
Divergent Synthesis of Four Monomeric Ellagitannins toward the Total Synthesis of an Oligomeric Ellagitannin, Nobotanin K

Abstract
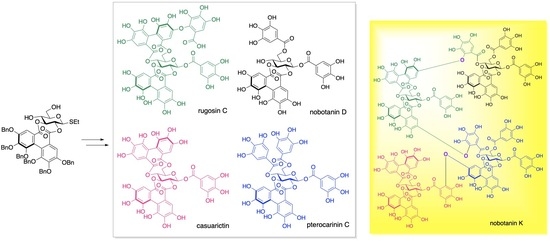
:
1. Introduction
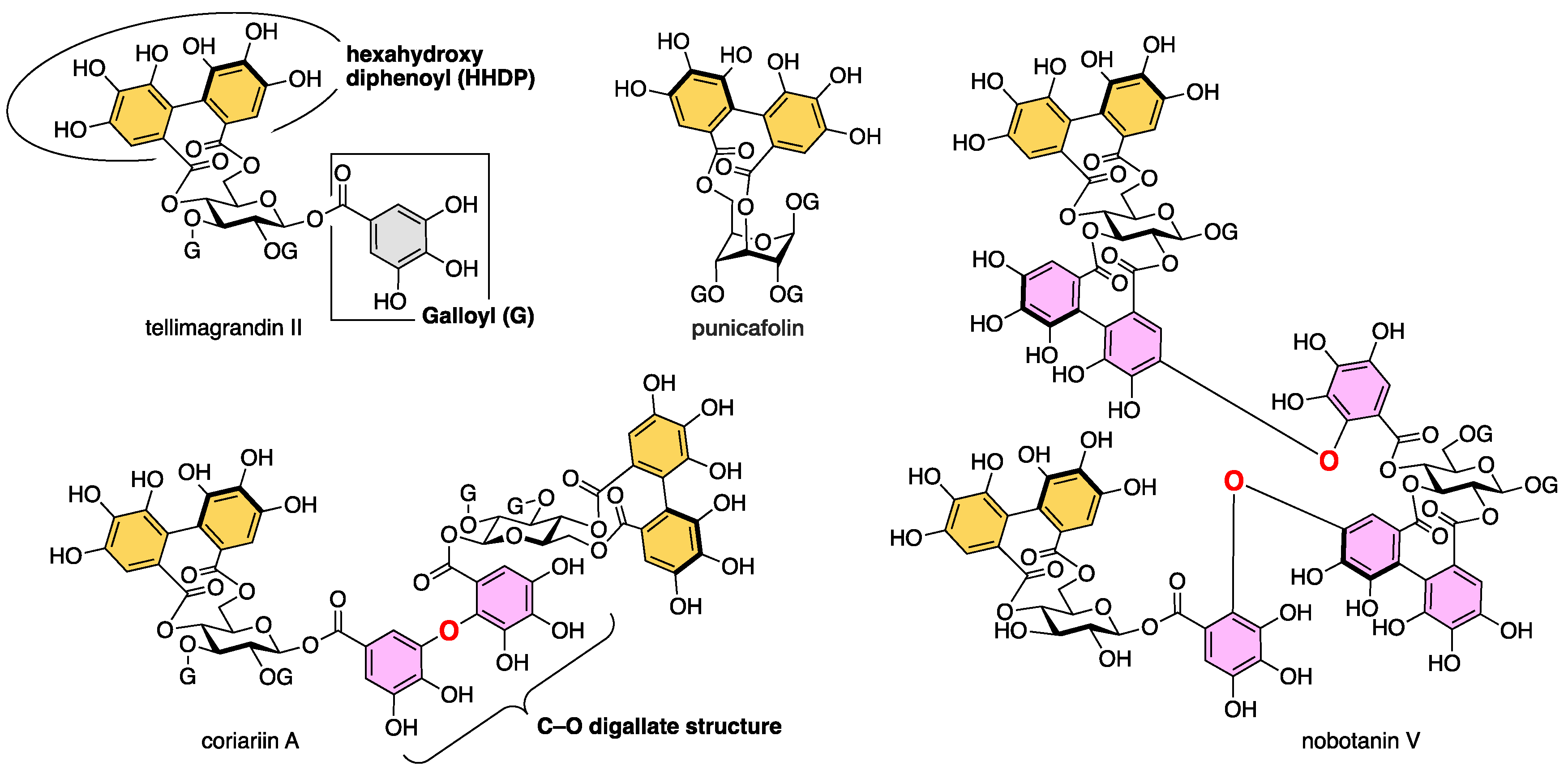
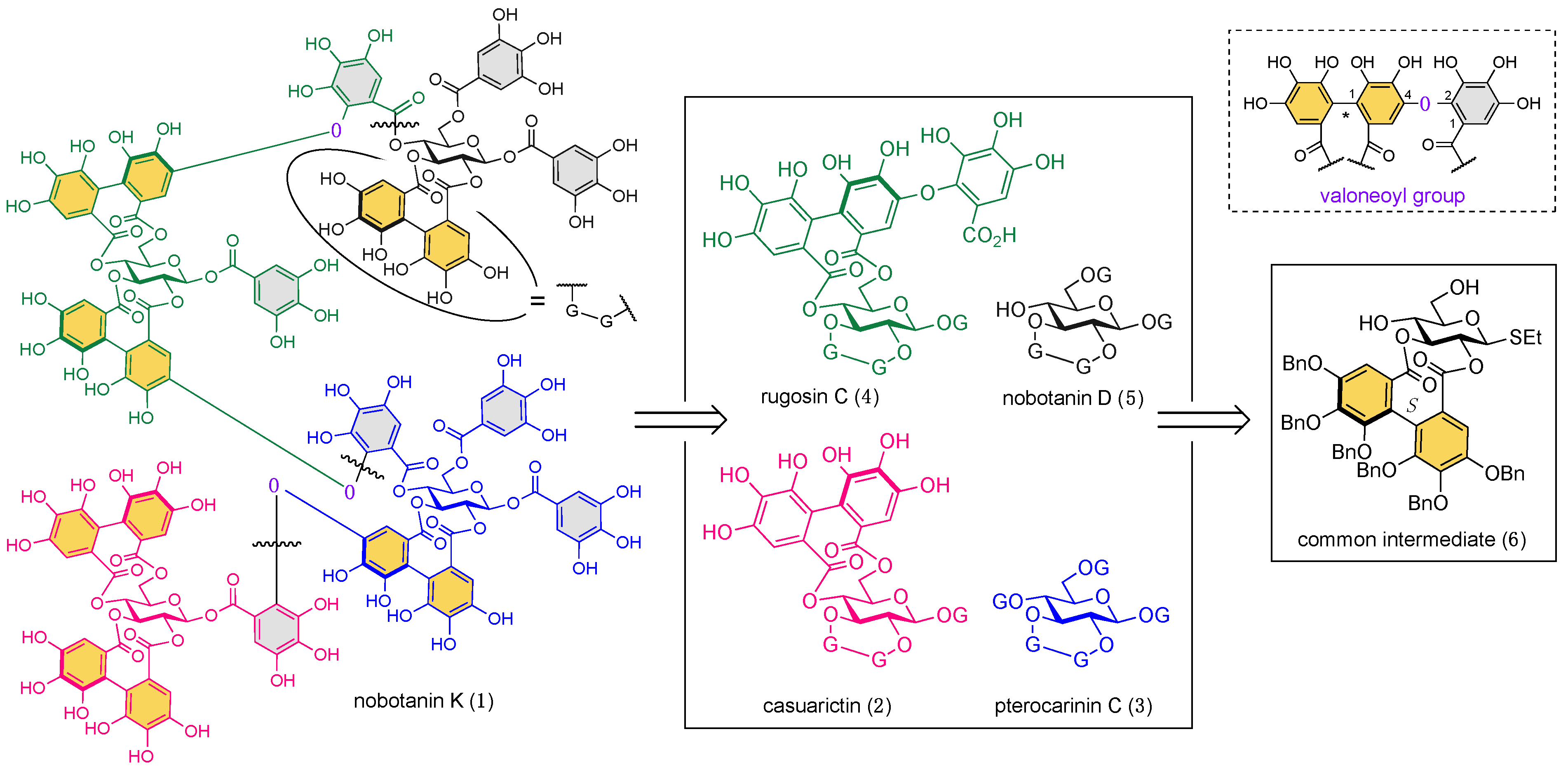
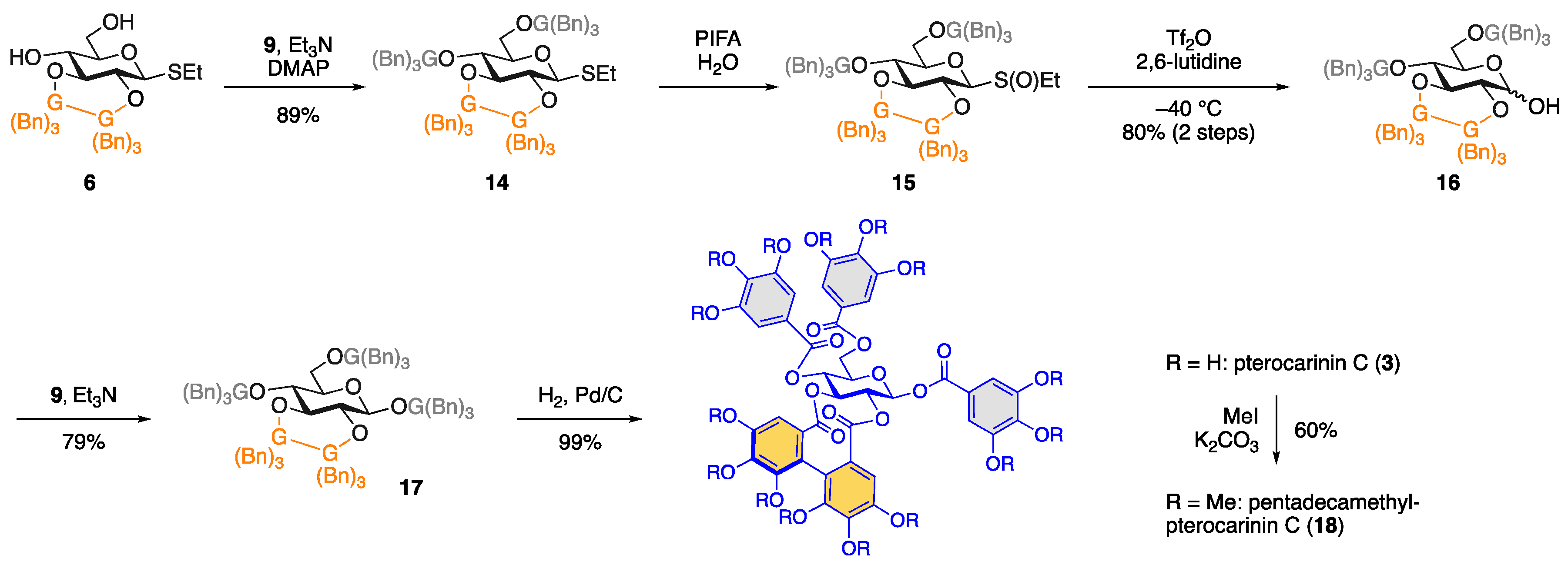
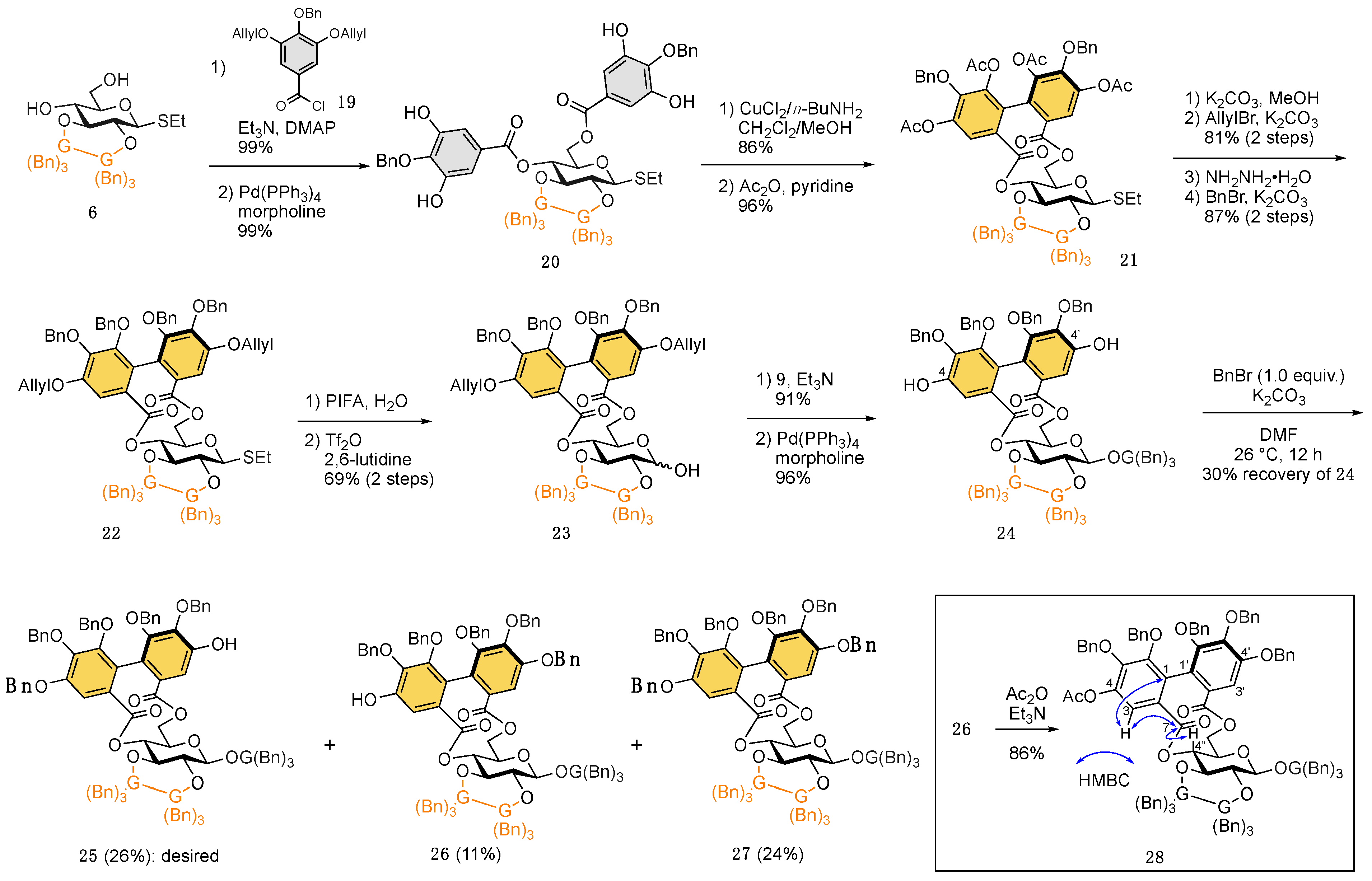
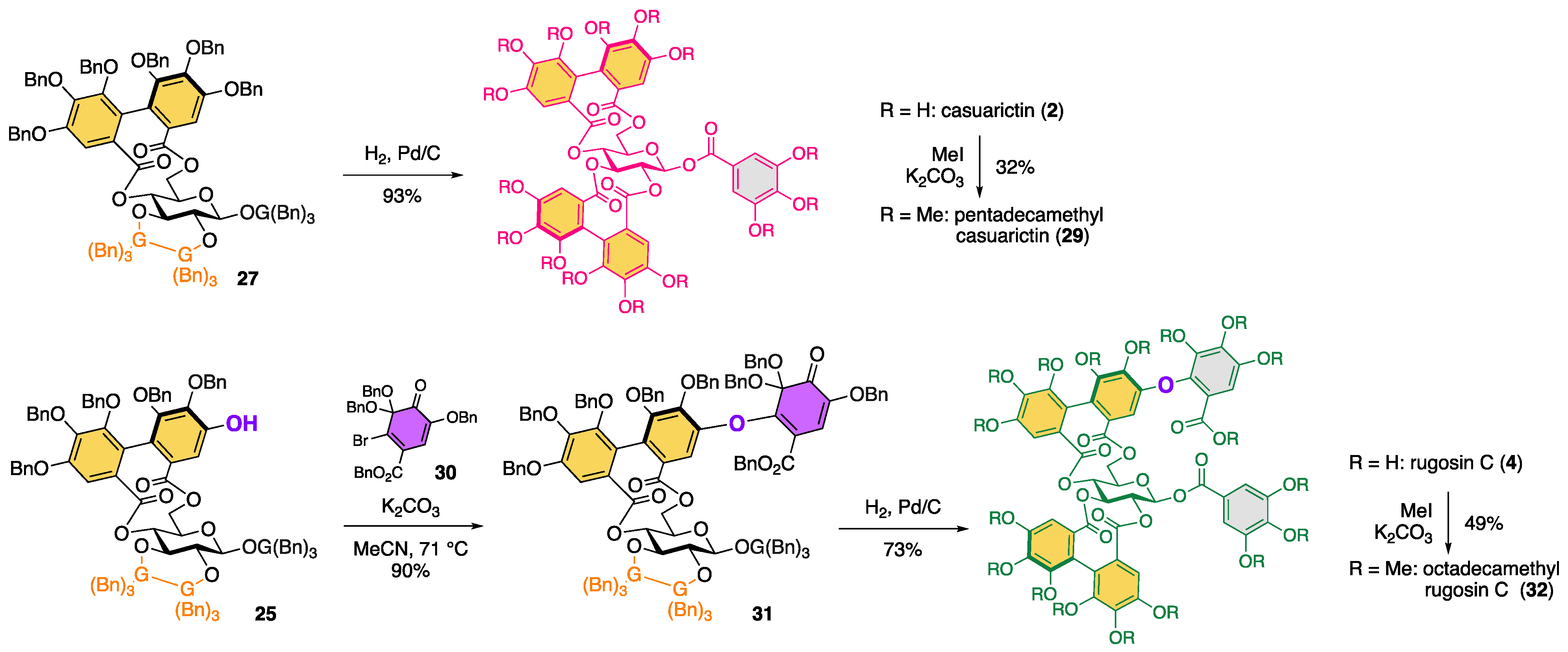
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H NMR Data for Nobotanin D (5) in Acetone-d6 + D2O | |||
|---|---|---|---|
| Assignment | Natural Product | Synthetic Product | Δ(Natural–Syn.) |
| δ | δ | δ | |
| galloyl | 7.13 | 7.13 | 0.00 |
| galloyl | 7.12 | 7.12 | 0.00 |
| HHDP | 6.70 | 6.71 | −0.01 |
| HHDP | 6.42 | 6.42 | 0.00 |
| H-1 | 6.17 | 6.16 | 0.00 |
| H-3 | 5.24 | 5.24 | 0.00 |
| H-2 | 5.06 | 5.06 | 0.00 |
| H-6 | 4.61 | 4.61 | −0.01 |
| H-6 | 4.46 | 4.46 | 0.00 |
| H-5 | 4.08 | 4.08 | 0.00 |
| H-4 | 4.00 | 4.00 | −0.01 |
| Specific optical rotation for nobotanin D (5) in MeOH (unit of c: mg/mL) | |||
| natural product (c = 0.08, 25 °C) | synthetic product (c = 0.09, 25 °C) | ||
| 18 | 20 | ||
| 1H NMR Data for Pterocarinin C (3) in Acetone-d6 | |||||
|---|---|---|---|---|---|
| Natural Product | Our Synthetic Product | Kawabata Group’s Synthetic Product [38] | Δ(Natural–Ours) | Δ(Kawa.–Ours) | |
| Assignment | δ | δ | δ | δ | δ |
| galloyl | 7.18 | 7.18 | 7.17 | 0.00 | −0.01 |
| galloyl | 7.17 | 7.17 | 7.17 | 0.00 | 0 |
| galloyl | 7.14 | 7.14 | 7.15 | 0.00 | 0.01 |
| HHDP | 6.47 | 6.46 | 6.46 | 0.01 | 0 |
| HHDP | 6.44 | 6.43 | 6.44 | 0.01 | 0.01 |
| H-1 | 6.36 | 6.36 | 6.35 | 0.00 | −0.01 |
| H-4 | 5.65–5.58 | 5.65–5.59 | 5.64–5.58 | 0–(−0.01) | (−0.01)–(−0.01) |
| H-3 | |||||
| H-2 | 5.21 | 5.22 | 5.21 | −0.01 | −0.01 |
| H-6 | 4.56 | 4.56 | 4.56 | 0.00 | 0 |
