
Pathophysiological Correlation between Cigarette Smoking and Amyotrophic Lateral Sclerosis


1
Spine Labs, St. George & Sutherland Clinical School, University of New South Wales, Kogarah, NSW 2217, Australia
2
Centre for Inflammation, School of Life Sciences, Faculty of Science, Centenary Institute and University of Technology Sydney, Sydney, NSW 2050, Australia
3
Spine Service, St. George Hospital, Kogarah, NSW 2217, Australia
*
Author to whom correspondence should be addressed.
NeuroSci 2021, 2(2), 120-134; https://0-doi-org.brum.beds.ac.uk/10.3390/neurosci2020008
Submission received: 20 February 2021
/
Revised: 6 April 2021
/
Accepted: 16 April 2021
/
Published: 20 April 2021
(This article belongs to the Special Issue Feature Papers in Neurosci 2021)
Abstract
:Cigarette smoke (CS) has been consistently demonstrated to be an environmental risk factor for amyotrophic lateral sclerosis (ALS), although the molecular pathogenic mechanisms involved are yet to be elucidated. Here, we propose different mechanisms by which CS exposure can cause sporadic ALS pathogenesis. Oxidative stress and neuroinflammation are widely implicated in ALS pathogenesis, with blood–spinal cord barrier disruption also recognised to be involved in the disease process. In addition, immunometabolic, epigenetic and microbiome alterations have been implicated in ALS recently. Identification of the underlying pathophysiological mechanisms that underpin CS-associated ALS will drive future research to be conducted into new targets for treatment.
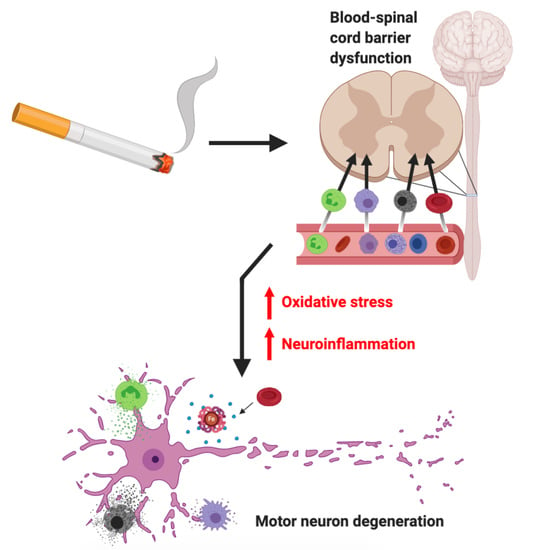
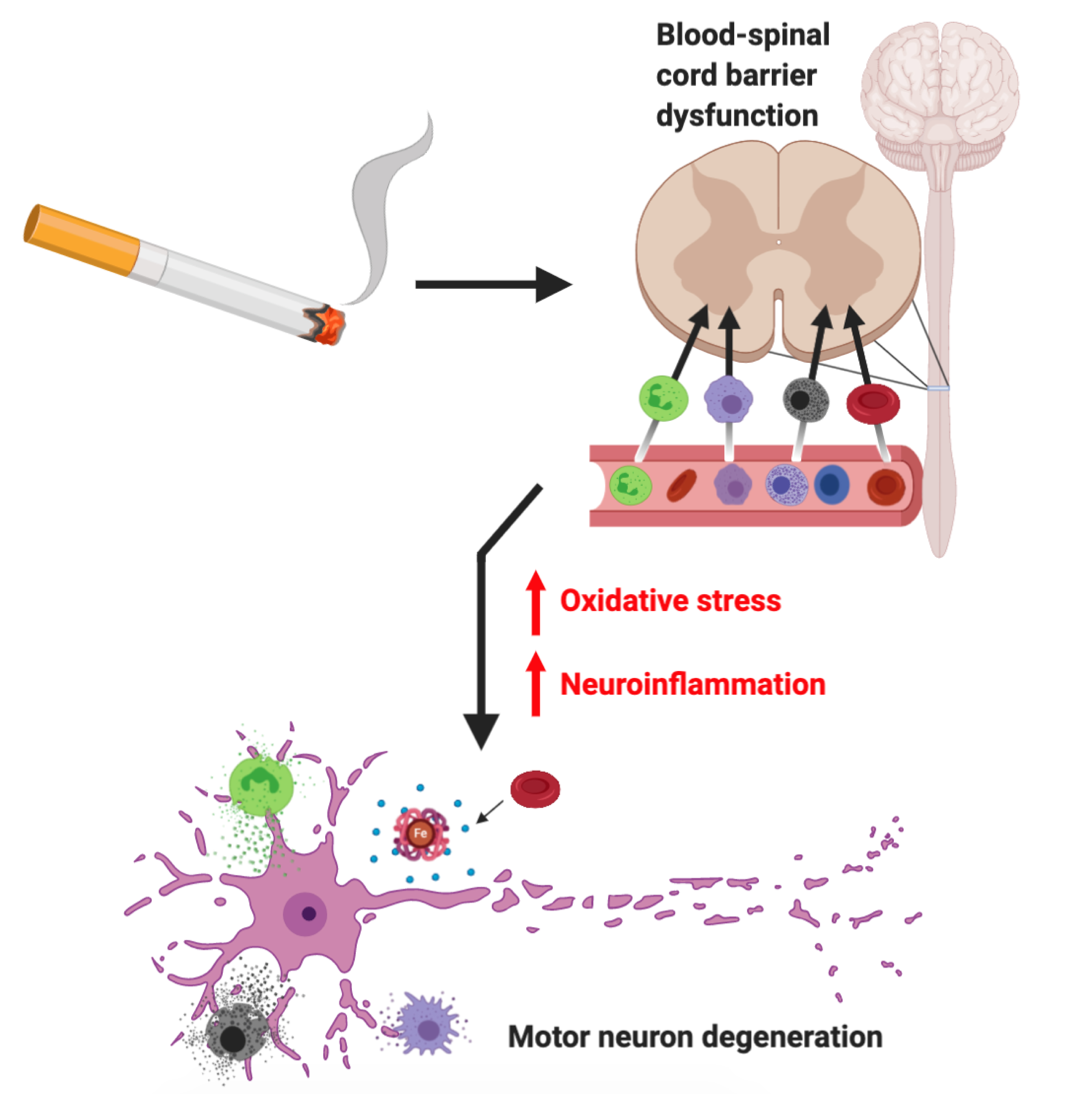
1. Background
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease affecting motor neurons in both the cerebral motor cortex and spinal cord, causing progressive paralysis and premature death typically within 3–5 years from the time of diagnosis. Clinical presentation varies, more commonly involving spinal-onset disease (~70%) characterised by limb muscle weakness, or more rarely bulbar-onset disease (~30%) involving disorders of speech [1,2]. The global prevalence of ALS has been estimated to be >220,000, with future predictions reaching 375,000 by 2040 owing to the aging population. These projections are likely underestimated due to improvements in healthcare and economic conditions, particularly among developing nations [3]. Around 90% of all diagnoses are sporadic ALS (sALS), while genetically linked familial ALS (fALS) is responsible for 5–10% of cases [4]. Much of our knowledge of ALS pathophysiology has derived from different cell and animal models carrying gene mutations linked to fALS, such as superoxide dismutase 1 (SOD1), chromosome 9 open reading frame 72 (C9orf72), Fused in Sarcoma (FUS), and TAR DNA-binding protein 43 (TDP-43) [5]. Despite numerous hypotheses being proposed regarding sALS pathogenesis [6], the exact cause remains uncertain. Available treatments for ALS have limited efficacy and therapeutic methods are largely restricted to symptomatic and palliative care [7]. Riluzole, an anti-glutamatergic agent, is a Therapeutic Goods Administration (TGA) approved drug for ALS in Australia that extends median survival by 2–3 months due to delaying respiratory dysfunction [8,9]. Recently, the antioxidant edaravone was approved for the treatment of ALS in the United States (US), although phase III studies revealed limited efficacy in the wider ALS population [10]. It is now well-recognized that ALS is a multifactorial disease, involving a complex interplay between genetic and environmental factors. Indeed, the actual disease process starts long before clinical symptoms manifest.
