; Logan, S.K.; Fisher, S.J. Adhesive interactions in peri-implantation morphogenesis and placentation. Reprod. Toxicol. 1997, 11, 367–375. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Sato, Y.; Horie, A.; Tani, H.; Miyazaki, Y.; Okunomiya, A.; Matsumoto, H.; Hamanishi, J.; Kondoh, E.; Mandai, M. Endovascular trophoblast expresses CD59 to evade complement-dependent cytotoxicity. Mol. Cell. Endocrinol. 2019, 490, 57–67. [Google Scholar] [CrossRef]
- Zhang, P. CD42b Immunostaining as a marker for placental fibrinoid in normal pregnancy and complications. Fetal. Pediatr. Pathol. 2020, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Genbacev, O.; Joslin, R.; Damsky, C.H.; Polliotti, B.M.; Fisher, S.J. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J. Clin. Investig. 1996, 97, 540–550. [Google Scholar] [CrossRef]
- Van den Driesche, S.; Myers, M.; Gay, E.; Thong, K.J.; Duncan, W.C. HCG up-regulates hypoxia inducible factor-1 alpha in luteinized granulosa cells: Implications for the hormonal regulation of vascular endothelial growth factor A in the human corpus luteum. Mol. Hum. Reprod. 2008, 14, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiden, U.; Eyth, C.P.; Majali-Martinez, A.; Desoye, G.; Tam-Amersdorfer, C.; Huppertz, B.; Ghaffari Tabrizi-Wizsy, N. Expression of matrix metalloproteinase 12 is highly specific for non-proliferating invasive trophoblasts in the first trimester and temporally regulated by oxygen-dependent mechanisms including HIF-1A. Histochem. Cell. Biol. 2018, 149, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.H.; Dunk, C.E.; Lye, S.J.; Aplin, J.D.; Harris, L.K.; Jones, R.L. Extravillous trophoblast and endothelial cell crosstalk mediates leukocyte infiltration to the early remodeling decidual spiral arteriole wall. J. Immunol. 2017, 198, 4115–4128. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.; Harris, L.K.; Innes, B.A.; Lash, G.E.; Aljunaidy, M.M.; Aplin, J.D.; Baker, P.N.; Robson, S.C.; Bulmer, J.N. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. 2012, 26, 4876–4885. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.K.; Clancy, O.H.; Myers, J.E.; Baker, P.N. Plasma from women with preeclampsia inhibits trophoblast invasion. Reprod. Sci. 2009, 16, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P. Expression of Wilm’s Tumor Gene in Endometrium with Potential Link to Gestational Vascular Transformation. Reprod. Med. 2020, 1, 17–31. Available online: https://0-www-mdpi-com.brum.beds.ac.uk/2673-3897/1/1/3 (accessed on 30 April 2020). [CrossRef]
- Harris, L.K.; Keogh, R.J.; Wareing, M.; Baker, P.N.; Cartwright, J.E.; Aplin, J.D.; Whitley, G.S. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am. J. Pathol. 2006, 169, 1863–1874. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.D.; Dunk, C.E.; Aplin, J.D.; Harris, L.K.; Jones, R.L. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 2009, 174, 1959–1971. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.K. Review: Trophoblast-vascular cell interactions in early pregnancy: How to remodel a vessel. Placenta 2010, 31, S93–S98. [Google Scholar] [CrossRef]
- Harris, L.K.; Smith, S.D.; Keogh, R.J.; Jones, R.L.; Baker, P.N.; Knöfler, M.; Cartwright, J.E.; Whitley, G.S.; Aplin, J.D. Trophoblast-and vascular smooth muscle cell-derived MMP-12 mediates elastolysis during uterine spiral artery remodeling. Am. J. Pathol. 2010, 177, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Knöfler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.K.J.B.; James, J. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell. Mol. Life Sci. 2019, 76, 3479–3496. [Google Scholar] [CrossRef] [Green Version]
- Nadkarni, S.; Smith, J.; Sferruzzi-Perri, A.N.; Ledwozyw, A.; Kishore, M.; Haas, R.; Mauro, C.; Williams, D.J.; Farsky, S.H.P.; Marelli-Berg, F.M.; et al. Neutrophils induce proangiogenic T cells with a regulatory phenotype in pregnancy. Proc. Natl. Acad. Sci. USA 2016, 113, E8415–E8424. [Google Scholar] [CrossRef] [Green Version]