| H-5 | 4.52 | 4.52 | 4.53–4.51 | 0.00 | 0 |
| H-6 | 4.39 | 4.39 | 4.40 | 0.00 | 0.01 |
| Specific optical rotation for pterocarinin C (3) in acetone (unit of c: mg/mL) | |||||
| natural product (c = 0.07, 25 °C) | Kawabata group [39] (c = 0.8, 20 °C) | ours (c = 1.0, 25 °C) | |||
| 18 | 59 | 56 | |||
| 1H NMR Data for Casuarictin (3) in Acetone-d6 | |||
|---|---|---|---|
| Assignment | Literature Data [25] | Synthetic Product | Δ(Lit.–Syn.) |
| δ | δ | δ | |
| galloyl | 7.18 | 7.18 | 0.00 |
| HHDP | 6.68 | 6.67 | 0.01 |
| HHDP | 6.55 | 6.54 | 0.01 |
| HHDP | 6.47 | 6.46 | 0.01 |
| HHDP | 6.38 | 6.37 | 0.01 |
| H-1 | 6.22 | 6.22 | 0.00 |
| H-3 | 5.45 | 5.45 | 0.00 |
| H-6 | 5.37 | 5.37 | 0.00 |
| H-4 | 5.18 | 5.19 | −0.01 |
| H-2 | 5.17 | 5.18 | −0.01 |
| H-5 | 4.50 | 4.51 | −0.01 |
| H-6 | 3.88 | 3.88 | 0.00 |
| Specific optical rotation for casuarictin (3) in MeOH (unit of c: mg/mL) | |||
| literature data [23] a (c = 0.2) | synthetic product (c = 0.12, 23 °C) | ||
| 35 | 26 | ||
| 1H NMR Data for Rugosin C (3) in Acetone-d6 | |||
|---|---|---|---|
| Assignment | Literature Data [23] | Synthetic Product | Δ(Lit.–Syn.) |
| δ | δ | δ | |
| galloyl | 7.15 | 7.15 | 0.00 |
| HHDP or valoneoyl | 7.14 | 7.14 | 0.00 |
| HHDP or valoneoyl | 6.54 | 6.54 | 0.00 |
| HHDP or valoneoyl | 6.46 | 6.45 | 0.01 |
| HHDP or valoneoyl | 6.40 | 6.40 | 0.00 |
| HHDP or valoneoyl | 6.34 | 6.38 | −0.04 |
| H-1 | 6.18 | 6.19 | −0.01 |
| H-3 | 5.44 | 5.44 | 0.00 |
| H-6 | 5.28 | 5.28 | 0.00 |
| H-4 | 5.14 | 5.14 | 0.00 |
| H-2 | 5.07 | 5.07 | 0.00 |
| H-5 | 4.46 | 4.46 | 0.00 |
| H-6 | 3.79 | 3.79 | 0.00 |
| 1H NMR Data for Permethylated Rugosin C (32) in Acetone-d6 | ||||
|---|---|---|---|---|
| Assignment | Number of Protons | Literature Data [3] | Synthetic Product | Δ(Lit.–Syn.) |
| δ | δ | δ | ||
| galloyl | 2 | 7.31 | 7.32 | −0.01 |