2. Cigarette Smoke and ALS
Current evidence of genetic and environmental risk factors associated with the onset of sALS is limited to observation and populational studies. Among many proposed risk factors for ALS, it has been suggested that cigarette smoking may be considered an established risk factor [11] despite conflicting epidemiological evidence. While several population-based and cohort studies report smokers having an increased risk of ALS, greater ALS-related mortality and disease severity [12,13,14,15], others found weak or no association between smoking and ALS risk (Table 1) [16,17]. These inconsistent results may be explained in part by general limitations of observational epidemiological studies [11] as well as small sample sizes and survival, recall and selection bias. In particular, recall bias becomes significantly problematic when smoking history is quantified using “pack years”, and further inconsistencies arise when considering different cigarettes contain varying concentrations of toxicants and their delivery is not consistent across all products [18,19,20]. Recently, a large case–control study as part of the Euro-MOTOR project concluded that smokers’ pack year history was positively associated with increased sALS risk [21]. An investigation of monozygotic twins with the C9orf72 mutation, a gene strongly associated with both familial and sALS [22], revealed that despite having similar environmental exposures, the ALS affected twin had a 10 year smoking history and a prior instance of head trauma, while the other twin remained asymptomatic [23].
The negative influence of cigarette smoke (CS) on health is well-established [30]. Several studies have revealed the association of CS and fine particulate air pollution with neurodegenerative disorders including Alzheimer’s disease, dementia, Parkinson’s and multiple sclerosis [31,32,33]. Indeed, this is supported by a recent study demonstrating CS to cause greater biomarker expression of oxidation, neurodegeneration and neuroinflammation in cerebrospinal fluid (CSF) [34]. CS contains >100,000 toxic compounds that may be inhaled directly (active smoking) or indirectly (passive smoking) [35]. Environmental case–control studies reveal that exposure to hazardous air pollutants, many of which are present in CS [36], increases the risk of sALS [37,38]. Analysis of CS contents demonstrates significant concentrations of heavy metals [39], which are associated with greater sALS risk [40]. Indeed, formaldehyde, another hazardous compound found in CS, has also been proposed to be associated with sALS in a dose–response relationship [12]. While the exact nature of the relationship between CS and ALS is unknown, many factors, including oxidative stress and neuroinflammation, have been suggested as putative risk factors [41]. Recent reports also suggest several new mechanisms that might be in play. Here, we describe different possible mechanisms (direct and indirect pathways) by which CS can affect ALS onset and progression.
3. CS-Induced ALS Pathogenesis: Direct Pathways
3.1. Oxidative Stress
Toxic compounds in CS have been shown to cause tissue damage through hypoxia and oxidative stress in the lungs [42], and other distal tissues [43,44]. It is likely that continuous CS exposure has similar effects on motor neurons and surrounding cells in the spinal cord, contributing to ALS pathogenesis. CS-mediated oxidative stress can also aggravate the onset and progression of symptoms in sALS by augmenting neurodegeneration. Reactive oxygen species (ROS) generated in response to CS has been shown to induce free radical damage in the form of lipid peroxidation of membrane unsaturated lipids, DNA strand breakage, protein oxidation, mitochondrial depolarisation, RNA oxidation and cell apoptosis, which are also some of the well-established markers of ALS [45,46,47].
Currently, oxidative stress is one of the most widely accepted hypotheses for ALS pathogenesis. Oxidative stress occurs when there is an imbalance between ROS production and elimination via protective anti-oxidant factors [48]. The proposed role of oxidative stress in ALS pathogenesis involves direct damage to motor neurons as well as the surrounding microenvironment [49]. Additional mechanisms of oxidative stress-induced neurodegeneration include the promotion of mitochondrial dysfunction, protein aggregation, and endoplasmic reticulum stress responses [50]. Indeed, as oxidative stress contributes to abnormal RNA processing, transcriptional dysregulation, impaired axonal transport and protein misfolding, it has been proposed as a central mechanism in proteostasis disruption observed in ALS [51]. Growing evidence suggests the causes of proteome dyshomeostasis involving imbalances in protein production and degradation may be the unifying mechanism in ALS pathogenesis [52]. Notably, Ash and colleagues [53] demonstrate that aryl hydrocarbon receptor transcription factor agonists, including several toxicant by-products of CS, increase levels of TAR DNA Binding Protein-43 (TP43). TDP-43 is a consistent feature in sALS cases and plays a key role in the pathological progression of ALS, including driving neurodegeneration [54].
Analyses conducted on sALS patients repeatedly demonstrate oxidative stress biomarkers present in CSF, urine and plasma [55]. Durazzo and colleagues [32] reveal greater biomarker expression of free radical-induced oxidative stress in smokers’ CSF samples compared to non-smokers. Additional studies on ALS also demonstrate alterations in markers of oxidative stress, such as glutathione and nuclear factor E2-related factor-2 [56,57,58]. Furthermore, post-mortem results in both sALS and fALS patients reveal DNA, lipid and protein damage secondary to oxidative stress [59,60,61,62,63]. Together, this evidence reinforces the likelihood that CS-mediated oxidative stress is a key driver of ALS pathogenesis.
3.2. Neuroinflammation
CS-induced oxidative stress can lead to inflammation in neurons and surrounding cells, which is another mediator of ALS pathogenesis. Neuroinflammation generates additional ROS which can further promote oxidative stress. Thus, smokers experience an increased and continual free radical load from both CS and subsequent inflammatory responses. Prolonged exposure to CS causes a combination of proinflammatory and immunosuppressive processes which impair humoral and cell-mediated immune responses [64]. It is likely that repeated CS exposure promotes a neuroinflammatory environment, facilitating ALS neurodegeneration. While this mechanism may involve the direct invasion of pro-inflammatory cells into the spinal cord, other intrinsic neuroinflammatory inducers such as activated glial cells may be contributing.
Neuroinflammation is characterised by microglial and astrocyte activation along with inflammatory cytokine overproduction [65,66]. Evidence of activated microglia and lymphocytic infiltrates within spinal cords of ALS patients suggests neuroinflammation occurs alongside motor neuron degeneration [67]. Analysis of CSF from ALS patients supports these results, revealing greater inflammatory mediators such as Interleukin (IL)-8 and eotaxin, responsible for inducing neutrophil and eosinophil chemotaxis and activation, respectively [68]. Analysis of astrocytes reveals that motor neuron damaging inflammatory mediators are secreted in both familial and sporadic ALS patients [69,70]. Further studies on ALS patients demonstrate a systemic pro-inflammatory state and an impaired antioxidant system, with increased levels of proinflammatory cytokines such as IL-6 and IL-8 [71]. Similarly, multiple human, animal and in vitro studies have concluded that CS exposure increases systemic markers of inflammatory cytokines, such as IL-6, 8 and 1β along with tumour necrosis factor-α (TNF-α) [72,73]. Together, these results strongly implicate CS-induced neuroinflammation in the pathogenesis of ALS.