- Dickson, G.; Smith, J.; Sferruzzi-Perri, A.N.; Ledwozyw, A.; Kishore, M.; Haas, R.; Mauro, C.; Williams, D.J.; Farsky, S.H.; Marelli-Berg, F.M.; et al. Enhanced myogenesis in NCAM-transfected mouse myoblasts. Nature 1990, 344, 348–351. [Google Scholar] [CrossRef]
- Lackie, P.M.; Zuber, C.; Roth, J. Expression of polysialylated N-CAM during rat heart development. Differentiation 1991, 47, 85–98. [Google Scholar] [CrossRef]
- Weledji, E.P.; Assob, J.C. The ubiquitous neural cell adhesion molecule (N-CAM). Ann. Med. Surg. (Lond.) 2014, 3, 77–81. [Google Scholar] [CrossRef]
- Galuska, C.E.; Lütteke, T.; Galuska, S.P. Is Polysialylated NCAM Not only a regulator during brain development but also during the formation of other organs? Biology (Basel) 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Hromatka, B.S.; Drake, P.M.; Kapidzic, M.; Stolp, H.; Goldfien, G.A.; Shih, I.-M.; Fisher, S.J. Polysialic acid enhances the migration and invasion of human cytotrophoblasts. Glycobiology 2013, 23, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhold, B.; Seidenfaden, R.; Rockle, I.; Muhlenhoff, M.; Schertzinger, F.; Conzelmann, S.; March, J.D.; Gerardy-Schahn, R.; Hildebrandt, H. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J. Biol. Chem. 2005, 280, 42971–42977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell. Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.E.; Rutishauser, U.; Magnuson, T. Targeted mutation of Ncam to produce a secreted molecule results in a dominant embryonic lethality. Proc. Natl. Acad. Sci. USA 1996, 93, 6421–6424. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J. Oxygen, the Janus gas; its effects on human placental development and function. J. Anat. 2009, 215, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Roberts, V.H.J.; Morgan, T.K.; Bednarek, P.; Morita, M.; Burton, G.J.; Lo, J.O.; Frias, A.E. Early first trimester uteroplacental flow and the progressive disintegration of spiral artery plugs: New insights from contrast-enhanced ultrasound and tissue histopathology. Hum. Reprod. 2017, 32, 2382–2393. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; van Patot, M.T.; Gardner, L.; Jauniaux, E.; Burton, G.J.; Charnock-Jones, D.S. Energy status and HIF signalling in chorionic villi show no evidence of hypoxic stress during human early placental development. Mol. Hum. Reprod. 2015, 21, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Tache, V.; Ciric, A.; Moretto-Zita, M.; Li, Y.; Peng, J.; Maltepe, E.; Milstone, D.S.; Parast, M. Hypoxia and trophoblast differentiation: A key role for PPARγ. Stem. Cells Dev. 2013, 22, 2815–2824. [Google Scholar] [CrossRef] [Green Version]
- Wakeland, A.K.; Soncin, F.; Moretto-ZIta, M.; Chang, C.-W.; Horii, M.; Pizzo, D.; Nelson, K.K.; Laurent, L.C.; Parast, M.M. Hypoxia directs human extravillous trophoblast differentiation in a hypoxia-inducible factor-dependent manner. Am. J. Pathol. 2017, 187, 767–780. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.W.; Wakeland, A.K.; Parast, M.M. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J. Endocrinol. 2018, 236, R43–R56. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, W.J.; Boyd, J.D. Development of the human placenta in the first three months of gestation. J. Anat. 1960, 94, 297–328. [Google Scholar] [PubMed]
- Dixon, H.G.; Robertson, W.B. Vascular changes in the placental bed. Pathol. Microbiol. (Basel) 1961, 24, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.J.; Boyd, J.D. Trophoblast in human utero-placental arteries. Nature 1966, 212, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P. Comparison of Decidual Vasculopathy in Central and Peripheral Regions of Placenta with Implication of Lateral Growth and Spiral Artery Remodeling. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.06.16.154484v1 (accessed on 16 June 2020).