| valoneoyl | 1 | 7.25 | 7.25 | 0.00 |
| HHDP and valoneoyl | 1 | 6.85 | 6.83 | 0.02 |
| 1 | 6.83 | 6.83 | 0.00 | |
| HHDP | 1 | 6.69 | 6.69 | 0.00 |
| valoneoyl | 1 | 6.50 | 6.50 | 0.00 |
| H-1 | 1 | 6.26 | 6.26 | 0.00 |
| H-3 | 1 | 5.55 | 5.54 | 0.01 |
| H-2 | 1 | 5.23 | 5.22 | 0.01 |
| H-6 | 1 | 5.15 | 5.16 | −0.01 |
| H-4 | 1 | 5.06 | 5.07 | −0.01 |
| H-5 | 1 | 4.39 | 4.42 | −0.03 |
| O-Me | 3 | 4.06 | 4.06 | 0.00 |
| O-Me | 3 | 3.90 | 3.90 | 0.00 |
| O-Me | 3 | 3.89 | 3.89 | 0.00 |
| O-Me | 3 | 3.89 | 0.00 | |
| O-Me | 6 | 3.87 | 3.87 | 0.00 |
| O-Me | 3 | 3.86 | 3.86 | 0.00 |
| O-Me | 3 | 3.85 | 3.86 | −0.01 |
| O-Me | 3 | 3.83 | 3.83 | 0.00 |
| O-Me | 3 | 3.80 | 3.82 | −0.02 |
| O-Me | 3 | 3.80 | 0.00 | |
| O-Me | 3 | 3.76 | 3.77 | −0.01 |
| O-Me | 3 | 3.76 | 0.00 | |
| O-Me | 3 | 3.76 | 0.00 | |
| O-Me | 3 | 3.74 | 3.74 | 0.00 |
| O-Me | 3 | 3.70 | 3.70 | 0.00 |
| O-Me | 3 | 3.66 | 3.65 | 0.01 |
| O-Me | 3 | 3.58 | 3.60 | −0.02 |
References
- Pouységu, L.; Deffieux, D.; Malik, G.; Natangelo, A.; Quideau, S. Synthesis of ellagitannin natural products. Nat. Prod. Rep. 2011, 28, 853–874. [Google Scholar] [CrossRef] [PubMed]
- Quideau, S. Chemistry and Biology of Ellagitannins—An Underestimated Class of Bioactive Plant Polyphenols; World Scientific: Singapore, 2009. [Google Scholar]
- Yoshida, T.; Ohbayashi, H.; Ishihara, K.; Ohwashi, W.; Haba, K.; Okano, Y.; Shingu, T.; Okuda, T. Tannins and related polyphenols of melastomataceous plants. I. hydrolyzable tannins from Tibouchina semidecandra COGN. Chem. Pharm. Bull. 1991, 39, 2233–2240. [Google Scholar] [CrossRef]
- Yoshida, T.; Ohwashi, W.; Haba, K.; Ohbayashi, H.; Ishihara, K.; Okano, Y.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomataceous Plants. II. Nobotanins B, C and E, Hydrolyzable Tannin Dimer and Trimers from Tibouchina Semidecandra COGN. Chem. Pharm. Bull. 1991, 39, 2264–2270. [Google Scholar] [CrossRef]