3.3. Immunometabolism
Microglia are resident macrophage-like cells in the central nervous system (CNS) that act as immune sentinels and maintain CNS homeostasis [74]. They are very sensitive to physiological changes in the local environment and can become “activated” in response to an insult, during which their number, morphology and activity change, with the release of more inflammatory cytokines and chemokines. Evidence of activated microglia has been reported in the motor cortex and spinal cord of sALS patients [75,76,77]. Studies in sALS animal models have provided valuable insights into the involvement of microglia in ALS; however, their dual role in ALS has emerged. Although reactive microglia are thought to have neurotoxic effects, a recent report indicates that they might play a protective role in ALS [78]. It is possible that microglia play a protective role early in the disease and transition to a toxic state upon repeated insults from environmental factors, such as CS exposure. Indeed, carcinogens from CS have been shown to induce microglia activation and neuronal damage [79].
Microglia activation is associated with metabolic reprogramming, where the microglia switch from mitochondrial oxidative phosphorylation to glycolysis when activated [80]. Several pro-inflammatory factors and oxidative stress have been implicated in this phenomenon. CS exposure likely induces such immunometabolic changes in microglia during activation, via direct oxidative stress in these cells as well as by promoting a neuroinflammatory environment involving surrounding cells. At a systemic level, CS exposure has been linked to various metabolic syndromes, such as insulin resistance and abnormal lipoprotein metabolism [81]. At cellular level, CS is known to induce the dysregulation of several metabolic pathways [82]. Thus, CS exposure can not only cause microglia activation but likely keep them locked in an activated state, which can further enhance ALS disease progression.
3.4. Epigenetic Alterations
Recent reports indicate aberrant epigenetic modifications as another possible cause of ALS [83]. Epigenetics refers to heritable changes in gene expression without modification to the genome sequence. The three main epigenetic mechanisms include DNA methylation, microRNAs (miRNAs) and histone post-translational modifications [84]. In post-mortem sALS spinal cords, global alternations in DNA methylation and hydroxymethylation were observed [85]. Here, key genes that were either upregulated or downregulated were highly associated with immune and inflammatory responses. Similarly, the dysregulation of miRNAs was observed in ALS patients, with concomitant dysregulation of mRNA targets that were previously implicated in ALS pathogenesis [86]. Furthermore, several histone modifications are associated with ALS [87]. Collectively, these studies indicate various possible roles of epigenetic changes in ALS pathogenesis.
CS accelerates epigenetic age and decreases life expectancy via DNA methylation changes that influence gene regulation and genome stability [88,89]. It is notable that, like many neurodegenerative diseases, aging is a major risk factor for ALS [90]. CS is also associated with unique miRNA signatures that cause systemic inflammation and immune cell activation [91]. Such inflammation is mediated by CS through histone modifications as well [92]. Given the strong links between CS and epigenetic modifications, it is likely that CS exposure causes ALS via the induction of pathologic epigenetic alterations in neuronal and surrounding cells.
4. CS-Induced ALS Pathogenesis: Indirect Pathways
4.1. Blood–Spinal Cord Barrier Dysfunction
The blood–spinal cord barrier (BSCB) maintains CNS homeostasis, providing a regulated microenvironment for cellular components of the spinal cord, and is often considered as a functional equivalent to the blood–brain barrier (BBB). Both the BSCB and BBB consist of neurovascular functional units, involving microvascular non-fenestrated endothelial cells (ECs) joined by cell-to-cell tight junction (TJ) proteins [93]. The BSCB is more permeable than the BBB, likely due to reduced expression of TJ proteins, exposing the spinal cord to greater amounts of potentially neurotoxic stimuli compared to the brain [94,95].
Over the last decade, there has been an accumulation of evidence demonstrating the BSCB to be involved in ALS aetiology and progression. Spinal cords of Sod1 transgenic mice reveal greater BSCB permeability of harmful blood-borne substances, microhaemorrhages, reduced expression of TJ proteins and basement membrane components along with decreased spinal cord blood flow and capillary length [96,97,98,99,100]. Importantly, many of these observed changes precede motor neuron degeneration and neuroinflammation, implicating BSCB impairment as the primary cause for subsequent neuronal damage. Additional analyses conducted on human post-mortem sALS and fALS spinal cord tissues support these results and also reveal severe intra/extra-cellular oedema, degenerated pericytes and astrocytic end-feet from ECs along with reduced TJ protein mRNA expression [101,102,103]. Notably, spontaneous and/or accelerated BSCB breakdown in Sod1 mice directly contributes to early motor neuron injury, while removing BSCB-derived sources of neuronal injury and/or restoring BSCB integrity delays motor neuron degeneration onset [99].
CS has been demonstrated to impair BBB viability and function over time through inflammatory and oxidative processes [104,105,106]. Hence, given the physiological similarities between the BSCB and BBB, it can be postulated that the oxidative and inflammatory processes involved in barrier disruption may not be BBB-specific, and a similar pathological process of degeneration may be taking place in the BSCB. Even though little is currently known regarding the cause of BSCB dysfunction, oxidative stress has been suggested as a likely mechanism contributing to the pathophysiological cascade of BSCB impairment [107]. Thus, it is likely that CS induces oxidative stress and inflammatory processes that damage BSCB components, initiating primary BSCB disruption. This mechanism may involve the promotion of EC inflammation, modulation of EC activity and their interactions, reductions in TJ protein expression, basal lamina disruption and impaired astrocyte/pericyte-EC communication (Figure 1).
4.2. Cell Influx
BSCB disruption after CS exposure likely increases spinal cord permeability to circulating cells such as erythrocytes and pro-inflammatory innate/adaptive immune cells, including mast cells, macrophages, neutrophils and T lymphocytes, producing a neurotoxic CNS environment. These cells are further activated by CS, increasing their pro-inflammatory behaviour [108,109]. When progressive BSCB disruption begins, erythrocytes, given their small cell diameter of 8 μm [110], may begin the process of cellular infiltration into the spinal cord. Following extravasation and lysis, haemoglobin-derived heme degradation results in local increases in bilirubin, carbon monoxide and iron [111]. Haemoglobin-derived iron has a direct toxic effect on motor neurons via iron-dependant oxidative pathways [112] and has been continually implicated in the ALS disease process [97,99,111,113]. In states of neuroinflammation, immune cells such as iron-containing macrophages also migrate into the CNS and release iron, contributing to this free radical-mediated oxidative damage pathway, and may further facilitate neurotoxicity [113].
Additionally, leukocyte-mediated release of pro-inflammatory cytokines at the BSCB also stimulates astrocyte production of chemokines, which promote extravasation of leukocytes into the CNS [114]. Pro-inflammatory cells generate neuroinflammatory cytokines and subsequent ROS that increase motor neuron oxidative stress, exacerbating neurodegenerative mechanisms in ALS. Here, a degenerative cascade of oxidative damage probably ensues, whereby greater ROS production within the neuron then crosses the cell membrane to activate surrounding microglia, which, in turn, release more cytokines and ROS [50]. These greater levels of inflammatory cytokines produced by microglia and pro-inflammatory cells further enhance the neuroinflammatory processes of motor neuron degeneration. Given the susceptibility of the CNS to oxidative stress, high rate of oxygen consumption and relatively low concentration of antioxidants [55], we posit that homeostatic pro-oxidative and anti-oxidative mechanisms are shifted to promote a pro-oxidative neuroenvironment. Furthermore, these oxidative and neuroinflammatory processes associated with the neurotoxic environment may extend back to the BSCB, leading to secondary BSCB damage. This facilitates further permeability leading to greater immune-cell migration, aiding the development and maintenance of a neurotoxic environment (Figure 2).