- Pugh, C.W.; Chang, G.W.; Cockman, M.; Epstein, A.C.; Gleadle, J.M.; Maxwell, P.H.; Nicholls, L.G.; O’Rourke, J.F.; Ratcliffe, P.J.; Raybould, E.C.; et al. Regulation of gene expression by oxygen levels in mammalian cells. Adv. Nephrol. Necker. Hosp. 1999, 29, 191–206. [Google Scholar]
- Ratcliffe, P.J.; O’Rourke, J.F.; Maxwell, P.H.; Pugh, C.W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 1998, 201, 1153–1162. [Google Scholar]
- Khong, T.Y.; De Wolf, F.; Robertson, W.B.; Brosens, I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br. J. Obstet. Gynaecol. 1986, 93, 1049–1059. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Dixon, G.; Robertson, W.B.; Brosens, I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta 1980, 1, 3–19. [Google Scholar] [CrossRef]
- Brosens, I.A.; Dixon, H.G.; Robertson, W.B. Fetal growth retardation and the arteries of the placental bed. BJOG 1977, 84, 656–664. [Google Scholar] [CrossRef]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Pijnenborg, R.; Bland, J.; Robertson, W.; Dixon, G.; Brosens, I. The pattern of interstitial trophoblastic invasion of the myometrium in early human pregnancy. Placenta 1981, 2, 303–316. [Google Scholar] [CrossRef]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: From spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Zeek, P.M.; Assali, N.S. Vascular changes in the decidua associated with eclamptogenic toxemia of pregnancy. Am. J. Clin. Pathol. 1950, 20, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, W.B.; Khong, T.Y.; Brosens, I.; De Wolf, F.; Sheppard, B.L.; Bonnar, J. The placental bed biopsy: Review from three European centers. Am. J. Obstet. Gynecol. 1986, 155, 401–412. [Google Scholar] [CrossRef]
- Rafii, S.; Carmeliet, P. VEGF-B improves metabolic health through vascular pruning of fat. Cell. Metab. 