- Yoshida, T.; Haba, K.; Nakata, F.; Okano, Y.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomataceous Plants. III. Nobotanins G, H and I, Dimeric Hydrolyzable Tannins from Heterocentron roseum. Chem. Pharm. Bull. 1992, 40, 66–71. [Google Scholar] [CrossRef]
- Yoshida, T.; Nakata, F.; Hosotani, K.; Nitta, A.; Okudat, T. Dimeric hydrolysable tannins from melastoma Malabathricum. Phytochemistry 1992, 31, 2829–2833. [Google Scholar] [CrossRef]
- Yoshida, T.; Haba, K.; Arata, R.; Nakata, F.; Shingu, T.; Okuda, T. Tannins and Related Polyphenols of Melastomataceous Plants. VII. Nobotanins J and K, Trimeric and Tetrameric Hydrolyzable Tannins from Heterocentron roseum. Chem. Pharm. Bull. 1995, 43, 1101–1106. [Google Scholar] [CrossRef]
- Yoshida, T.; Nakata, F.; Okuda, T. Tannins and Related Polyphenols of Melastomataceous Plants. VIII. Nobotanins L, M and N, Trimeric Hydrolyzable Tannins from Tibouchina semidecandra. Chem. Pharm. Bull. 1999, 47, 824–827. [Google Scholar] [CrossRef]
- Yoshida, T.; Amakura, Y.; Yokura, N.; Ito, H.; Isaza, J.H.; Ramirez, S.; Pelaez, D.P.; Renner, S.S. Oligomeric hydrolyzable tannins from Tibouchina multiflora. Phytochemistry 1999, 52, 1661–1666. [Google Scholar] [CrossRef]
- Ito, H.; Isaza, J.H.; Amakusa, Y.; Yoshida, T. Ellagitannin Oligomers from Melastomataceou Plants. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu 2001, 43, 175–180. [Google Scholar]
- Isaza, M.J.H.; Ito, H.; Yoshida, T. Tetrameric and pentameric ellagitannins from monochaetum multiflorum. Heterocycles 2001, 55, 29–32. [Google Scholar]