CS exposure increases both the recruitment and degranulation of mast cells in different tissues, including lungs and skin [108,115]. Similarly, CS can activate resident mast cells near the BSCB, which along with activated mast cells from other tissues can migrate into the spinal cord following BSCB disruption and cause neurodegeneration. In fact, mast cells have been implicated in both neuroinflammation and neurodegeneration through their ability to release inflammatory cytokines (TNF-α, IL-6), histamine and proteases [66]. These cytokines, along with the release of chemoattractants, result in the recruitment of other leukocytes such as neutrophils, further exacerbating the inflammatory environment. The release of cytokines and proteases from mast cells can degrade TJ proteins and extracellular matrix components to facilitate greater BSCB permeability. This is supported by the analysis of ALS patients, which revealed the presence of mast cells expressing IL-17 in spinal cord tissue [116] along with elevated serum and CSF levels of the proinflammatory cytokine IL-15, an NK cell chemoattractant [117]. Finally, CS can cause metabolic reprogramming in these cells, as described before in regard to microglia, and render them into a more pro-inflammatory state.
4.3. Microbiome
The gut microbiota represents a key interface between the environment and the immune system, being a major site of toxin and intrinsic antigen production. Given its ability to influence myelination, the BBB, synaptic plasticity and neuronal transmission, gut microbiome imbalance (dysbiosis) clearly exerts a pathogenic influence on the CNS [118,119,120]. Recently, the gut microbiome has been implicated in the pathogenesis of multiple neurological disorders, including ALS [121,122,123,124]. Analysis of Sod1 transgenic mice reveals altered gut microbiota composition, along with exacerbations in motor abnormalities and significant reductions in motor neuron cell counts following microbiome depletion using broad-spectrum antibiotics [121]. Studies of ALS patients demonstrate gut microbiome variations, especially during the disease course, as well as an imbalance in potentially protective and neurotoxic/proinflammatory microbial groups [122,125]. Furthermore, a population-based case–control study of 2484 ALS patients revealed an association between antibiotic use, particularly repeated use, and increased ALS risk [126].
Active and passive smoking are known to impair immune system functioning through a range of immunosuppressive mechanisms and have been continually demonstrated to influence the microbiome. These oral, lung and gut microbiome modifications are associated with various diseases such as chronic obstructive pulmonary disease (COPD), asthma, ulcerative colitis and even cancers [127,128,129,130]. In particular, even after smoking cessation, many changes initiating gut dysbiosis persist for prolonged periods of time [129]. While multiple theories exist regarding smoking’s impact on the microbiome, including immunosuppression and oxygen deprivation, the exact mechanisms involved are yet to be established. Importantly, the gut microbiome has been reported to play an important role in BBB permeability, serving as a mechanism that facilitates the direct and indirect transmission of microbial signals from the gut to the CNS [119,131]. Thus, it is possible that CS-induced microbiome modifications may play a key role in shifting the balance between protective and pathogenic immune responses that are responsible for BSCB disruption. This mechanism may involve gut microbiota modifications promoting pro-inflammatory immune cell–BSCB interactions, leading to the development of a neurotoxic environment associated with ALS pathogenesis.
5. Conclusions
CS is one of the major risk factors associated with sALS; however, the exact aetiology remains elusive. While CS may not be the only cause for ALS pathogenesis, it may aggravate or effectuate other predisposing factors such as genetic susceptibility or unknown environmental factors. Here, we propose several key mechanisms by which CS may contribute to ALS pathogenesis and pathophysiological progression. Neurodegeneration in ALS can be facilitated by CS via direct or indirect pathways. These pathways are likely not mutually exclusive and may indeed involve a complex interplay between one another. Direct mechanisms involve oxidative stress, neuroinflammation, immunometabolic changes and epigenetic alterations caused by toxic by-products in CS. Indirect mechanisms involve primary/secondary disruption of the BSCB due to CS and altered microbiomes, which may, in turn, promote immune cell influx and their activation. Both these proposed pathways create a pro-oxidative and pro-inflammatory CNS environment that enhances the pathological progression of ALS. Indeed, it is highly likely that exposures to air pollution, e-cigarettes and bushfire smoke may also contribute to ALS pathogenesis via similar pathways [38,132].
The pathophysiology of ALS has been extensively studied in animals that carry genetic mutations linked to fALS [133]. Since the aetiology of sALS is not yet defined, much of the human analyses have been performed in post-mortem ALS patients. Given the established link between CS and ALS, we propose using animal models of CS exposure to investigate whether ALS symptoms appear in these animals. Several mouse models of CS exposure have recently been reported [108,134]. Although these models have been primarily used to assess lung function, it will be valuable to analyse their spinal cord as well. We should also take into account the age of the animals as it plays an important role in ALS onset. Thus, comparisons regarding the effects of CS in mice at different ages should be performed. Finally, it will be interesting to test if CS exposure in fALS mouse models (SOD1-G93A, TDP43, FUS, C9orf72) accelerates disease onset and progression and aggravates symptoms. Establishing these new models of environmental risk factor-mediated ALS will certainly augment and improve our knowledge of the disease process.
From a therapeutic perspective, antioxidant and anti-inflammatory treatments are continually being explored to mitigate the oxidative and neuroinflammatory degenerative effects present in ALS. However, there is insufficient evidence of the efficacy of these treatments in ALS patients [135]. Therefore, new targets and strategies need to be explored to develop more effective ALS therapies. One such evolving target is immune cells. Notably, Masitinib, a tyrosine kinase inhibitor modulating neuroinflammatory processes of multiple immune cells, slows the progression of ALS in combination with riluzole [136]. Considering our proposed hypotheses, new therapeutic strategies may arise in relation to (a) targeting metabolic pathways in immune cells to inhibit activation/inflammation, (b) epigenetic therapy, (c) maintaining BSCB integrity such as limiting mast cell-mediated disruption of the BSCB, and (d) altering the microbiome using antibiotics, probiotics or faecal matter transplantation. We suggest that future studies for sALS treatment should be conducted using CS-exposed animal models. Future studies should also examine the relationship between bushfire exposure, air pollution and vaping with ALS risk and uncover underlying pathophysiological relationships.
Author Contributions
Conceptualization, S.M. and A.D.; writing—original draft preparation, S.M. and A.D.; figure and table preparations, S.M.; writing—review and editing, S.M., P.M.H., A.D.D. and A.D. All authors have read and agreed to the published version of the manuscript.
