
The Structural Flexibility of Cold- and Warm-Adapted Enzymes (Endonucleases I) by Molecular Dynamics Simulation †

Abstract
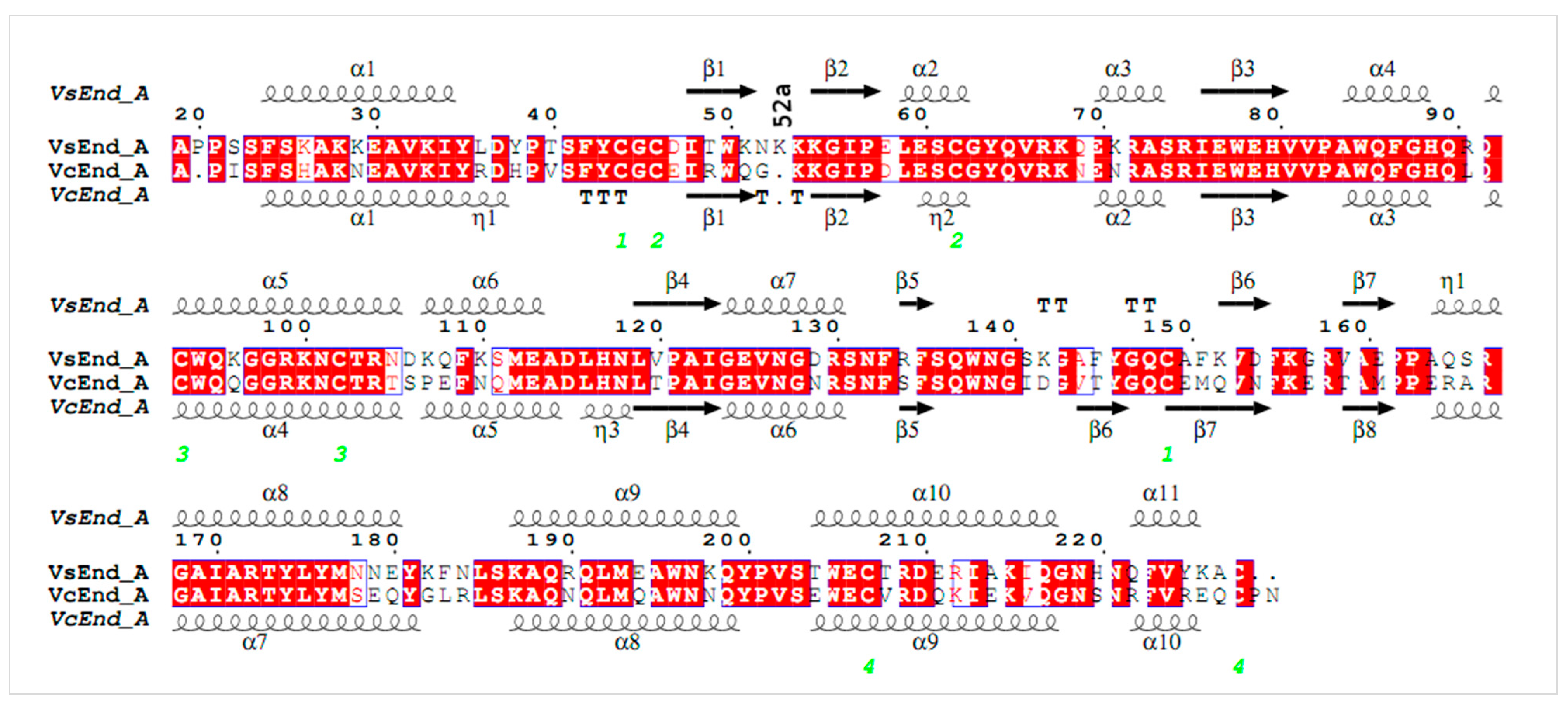