- Isaza, M.J.H.; Ito, H.; Yoshida, T. Oligomeric hydrolyzable tannins from Monochaetum multiflorum. Phytochemistry 2004, 65, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Wakamori, S.; Hirokane, T.; Ikeuchi, K.; Matsumoto, S. Structural revisions in natural ellagitannins. Molecules 2018, 23, 1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, N.; Hattori, M.; Namba, T.; Nishizawa, M.; Yamagishi, T.; Okuda, T. Inhibitory Effect of Tannins on Reverse Transcriptase from RNA Tumor Virus. J. Nat. Prod. 1985, 48, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Kishi, N.; Koshiura, R.; Yoshida, T.; Hatano, T.; Okuda, T. Relationship between the structures and the antitumor activities of tannins. Chem. Pharm. Bull. 1987, 35, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Nishimura, K.; Abe, H.; Maruta, H.; Sakagami, H.; Hatano, T.; Okuda, T.; Yoshida, T.; Tsai, Y.-J.; Uchiumi, F.; et al. Novel inhibitors of poly(ADP-ribose) glycohydrolase. Biochim. Biophys. Acta (BBA) Gen. Subj. 1993, 1158, 251–256. [Google Scholar] [CrossRef]
- Nakashima, H.; Murakami, T.; Yamamoto, N.; Sakagami, H.; Tanuma, S.-I.; Hatano, T.; Yoshida, T.; Okuda, T. Inhibition of human immunodeficiency viral replication by tannins and related compounds. Antivir. Res. 1992, 18, 91–103. [Google Scholar] [CrossRef]
- Yasuda, M.; Ikeoka, M.; Kondo, S.-I. Skin-related enzyme inhibitory activity by hydrolyzable polyphenols in water chestnut (Trapa natans) husk. Biosci. Biotechnol. Biochem. 2021, 85, 666–674. [Google Scholar] [CrossRef]
- Feldman, K.S.; Lawlor, M.D. Ellagitannin Chemistry. The First Total Synthesis of a Dimeric Ellagitannin, Coriariin A. J. Am. Chem. Soc. 2000, 122, 7396–7397. [Google Scholar] [CrossRef]
- Hirokane, T.; Ikeuchi, K.; Yamada, H. Total syntheses of laevigatins A and E. Eur. J. Org. Chem. 2015, 33, 7352–7359. [Google Scholar] [CrossRef]