Funding
P.M.H. is funded by a Fellowship and grants from the National Health and Medical Research Council (NHMRC) of Australia (1079187, 1175134) and by UTS.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Stegmann, G.M.; Hahn, S.; Liss, J.; Shefner, J.; Rutkove, S.; Shelton, K.; Duncan, C.J.; Berisha, V. Early detection and tracking of bulbar changes in ALS via frequent and remote speech analysis. NPJ Digit. Med. 2020, 3, 132. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.C.A.; Traynor, K.C.A.B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Le Masson, G. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-F.; Wu, Z.-Y. Genotype-phenotype correlations of amyotrophic lateral sclerosis. Transl. Neurodegener. 2016, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Compre-hensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Agosta, F.; Lunetta, C.; Filippi, M.; Quattrini, A. Recent advances in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Le, W.D.; Xie, Y.-Y.; Wang, X.-P. Current Therapy of Drugs in Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2016, 14, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L.-N. The efficacy and safety of riluzole for neurodegenerative movement disorders: A systematic review with meta-analysis. Drug Deliv. 2018, 25, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2017, 11. [Google Scholar] [CrossRef]
- Armon, C. Smoking may be considered an established risk factor for sporadic ALS. Neurology 2009, 73, 1693–1698. [Google Scholar] [CrossRef]
- Gallo, V.; Bueno-De-Mesquita, H.B.; Vermeulen, R.; Andersen, P.M.; Kyrozis, A.; Linseisen, J.; Kaaks, R.; Allen, N.E.; Roddam, A.W.; Boshuizen, H.C.; et al. Smoking and risk for amyotrophic lateral sclerosis: Analysis of the EPIC cohort. Ann. Neurol. 2009, 65, 378–385. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, É.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Thun, M.J.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Smoking and risk of amyotrophic lateral sclerosis: A pooled analysis of 5 prospective cohorts. Arch. Neurol. 2011, 68, 207–213. [Google Scholar] [CrossRef]
- Calvo, A.; Canosa, A.; Bertuzzo, D.; Cugnasco, P.; Solero, L.; Clerico, M.; De Mercanti, S.; Bersano, E.; Cammarosano, S.; Ilardi, A.; et al. Influence of cigarette smoking on ALS outcome: A population-based study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1229–1233. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Fang, F. Smoking and amyotrophic lateral sclerosis: A mendelian randomization study. Ann. Neurol. 2019, 85, 482–484. [Google Scholar] [CrossRef]
- Fang, F.; Bellocco, R.; Hernán, M.A.; Ye, W. Smoking, Snuff Dipping and the Risk of Amyotrophic Lateral Sclerosis—A Prospective Cohort Study. Neuroepidemiology 2006, 27, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Ward, E.C. Smoking Is Not a Risk Factor for Sporadic Amyotrophic Lateral Sclerosis in an Australian Population. Neuroepidemiology 2012, 38, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bhartiya, D.; Kaur, J.; Kumari, S.; Singh, H.; Saraf, D.; Sinha, D.N.; Mehrotra, R. Regulation of toxic contents of smokeless tobacco products. Indian J. Med. Res. 2018, 148, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.; O’Connor, R.J. Constituents in tobacco and smoke emissions from Canadian cigarettes. Tob. Control. 2008, 17 (Suppl. 1), i24–i31. [Google Scholar] [CrossRef]
- Mendel, J.R.; Baig, S.A.; Hall, M.G.; Jeong, M.; Byron, M.J.; Morgan, J.C.; Noar, S.M.; Ribisl, K.M.; Brewer, N.T. Brand switching and toxic chemicals in cigarette smoke: A national study. PLoS ONE 2018, 13, e0189928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.; Visser, A.E.; D’Ovidio, F.; Vlaanderen, J.; Portengen, L.; Beghi, E.; Chio, A.; Logroscino, G.; Hardiman, O.; Pupillo, E.; et al. Effect modification of the association between total cigarette smoking and ALS risk by intensity, duration and time-since-quitting: Euro-MOTOR. J. Neurol. Neurosurg. Psychiatry 2019, 91, 33–39. [Google Scholar] [CrossRef]
- Van Mossevelde, S.; van der Zee, J.; Cruts, M.; Van Broeckhoven, C. Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 2017, 44, 117–124. [Google Scholar] [CrossRef]
- Xi, Z.; Yunusova, Y.; Van Blitterswijk, M.; Dib, S.; Ghani, M.; Moreno, D.; Sato, C.; Liang, Y.; Singleton, A.; Robertson, J.; et al. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology 2014, 83, 1476–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisskopf, M.G.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Cudkowicz, M.; Ascherio, A. Prospective Study of Cigarette Smoking and Amyotrophic Lateral Sclerosis. Am. J. Epidemiol. 2004, 160, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sutedja, N.A.; Veldink, J.H.; Fischer, K.; Kromhout, H.; Wokke, J.; Huisman, M.H.; Heederik, D.J.; Berg, L.H.V.D. Lifetime occupation, education, smoking, and risk of ALS. Neurology 2007, 69, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernán, M.A. Association of smoking with amyotrophic lateral sclerosis risk and survival in men and women: A prospective study. BMC Neurol. 2010, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- De Jong, S.W.; Huisman, M.H.B.; Sutedja, N.A.; Van Der Kooi, A.J.; De Visser, M.; Schelhaas, H.J.; Fischer, K.; Veldink, J.H.; Berg, L.H.V.D. Smoking, Alcohol Consumption, and the Risk of Amyotrophic Lateral Sclerosis: A Population-based Study. Am. J. Epidemiol. 2012, 176, 233–239. [Google Scholar] [CrossRef]
- Opie-Martin, S.; Jones, A.; Iacoangeli, A.; Al-Khleifat, A.; Oumar, M.; Shaw, P.J.; Shaw, C.E.; Morrison, K.E.; Wootton, R.E.; Davey-Smith, G.; et al. UK case control study of smoking and risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Opie-Martin, S.; Wootton, R.E.; Budu-Aggrey, A.; Shatunov, A.; Jones, A.R.; Iacoangeli, A.; Al Khleifat, A.; Davey-Smith, G.; Al-Chalabi, A. Relationship between smoking and ALS: Mendelian randomisation interrogation of causality. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1312–1315. [Google Scholar] [CrossRef]
- Onor, I.O.; Stirling, D.L.; Williams, S.R.; Bediako, D.; Borghol, A.; Harris, M.B.; Darensburg, T.B.; Clay, S.D.; Okpechi, S.C.; Sarpong, D.F. Clinical Effects of Cigarette Smoking: Epidemiologic Impact and Review of Pharmacotherapy Options. Int. J. Environ. Res. Public Health 2017, 14, 1147. [Google Scholar] [CrossRef] [Green Version]
- Kioumourtzoglou, M.-A.; Schwartz, J.D.; Weisskopf, M.G.; Melly, S.J.; Wang, Y.; Dominici, F.; Zanobetti, A. Long-term PM 2.5 Exposure and Neurological HospitalAdmissions in the Northeastern United States. Environ. Health Perspect. 2016, 124, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W.; Korecka, M.; Trojanowski, J.Q.; Shaw, L.M. History of cigarette smoking in cognitively-normal elders is associated with elevated cerebrospinal fluid biomarkers of oxidative stress. Drug Alcohol Depend. 2014, 142, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, M.; Chitnis, T. Association Between Cigarette Smoking and Multiple Sclerosis: A Review. JAMA Neurol. 2020, 77, 245. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Wang, J.; Xue, Q.; Yang, X.; Kang, Y.; Li, M.; Xu, J.; Li, G.; Li, C.; et al. Association of Cigarette Smoking with Cerebrospinal Fluid Biomarkers of Neurodegeneration, Neuroinflammation, and Oxidation. JAMA Netw. Open 2020, 3, e2018777. [Google Scholar] [CrossRef] [PubMed]