:1. Introduction
2. Materials and Methods
3. Results and Discussion
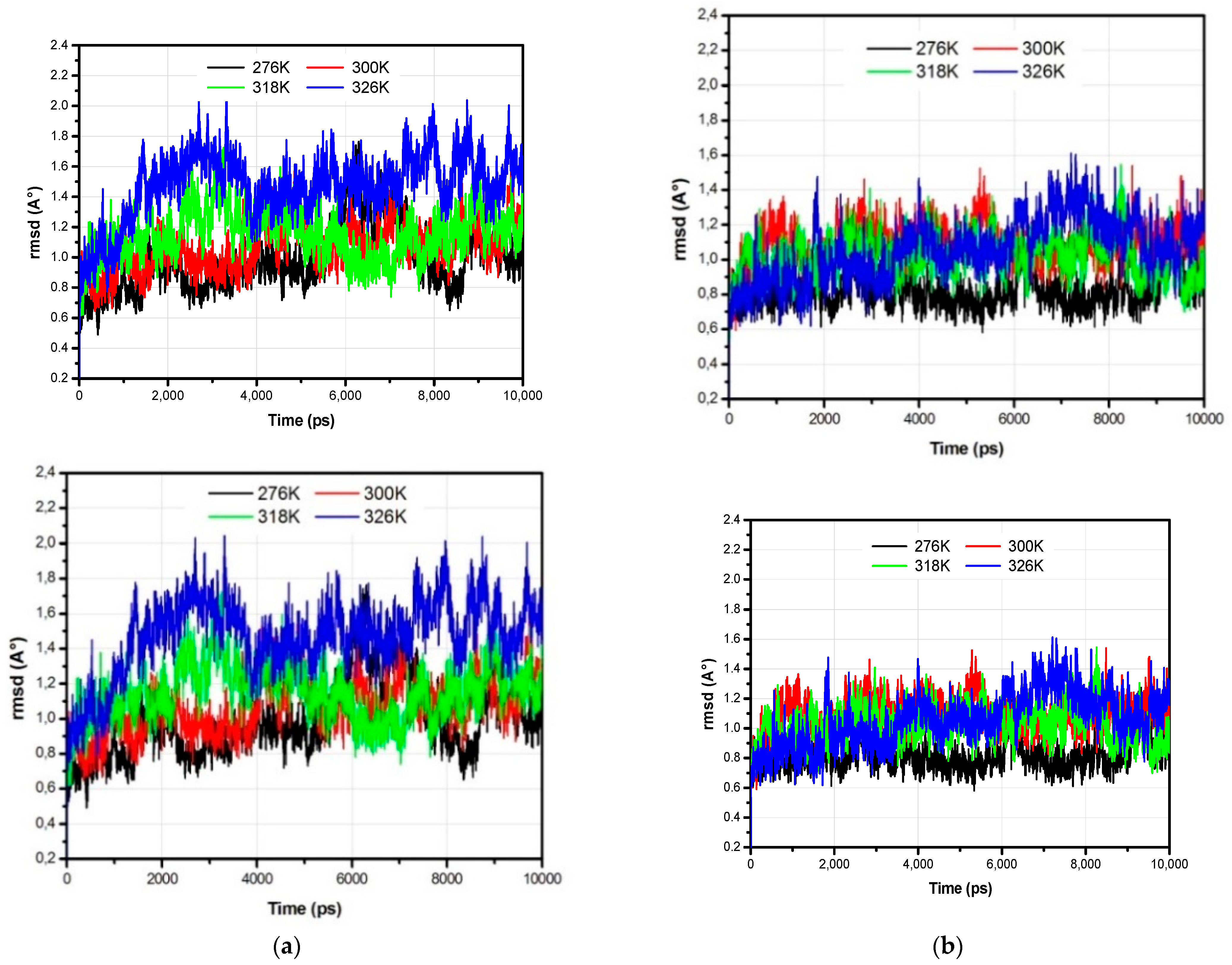