- Yoshida, T.; Ito, H.; Hipolito, I.J. Pentameric ellagitannin oligomers in melastomataceous plants—chemotaxonomic signifi-cance. Phytochemistry 2005, 66, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Hirokane, T.; Hirata, Y.; Ishimoto, T.; Nishii, K.; Yamada, H. A unified strategy for the synthesis of highly oxygenated diaryl ethers featured in ellagitannins. Nat. Commun. 2014, 5, 3478. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Hatano, T.; Yazaki, K.; Ogawa, N. Rugosin A, B, C and praecoxin A, tannins having a valoneoyl group. Chem. Pharm. Bull. 1982, 30, 4230–4233. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ikeda, Y.; Ohbayashi, H.; Ishihara, K.; Ohwashi, W.; Shingu, T.; Okuda, T. Dimeric ellagitannins in plants of melastomataceae. Chem. Pharm. Bull. 1986, 34, 2676–2679. [Google Scholar] [CrossRef]
- Okuda, T.; Yoshida, T.; Ashida, M.; Yazaki, K. Tannis of Casuarina and Stachyurus species. Part 1. Structures of pendunculagin, casuarictin, strictinin, casuarinin, casuariin, and stachyurin. J. Chem. Soc. Perkin Trans. 1 1983, 1765–1772. [Google Scholar] [CrossRef]
- Saijo, R.; Nonaka, G.; Nishioka, I. Tannins and related compounds. LXXXIV: Isolation and characterization of five new hydrolyzable tannins from the bark of mallotus japonicus. Chem. Pharm. Bull. 1989, 37, 2063–2070. [Google Scholar] [CrossRef]
- Wakamori, S.; Matsumoto, S.; Kusuki, R.; Ikeuchi, K.; Yamada, H. Total Synthesis of Casuarinin. Org. Lett. 2020, 22, 3392–3396. [Google Scholar] [CrossRef]
- Yamada, H.; Nagao, K.; Dokei, K.; Kasai, Y.; Michihata, N. Total synthesis of (−)-corilagin. J. Am. Chem. Soc. 2008, 130, 7566–7567. [Google Scholar] [CrossRef]
- Ashibe, S.; Ikeuchi, K.; Kume, Y.; Wakamori, S.; Ueno, Y.; Iwashita, T.; Yamada, H. Non-enzymatic Oxidation of a Pentagal-loylglucose Analog to Ellagitannins. Angew. Chem. Int. Ed. 2017, 56, 15402–15406. [Google Scholar] [CrossRef]