- Brokl, M.; Bishop, L.; Wright, C.G.; Liu, C.; McAdam, K.; Focant, J. Analysis of mainstream tobacco smoke particulate phase using comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. J. Sep. Sci. 2013, 36, 1037–1044. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; Van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous Compounds in Tobacco Smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Malek, A.M.; Barchowsky, A.; Bowser, R.; Heiman-Patterson, T.; Lacomis, D.; Rana, S.; Youk, A.; Talbott, E.O. Exposure to hazardous air pollutants and the risk of amyotrophic lateral sclerosis. Environ. Pollut. 2015, 197, 181–186. [Google Scholar] [CrossRef]
- Seelen, M.; Campos, R.A.T.; Veldink, J.H.; Visser, A.E.; Hoek, G.; Brunekreef, B.; Van Der Kooi, A.J.; De Visser, M.; Raaphorst, J.; Berg, L.H.V.D.; et al. Long-Term Air Pollution Exposure and Amyotrophic Lateral Sclerosis in Netherlands: A Population-based Case–control Study. Environ. Health Perspect. 2017, 125, 097023. [Google Scholar] [CrossRef] [Green Version]
- Benson, N.U.; Anake, W.U.; Adedapo, A.E.; Fred-Ahmadu, O.H.; Ayejuyo, O.O. Toxic metals in cigarettes and human health risk assessment associated with inhalation exposure. Environ. Monit. Assess. 2017, 189, 619. [Google Scholar] [CrossRef]
- Wang, M.-D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and pro-gression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef]
- Oskarsson, B.; Horton, D.K.; Mitsumoto, H. Potential Environmental Factors in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.; Donovan, C.; Liu, G.; Gomez, H.M.; Chimankar, V.; Harrison, C.L.; Wiegman, C.H.; Adcock, I.M.; Knight, D.A.; Hirota, J.A.; et al. Animal models of COPD: What do they tell us? Respirology 2017, 22, 21–32. [Google Scholar] [CrossRef]
- Fricker, M.; Goggins, B.J.; Mateer, S.; Jones, B.; Kim, R.Y.; Gellatly, S.L.; Jarnicki, A.G.; Powell, N.; Oliver, B.G.; Radford-Smith, G.; et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, W.; Yan, Z.; Yuan, X. Smoking increases the risk of diabetic foot amputation: A meta-analysis. Exp. Ther. Med. 2017, 15, 1680–1685. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, P.; Tierney, W.; Hossain, M.; Puvenna, V.; Janigro, D.; Cucullo, L. Pathophysiological Impact of Cigarette Smoke Exposure on the Cerebrovascular System with a Focus on the Blood-brain Barrier: Expanding the Awareness of Smoking Toxicity in an Underappreciated Area. Int. J. Environ. Res. Public Health 2010, 7, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco Smoke: Involvement of Reactive Oxygen Species and Stable Free Radicals in Mechanisms of Oxidative Damage, Carcinogenesis and Synergistic Effects with Other Respirable Particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Riancho, J.; Gonzalo, I.; Ruiz-Soto, M.; Berciano, J. Why do motor neurons degenerate? Actualisation in the pathogenesis of amyotrophic lateral sclerosis. Neurología 2019, 34, 27–37. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Carrã, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Ballance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.; Mitsumoto, H.; Shungu, D. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1G93A mice model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neu-ron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Körner, S.; Böselt, S.; Thau, N.; Rath, K.J.; Dengler, R.; Petri, S. Differential Sirtuin Expression Patterns in Amyotrophic Lateral Sclerosis (ALS) Postmortem Tissue: Neuroprotective or Neurotoxic Properties of Sirtuins in ALS? Neurodegener. Dis. 2012, 11, 141–152. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carrì, M. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Fitzmaurice, P.; Shaw, I.; Kleiner, H.; Miller, R.; Monks, T.; Lau, S.; Mitchell, J.; Lynch, P. Evidence for DNA damage in amy-otrophic lateral sclerosis. Mus. Nerve 1996, 19, 797–798. [Google Scholar]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Turner, M.R.; Bowser, R.; Bruijn, L.; Dupuis, L.; Ludolph, A.; McGrath, M.; Manfredi, G.; Maragakis, N.; Miller, R.G.; Pullman, S.L.; et al. Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Liang, C.-L.; Liu, H.; Zeng, Y.-Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune re-sponsiveness: Up and down or upside down? Oncotarget 2017, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implica-tions. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.K.; Nair, A.; Gupta, M. Mast Cells in Neurodegenerative Disease. Front. Cell. Neurosci. 2019, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2003, 55, 221–235. [Google Scholar] [CrossRef]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Abdel-Moaty, H.; Hunter, J.; Mhatre, M.; Mou, S.; Nguyen, K.; Potapova, T.; Pye, Q.N.; Qi, M.; Rice, H.; et al. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J. Neuroinflamm. 2006, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Vaart, H.; Postma, D.S.; Timens, W.; Hacken, N.H.T.T. Acute effects of cigarette smoke on inflammation and oxidative stress: A review. Thorax 2004, 59, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A.; Lawyer, G.; McDonough, S.; Wang, Q.; Kassem, N.O.; Kas-Petrus, F.; Ye, D.; Singh, K.P.; Kassem, N.O.; Rahman, I. Systemic biomarkers of inflammation, oxidative stress and tissue injury and repair among waterpipe, cigarette and dual tobacco smokers. Tob. Control. 2020, 29, s102–s109. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e829. [Google Scholar] [CrossRef]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.-Y.; Trojanowski, J.Q. Microglial Activation Correlates with Disease Progression and Upper Motor Neuron Clinical Symptoms in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [Green Version]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Ghosh, D.; Mishra, M.K.; Das, S.; Kaushik, D.K.; Basu, A. Tobacco carcinogen induces microglial activation and subsequent neuronal damage. J. Neurochem. 2009, 110, 1070–1081. [Google Scholar] [CrossRef]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Liu, J.; Ning, G. Active Smoking and Risk of Metabolic Syndrome: A Meta-Analysis of Prospective Studies. PLoS ONE 2012, 7, e47791. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yang, D.C.; Chen, C.-H. Metabolic reprogramming: A driver of cigarette smoke-induced inflammatory lung diseases. Free Radic. Biol. Med. 2021, 163, 392–401. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Sakowski, S.A.; Goutman, S.A.; Feldman, E.L. Amyotrophic lateral sclerosis: Mechanisms and therapeutics in the epigenomic era. Nat. Rev. Neurol. 2015, 11, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Q.; Javed, R.; Zhong, J.; Gao, H.; Liang, H. Effect of tobacco smoking on the epigenetic age of human respiratory organs. Clin. Epigenetics 2019, 11, 183. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, X.; Just, A.C.; Colicino, E.; Wang, C.; Coull, B.A.; Hou, L.; Zheng, Y.; Vokonas, P.; Schwartz, J.; et al. Smoking-Related DNA Methylation is Associated with DNA Methylation Phenotypic Age Acceleration: The Veterans Affairs Normative Aging Study. Int. J. Environ. Res. Public Health 2019, 16, 2356. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, T.; Partridge, L.; Isaacs, A.M. Ageing as a risk factor for ALS/FTD. Hum. Mol. Genet. 2017, 26, R105–R113. [Google Scholar] [CrossRef]
- Willinger, C.M.; Rong, J.; Tanriverdi, K.; Courchesne, P.L.; Huan, T.; Wasserman, G.A.; Lin, H.; Dupuis, J.; Joehanes, R.; Jones, M.R.; et al. MicroRNA Signature of Cigarette Smoking and Evidence for a Putative Causal Role of MicroRNAs in Smoking-Related Inflammation and Target Organ Damage. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Liu, X.; Li, J.; Ouyang, R.; Chen, P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenetics Chromatin 2019, 12, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartanusz, V.; Jezova, D.; Alajajian, B.; Digicaylioglu, M. The blood-spinal cord barrier: Morphology and Clinical Implications. Ann. Neurol. 2011, 70, 194–206. [Google Scholar] [CrossRef] [PubMed]