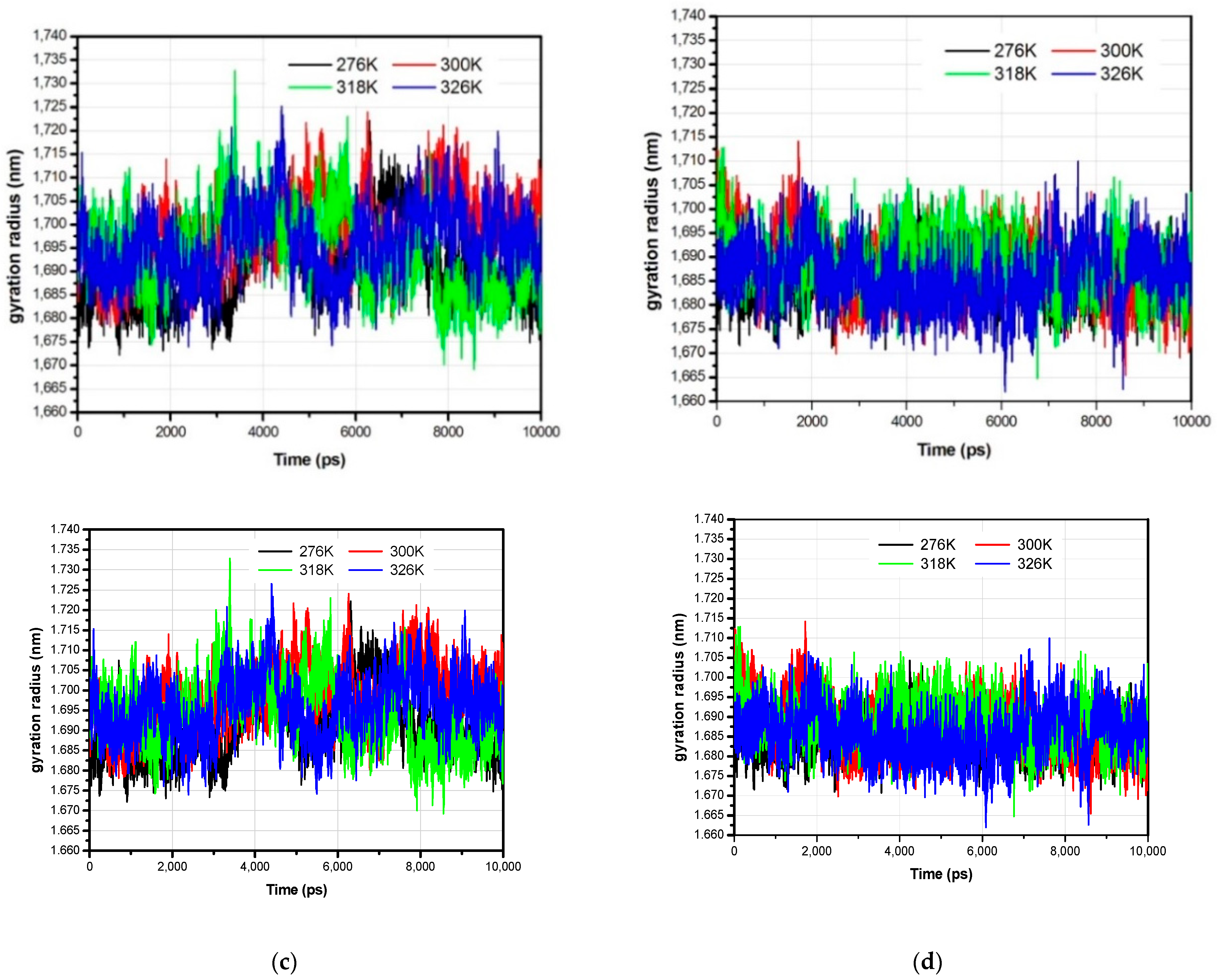
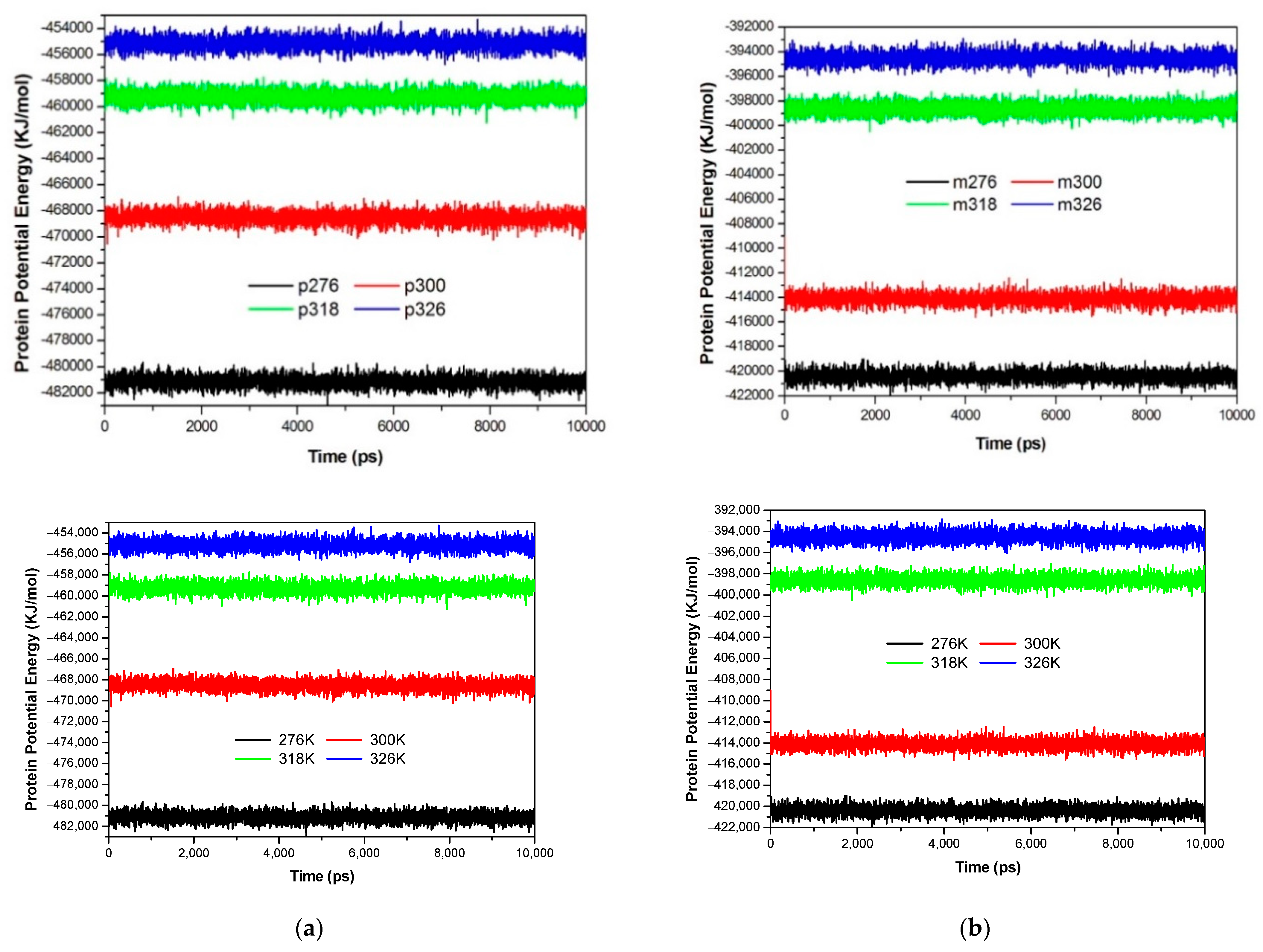