- Asakura, N.; Fujimoto, S.; Michihata, N.; Nishii, K.; Imagawa, H.; Yamada, H. Synthesis of chiral and modifiable hexahy-droxydiphenoyl compounds. J. Org. Chem. 2011, 76, 9711–9719. [Google Scholar] [CrossRef]
- Malik, G.; Natangelo, A.; Charris, J.; Pouységu, L.; Manfredini, S.; Cavagnat, D.; Buffeteau, T.; Deffieux, D.; Quideau, S. Synthetic Studies toward C-Glucosidic Ellagitannins: A Biomimetic Total Synthesis of 5-O-Desgalloylepipunicacortein A. Chem. Eur. J. 2012, 18, 9063–9074. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Seitz, S.P.; Papahatjis, D.P. A mild and general method for the synthesis of O-glycosides. J. Am. Chem. Soc. 1983, 105, 2430–2434. [Google Scholar] [CrossRef]
- Konradsson, P.; Udodong, U.E.; Fraser-Reid, B. Iodonium promoted reactions of disarmed thioglycosides. Tetrahedron Lett. 1990, 31, 4313–4316. [Google Scholar] [CrossRef]
- Aloui, M.; Fairbanks, A.J. N-Iodosaccharin: A potent new activator of thiophenylglycosides. Synlett 2001, 6, 797–799. [Google Scholar] [CrossRef]
- Ferrier, R.J.; Hay, R.W.; Vethaviyasar, N. A potentially versatile synthesis of glycosides. Carbohydr. Res. 1973, 27, 55–61. [Google Scholar] [CrossRef]
- Bols, M.; Hansen, H.C. Simple Synthesis of beta-D-Glucosyl Esters. Acta Chem. Scand. 1993, 47, 818–822. [Google Scholar] [CrossRef]
- Fukase, K.; Hasuoka, A.; Kinoshita, I.; Kusumoto, S. Iodosobenzene-triflic anhydride as an efficient promoter for glycosidation reaction using thioglycosides as donors. Tetrahedron Lett. 1992, 33, 7165–7168. [Google Scholar] [CrossRef]
- Takeuchi, H.; Ueda, Y.; Furuta, T.; Kawabata, T. Total Synthesis of Ellagitannins via Sequential Site-Selective Functionalization of Unprotected D-Glucose. Chem. Pharm. Bull. 2017, 65, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Errata for Chemical and Pharmaceutical Bulletin. Chem. Pharm. Bull. 2022, 70, 505. [CrossRef]