- DosSantos, M.F.; Holanda-Afonso, R.C.; Lima, R.L.; DaSilva, A.F.; Moura-Neto, V. The role of the blood–brain barrier in the development and treatment of migraine and other pain disorders. Front. Cell. Neurosci. 2014, 8, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.; Pachter, J.S. Isolation and culture of microvascular endothelial cells from murine spinal cord. J. Neuroimmunol. 2006, 177, 209–214. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of Compromised Blood-Spinal Cord Barrier in Early and Late Symptomatic SOD1 Mice Modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [Green Version]
- Nicaise, C.; Mitrecic, D.; Demetter, P.; De Decker, R.; Authelet, M.; Boom, A.; Pochet, R. Impaired blood–brain and blood–spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009, 1301, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neu-rovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Bowser, R.; Appel, S.H. Decreased Mrna Expression of Tight Junction Proteins in Lumbar Spinal Cords of Patients with ALS. Neurology 2009, 72, 1614–1616. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood–brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Alterations of the blood-spinal cord barrier in sporadic amyotrophic lateral sclerosis. Neuropathology 2015, 35, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Fofaria, N.; Prasad, S.; Sajja, R.K.; Weksler, B.; Couraud, P.-O.; Romero, I.A.; Cucullo, L. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: Is smoking reduced or nicotine-free products really safe? BMC Neurosci. 2014, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: A microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzova-Davis, S.; Woods, R.L., III; Louis, M.K.; Zesiewicz, T.A.; Kuzmin-Nichols, N.; Sullivan, K.L.; Miller, A.M.; Her-nandez-Ontiveros, D.G.; Sanberg, P.R. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE 2010, 5, e10614. [Google Scholar] [CrossRef]
- Beckett, E.L.; Stevens, R.L.; Jarnicki, A.G.; Kim, R.Y.; Hanish, I.; Hansbro, N.G.; Deane, A.; Keely, S.; Horvat, J.C.; Yang, M.; et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013, 131, 752–762.e7. [Google Scholar] [CrossRef] [Green Version]
- Sopori, M.L. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef]
- Diez-Silva, M.; Dao, M.; Han, J.; Lim, C.-T.; Suresh, S. Shape and Biomechanical Characteristics of Human Red Blood Cells in Health and Disease. MRS Bull. 2010, 35, 382–388. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 125, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Rifkind, J.M.; Mohanty, J.G.; Enagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2015, 5, 500. [Google Scholar] [CrossRef] [Green Version]
- Eve, D.J.; Steiner, G.; Mahendrasah, A.; Sanberg, P.R.; Kurien, C.; Thomson, A.; Borlongan, C.V.; Garbuzova-Davis, S. Reduction of microhemorrhages in the spinal cord of symptomatic ALS mice after intravenous human bone marrow stem cell trans-plantation accompanies repair of the blood-spinal cord barrier. Oncotarget 2018, 9, 10621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.H.; Johnsen, K.B.; Moos, T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell. Mol. Life Sci. 2014, 71, 1607–1622. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Ge, S.; Lemire, Y.; Jellison, E.R.; Serwanski, D.R.; Ruddle, N.H.; Pachter, J.S. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J. Neuroinflamm. 2014, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Hansbro, P.M.; Hamilton, M.J.; Fricker, M.; Gellatly, S.L.; Jarnicki, A.G.; Zheng, D.; Frei, S.M.; Wong, G.W.; Hamadi, S.; Zhou, S.; et al. Importance of Mast Cell Prss31/Transmembrane Tryptase/Tryptase-γ in Lung Function and Experimental Chronic Obstructive Pulmonary Disease and Colitis. J. Biol. Chem. 2014, 289, 18214–18227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiala, M.; Chattopadhay, M.; La Cava, A.; Tse, E.; Liu, G.; Lourenco, E.; Eskin, A.; Liu, P.T.; Magpantay, L.; Tse, S.; et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflamm. 2010, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Rentzos, M.; Rombos, A.; Nikolaou, C.; Zoga, M.; Zouvelou, V.; Dimitrakopoulos, A.; Alexakis, T.; Tsoutsou, A.; Samakovli, A.; Michalopoulou, M. Interleukin-15 and interleukin-12 are elevated in serum and cerebrospinal fluid of patients with amyo-trophic lateral sclerosis. Eur. Neurol. 2010, 63, 285–290. [Google Scholar] [CrossRef]
- Stilling, R.M.; Dinan, T.G.; Cryan, J.F. Microbial genes, brain & behaviour—Epigenetic regulation of the gut-brain axis. Genes Brain Behav. 2014, 13, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 2016, 165, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, D.; Cionci, N.B.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggìa, F.; Lucenti, M.A.; Bersano, E.; Cantello, R.; et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [Green Version]
- Sharon, G.; Cruz, N.J.; Kang, D.-W.; Gandal, M.J.; Wang, B.; Kim, Y.-M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhan, Y.; Mariosa, D.; Larsson, H.; Almqvist, C.; Ingre, C.; Zagai, U.; Pawitan, Y.; Fang, F. Antibiotics use and risk of amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2019, 26, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shi, G. Smoking and microbiome in oral, airway, gut and some systemic diseases. J. Transl. Med. 2019, 17, 225. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Peters, B.A.; Dominianni, C.; Zhang, Y.; Pei, Z.; Yang, L.; Ma, Y.; Purdue, M.P.; Jacobs, E.J.; Gapstur, S.M.; et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016, 10, 2435–2446. [Google Scholar] [CrossRef]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut–lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood–brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2017, 243, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dennekamp, M.; Abramson, M.J. The effects of bushfire smoke on respiratory health. Respirology 2011, 16, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Robberecht, W.; Bosch, L.V.D. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, M.; Maes, K.; De Vleeschauwer, S.; Thomas, D.; Verbeken, E.K.; Decramer, M.; Janssens, W.; Gayan-Ramirez, G.N. Long-term nose-only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis. Model. Mech. 2012, 5, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orrell, R.W.; Lane, R.J.M.; Ross, M. A systematic review of antioxidant treatment for amyotrophic lateral sclerosis/motor neuron disease. Amyotroph. Lateral Scler. 2008, 9, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernandez, M.; Marin, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical tria. Amyotroph. Lateral Scler. Frontotemporal Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Proposed mechanism of direct blood–spinal cord barrier (BSCB) disruption caused by cigarette smoking. (A) Normal BSCB components including endothelial cells, tight junction proteins, pericytes and astrocytic end feet processes. (B) Cigarette smoke induced BSCB damage through oxidative and inflammatory processes. Leukocytes such as mast cells, macrophages and neutrophils release inflammatory cytokines and subsequently reactive oxygen species (ROS) leading to an impairment and reduced expression of tight junction proteins. This facilitates further damage to BSCB components including disrupted astrocyte/pericyte-endothelial communication, modulated endothelial cell activity and endothelial basement membrane disruptions. Here, a neurotoxic environment surrounding the BSCB is established and maintained by the constant influx of inflammatory cytokines and ROS from leukocytes, resulting in further BSCB damage.