2016, 23, 571–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, H.G.; Robertson, W.B. A study of the vessels of the placental bed in normotensive and hypertensive women. J. Obstet. Gynaecol. Br. Emp. 1958, 65, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Raff, M.C. Size control: The regulation of cell numbers in animal development. Cell 1996, 86, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Raff, M.C.; Durand, B.; Gao, F.B. Cell number control and timing in animal development: The oligodendrocyte cell lineage. Int. J. Dev. Biol. 1998, 42, 263–267. [Google Scholar]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Khong, T.Y.; Chambers, H.M. Alternative method of sampling placentas for the assessment of uteroplacental vasculature. J. Clin. Pathol. 1992, 45, 925–927. [Google Scholar] [CrossRef] [Green Version]
- Hastie, N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef] [Green Version]
- Haber, D.A.; Housman, D.E. The genetics of Wilms’ tumor. Adv. Cancer Res. 1992, 59, 41–68. [Google Scholar] [PubMed]
- Van den Heuvel-Eibrink, M.M. Wilms Tumor; Codon Publications: Brisbane, Australia, 2016. [Google Scholar]
- Gurates, B.; Amsterdam, A.; Tamura, M.; Yang, S.; Zhou, J.; Fang, Z.; Amin, S.; Sebastian, S.; Bulun, S.E. WT1 and DAX-1 regulate SF-1-mediated human P450arom gene expression in gonadal cells. Mol. Cell. Endocrinol. 2003, 208, 61–75. [Google Scholar] [CrossRef]
- Wilhelm, D.; Englert, C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 2002, 16, 1839–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, A.J.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, Y.-Y.; Brownstein, D.; Mjoseng, H.; Lee, W.-C.; Buza-Viads, N.; Nerlov, C.; Eirik, S.; Jacobsen, E.; Perry, P.; Berry, R.; et al. Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet. 2011, 7, e1002404. [Google Scholar] [CrossRef] [Green Version]
- Chau, Y.-Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell. Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [Green Version]
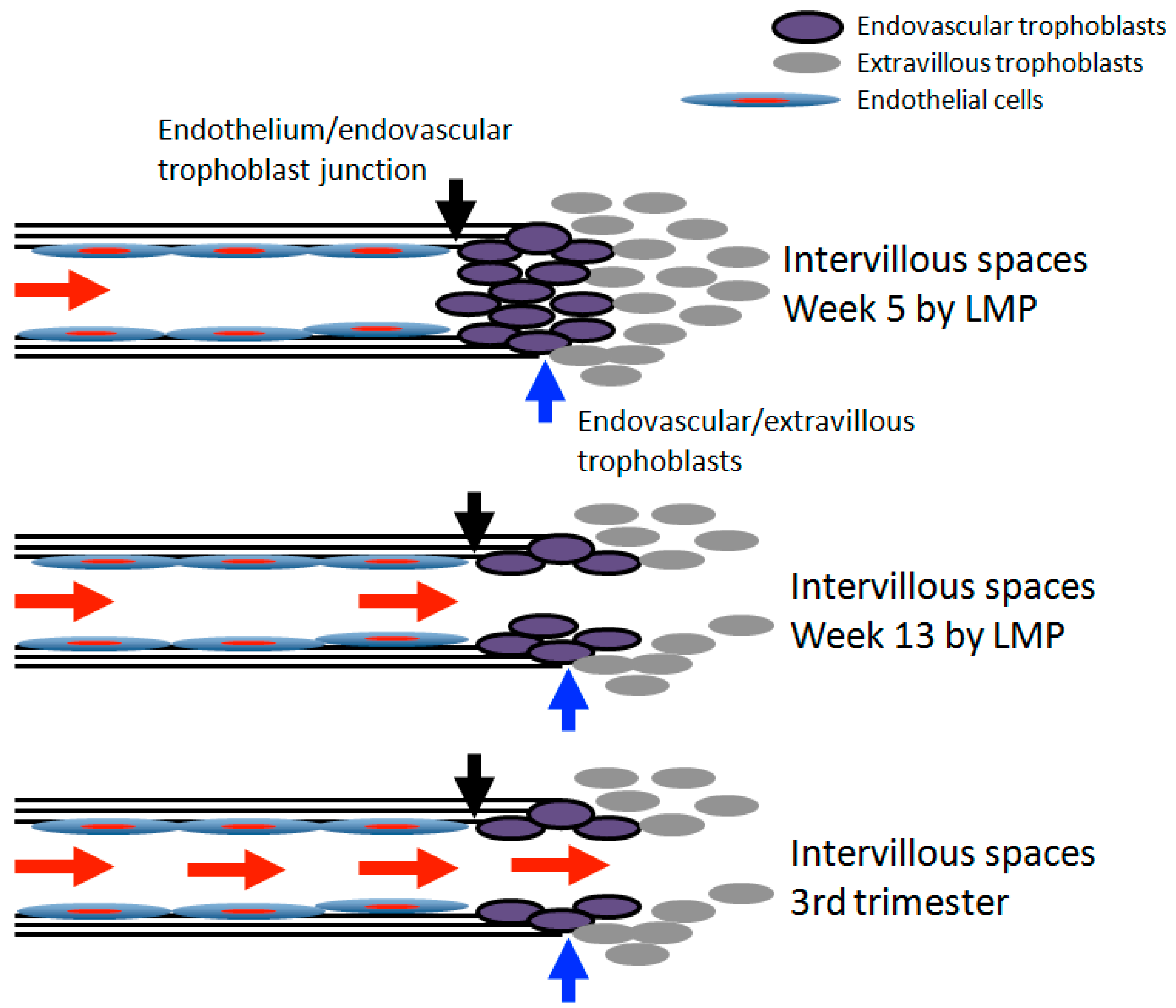
Figure 1.
Schematic representation of extravillous trophoblastic invasion of spiral artery with “plugging” and “deplugging’ between the week 5 week 13 by last menstrual period (LMP).
Figure 1.
Schematic representation of extravillous trophoblastic invasion of spiral artery with “plugging” and “deplugging’ between the week 5 week 13 by last menstrual period (LMP).