- Yamaguchi, S.; Ashikaga, Y.; Nishii, K.; Yamada, H. Total Synthesis of the Proposed Structure of Roxbin B; the Nonidentical Outcome. Org. Lett. 2012, 14, 5928–5931. [Google Scholar] [CrossRef]
- Konishi, H.; Hirokane, T.; Hashimoto, H.; Ikeuchi, K.; Matsumoto, S.; Wakamori, S.; Yamada, H. Synthesis of diaryl ether components of ellagitannins using ortho-quinone with consonant mesomeric effects. Chem. Commun. 2020, 56, 3991–3994. [Google Scholar] [CrossRef]
- Hashimoto, H.; Ishimoto, T.; Konishi, H.; Hirokane, T.; Wakamori, S.; Ikeuchi, K.; Yamada, H. Synthesis of an Ellagitannin Component, the Macaranoyl Group with a Tetra-ortho-Substituted Diaryl Ether Structure. Org. Lett. 2020, 22, 6729–6733. [Google Scholar] [CrossRef]
- Matsumoto, S.; Aoyama, A.; Wakamori, S.; Yamada, H. Total Synthesis of Macaranin B. Biosci. Biotechnol. Biochem. 2021, 85, 1937–1944. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, H.; Wakamori, S.; Ikeuchi, K.; Yamada, H. Divergent Synthesis of Four Monomeric Ellagitannins toward the Total Synthesis of an Oligomeric Ellagitannin, Nobotanin K. Organics 2022, 3, 293-303. https://0-doi-org.brum.beds.ac.uk/10.3390/org3030022
Hashimoto H, Wakamori S, Ikeuchi K, Yamada H. Divergent Synthesis of Four Monomeric Ellagitannins toward the Total Synthesis of an Oligomeric Ellagitannin, Nobotanin K. Organics. 2022; 3(3):293-303. https://0-doi-org.brum.beds.ac.uk/10.3390/org3030022
Chicago/Turabian StyleHashimoto, Hajime, Shinnosuke Wakamori, Kazutada Ikeuchi, and Hidetoshi Yamada. 2022. "Divergent Synthesis of Four Monomeric Ellagitannins toward the Total Synthesis of an Oligomeric Ellagitannin, Nobotanin K" Organics 3, no. 3: 293-303. https://0-doi-org.brum.beds.ac.uk/10.3390/org3030022