Figure 1.
Proposed mechanism of direct blood–spinal cord barrier (BSCB) disruption caused by cigarette smoking. (A) Normal BSCB components including endothelial cells, tight junction proteins, pericytes and astrocytic end feet processes. (B) Cigarette smoke induced BSCB damage through oxidative and inflammatory processes. Leukocytes such as mast cells, macrophages and neutrophils release inflammatory cytokines and subsequently reactive oxygen species (ROS) leading to an impairment and reduced expression of tight junction proteins. This facilitates further damage to BSCB components including disrupted astrocyte/pericyte-endothelial communication, modulated endothelial cell activity and endothelial basement membrane disruptions. Here, a neurotoxic environment surrounding the BSCB is established and maintained by the constant influx of inflammatory cytokines and ROS from leukocytes, resulting in further BSCB damage.

Figure 2.
Blood–spinal cord barrier (BSCB) leak establishes a neurotoxic environment for motor neurons. Primary damage to BSCB components by cigarette smoke (CS) exposure increases the permeability to pro-inflammatory immune cells such as mast cells, macrophages and neutrophils along with red blood cells that give rise to iron-containing haemoglobin. This leads to the generation of ROS and neuroinflammatory cytokines that increase motor neuron oxidative stress and exacerbate other neurodegenerative mechanisms in ALS. These include protein aggregation, alterations in axonal transport, endoplasmic reticulum stress responses and mitochondrial dysfunction. These pro-inflammatory cytokines and ROS also activate surrounding microglia which, in turn, release cytokines and ROS, further contributing to neurodegeneration. The inflammatory cytokines produced by microglia and pro-inflammatory cells further increase neuroinflammatory processes of motor neuron degeneration. Additionally, secondary BSCB dysfunction may occur concurrently with primary BSCB dysfunction. Here, oxidative and neuroinflammatory processes associated with the neurotoxic environment may extend back to the BSCB, facilitating secondary damage. This augments permeability and increases immune-cell migration, aiding the development and maintenance of a neurotoxic environment.
Figure 2.
Blood–spinal cord barrier (BSCB) leak establishes a neurotoxic environment for motor neurons. Primary damage to BSCB components by cigarette smoke (CS) exposure increases the permeability to pro-inflammatory immune cells such as mast cells, macrophages and neutrophils along with red blood cells that give rise to iron-containing haemoglobin. This leads to the generation of ROS and neuroinflammatory cytokines that increase motor neuron oxidative stress and exacerbate other neurodegenerative mechanisms in ALS. These include protein aggregation, alterations in axonal transport, endoplasmic reticulum stress responses and mitochondrial dysfunction. These pro-inflammatory cytokines and ROS also activate surrounding microglia which, in turn, release cytokines and ROS, further contributing to neurodegeneration. The inflammatory cytokines produced by microglia and pro-inflammatory cells further increase neuroinflammatory processes of motor neuron degeneration. Additionally, secondary BSCB dysfunction may occur concurrently with primary BSCB dysfunction. Here, oxidative and neuroinflammatory processes associated with the neurotoxic environment may extend back to the BSCB, facilitating secondary damage. This augments permeability and increases immune-cell migration, aiding the development and maintenance of a neurotoxic environment.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Epidemiological evidence regarding the association between cigarette smoking and amyotrophic lateral sclerosis (ALS).
Table 1.
Epidemiological evidence regarding the association between cigarette smoking and amyotrophic lateral sclerosis (ALS).
| Source | Study Design | Participants with ALS included in Patient Population | Findings |
|---|---|---|---|
| Weisskopf et al., 2004 [24] | Prospective cohort study | 330 | Current smokers had an increased risk of ALS and ALS-related death in females but not males, no increased risk associated with pack years or cigarettes smokes per day. |
| Fang et al., 2006 [16] | Prospective cohort study | 160 | No strong evidence was found between smokers and ALS risk. |
| Sutedja et al., 2007 [25] | Case–control study | 364 | Current smokers had an increased risk of ALS when compared to non-smokers. |
| Gallo et al., 2009 [12] | Prospective cohort study | 118 | Pack years associated with increased risk of ALS, current smokers had greater risk of ALS-related mortality, number of years since quitting associated with decreased ALS risk. |
| Alonso et al., 2010 [26] | Population-based case–control study | 1143 | Female ever smokers a were associated with an increased risk of ALS but not men, smoking is a predictor in female ALS-related mortality not men. |
| Wang et al., 2011 [13] | Pooled analysis of 5 prospective cohorts | 832 | Ever smokers had an increased risk of developing ALS when compared to never smokers, mean number of daily cigarettes smoked and smoking duration were positively associated with ALS, younger age of smoking initiation associated with higher risk of ALS. |
| de Jong et al., 2012 [27] | Population-based case–control study | 494 | Current smokers had an increased risk of ALS and a worse prognosis following ALS diagnosis with a shorter survival. |
| Pamphlett et al., 2012 [17] | Case–control study | 631 | Smoking status, pack years and age when smoking began were not associated with an increased risk of ALS. |
| Calvo et al., 2016 [14] | Population-based cohort study | 650 | Patients who were currently smoking had a younger age of ALS onset, current and former smokers had a significantly shorter median survival compared to never smokers, smoking status significantly correlated with mean monthly decline in ALS disease severity. |
| Zhan et al., 2019 [15] | Mendelian randomisation population-based study | 12,577 | Ever smokers had a higher risk for ALS when compared to never smokers. |
| Peters et al., 2020 [21] | Population-based case–control study | 1410 | Smoking pack years positively associated with ALS risk when compared to never smokers, increased risk associated strongly with smoking duration rather than smoking intensity, inverse relationship observed between ALS risk and time-since-quitting smoking. |
| Opie-Martin et al., 2020 [28] | Retrospective case–control study | 202 | Weak association between current smoking and risk of ALS. |
| Opie-Martin et al., 2020 [29] | Mendelian randomisation population-based study | 20,806 | No strong evidence was found between ever smokers and ALS risk. |
a Ever smokers defined as both former and current cigarette smokers.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Menounos, S.; Hansbro, P.M.; Diwan, A.D.; Das, A. Pathophysiological Correlation between Cigarette Smoking and Amyotrophic Lateral Sclerosis. NeuroSci 2021, 2, 120-134. https://0-doi-org.brum.beds.ac.uk/10.3390/neurosci2020008
AMA Style
Menounos S, Hansbro PM, Diwan AD, Das A. Pathophysiological Correlation between Cigarette Smoking and Amyotrophic Lateral Sclerosis. NeuroSci. 2021; 2(2):120-134. https://0-doi-org.brum.beds.ac.uk/10.3390/neurosci2020008
Chicago/Turabian StyleMenounos, Spiro, Philip M. Hansbro, Ashish D. Diwan, and Abhirup Das. 2021. "Pathophysiological Correlation between Cigarette Smoking and Amyotrophic Lateral Sclerosis" NeuroSci 2, no. 2: 120-134. https://0-doi-org.brum.beds.ac.uk/10.3390/neurosci2020008