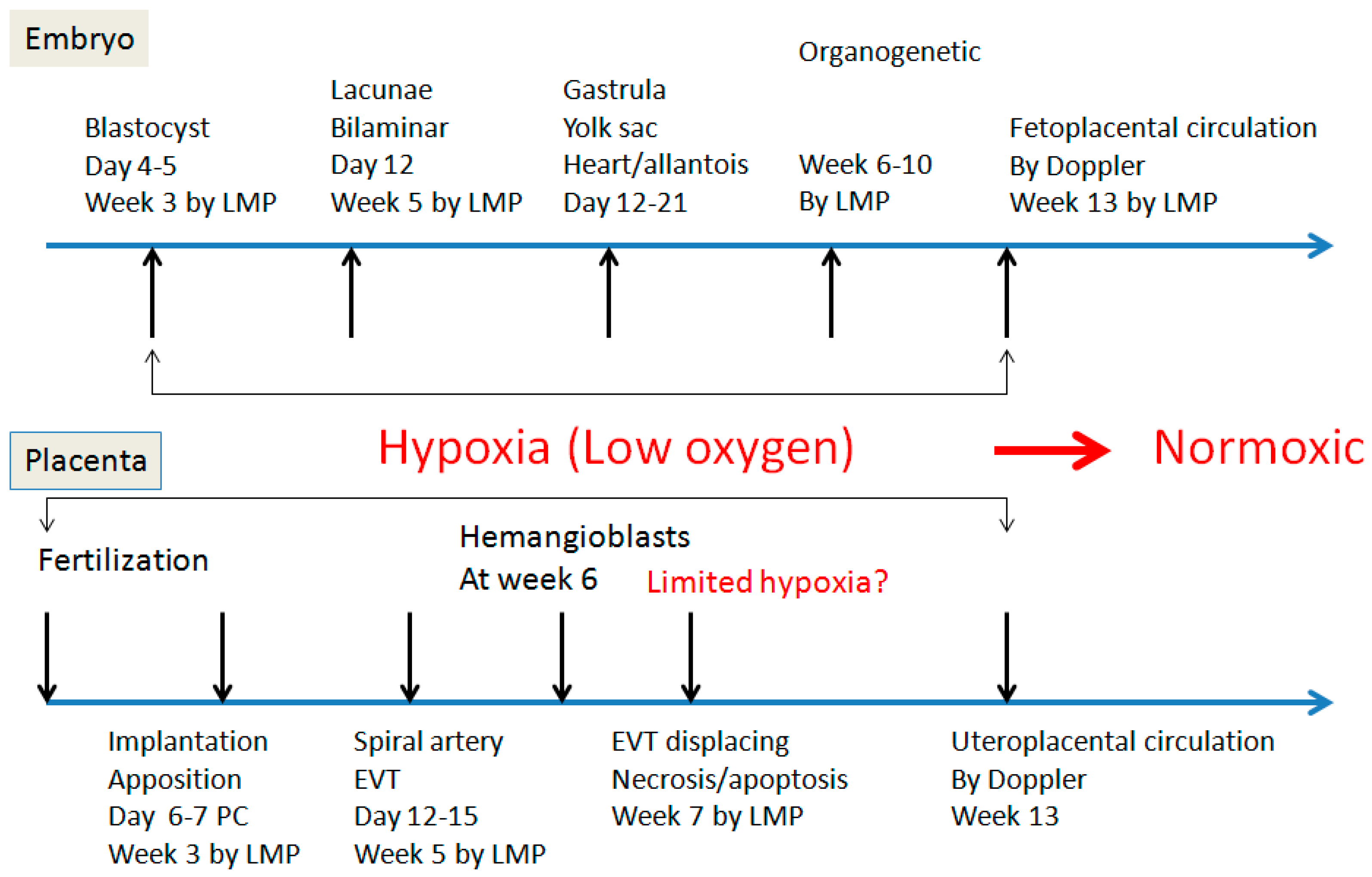
Figure 2.
Schematic representation of embryonic and placental development from fertilization to 13 weeks with establishment of fetoplacental and uteroplacental circulations. LMP: last menstrual period. PC: post conception.
Figure 2.
Schematic representation of embryonic and placental development from fertilization to 13 weeks with establishment of fetoplacental and uteroplacental circulations. LMP: last menstrual period. PC: post conception.

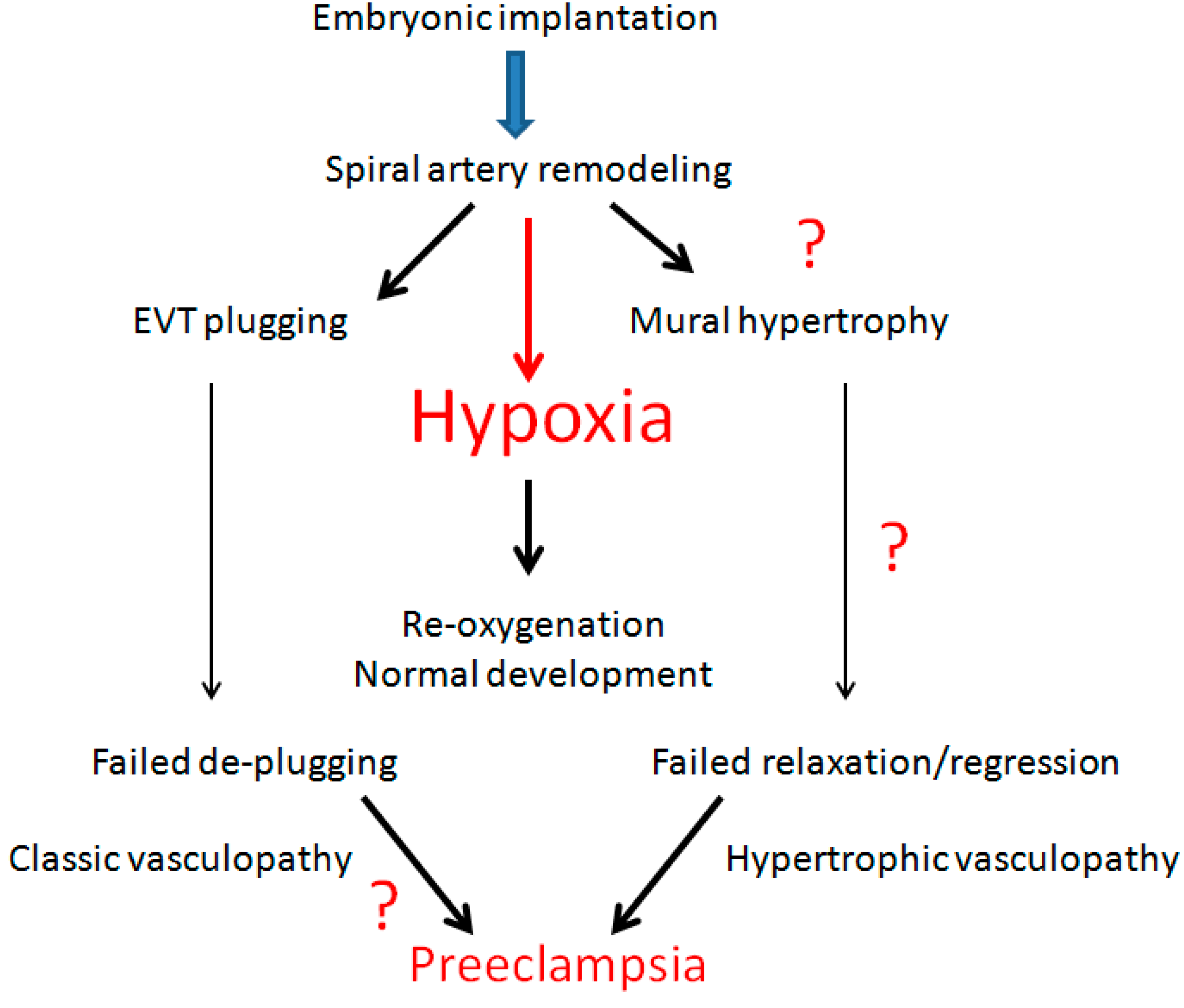
Figure 3.
Schematic representation of hypoxia and re-oxygenation sequence with failure of re-oxygenation and potential pathogenesis of preeclampsia.
Figure 3.
Schematic representation of hypoxia and re-oxygenation sequence with failure of re-oxygenation and potential pathogenesis of preeclampsia.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, P. Decidual Vasculopathy and Spiral Artery Remodeling Revisited III: Hypoxia and Re-oxygenation Sequence with Vascular Regeneration. Reprod. Med. 2020, 1, 77-90. https://0-doi-org.brum.beds.ac.uk/10.3390/reprodmed1020006
AMA Style
Zhang P. Decidual Vasculopathy and Spiral Artery Remodeling Revisited III: Hypoxia and Re-oxygenation Sequence with Vascular Regeneration. Reproductive Medicine. 2020; 1(2):77-90. https://0-doi-org.brum.beds.ac.uk/10.3390/reprodmed1020006
Chicago/Turabian StyleZhang, Peilin. 2020. "Decidual Vasculopathy and Spiral Artery Remodeling Revisited III: Hypoxia and Re-oxygenation Sequence with Vascular Regeneration" Reproductive Medicine 1, no. 2: 77-90. https://0-doi-org.brum.beds.ac.uk/10.3390/reprodmed1020006