3.1. The Global Structural Stability
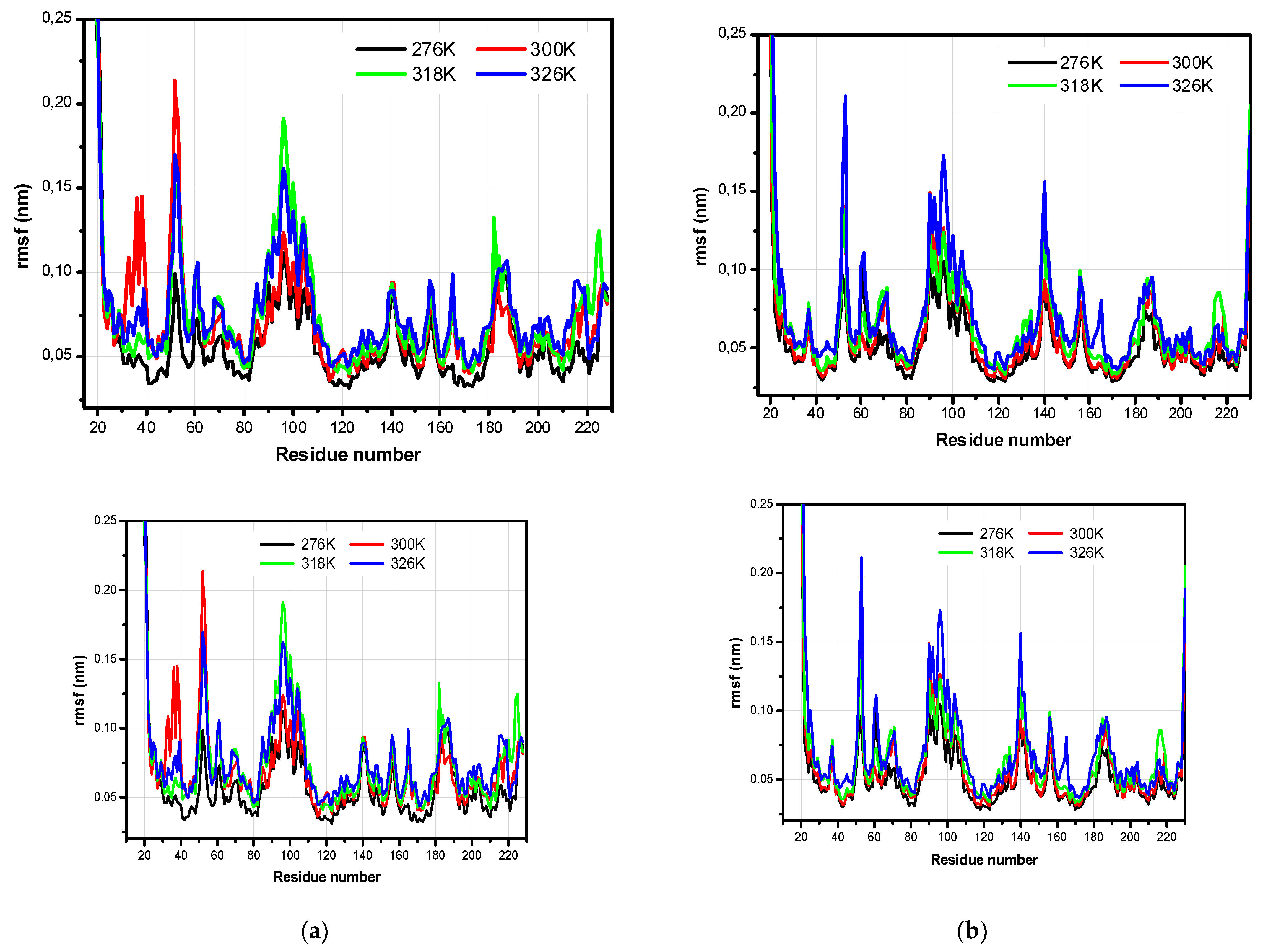
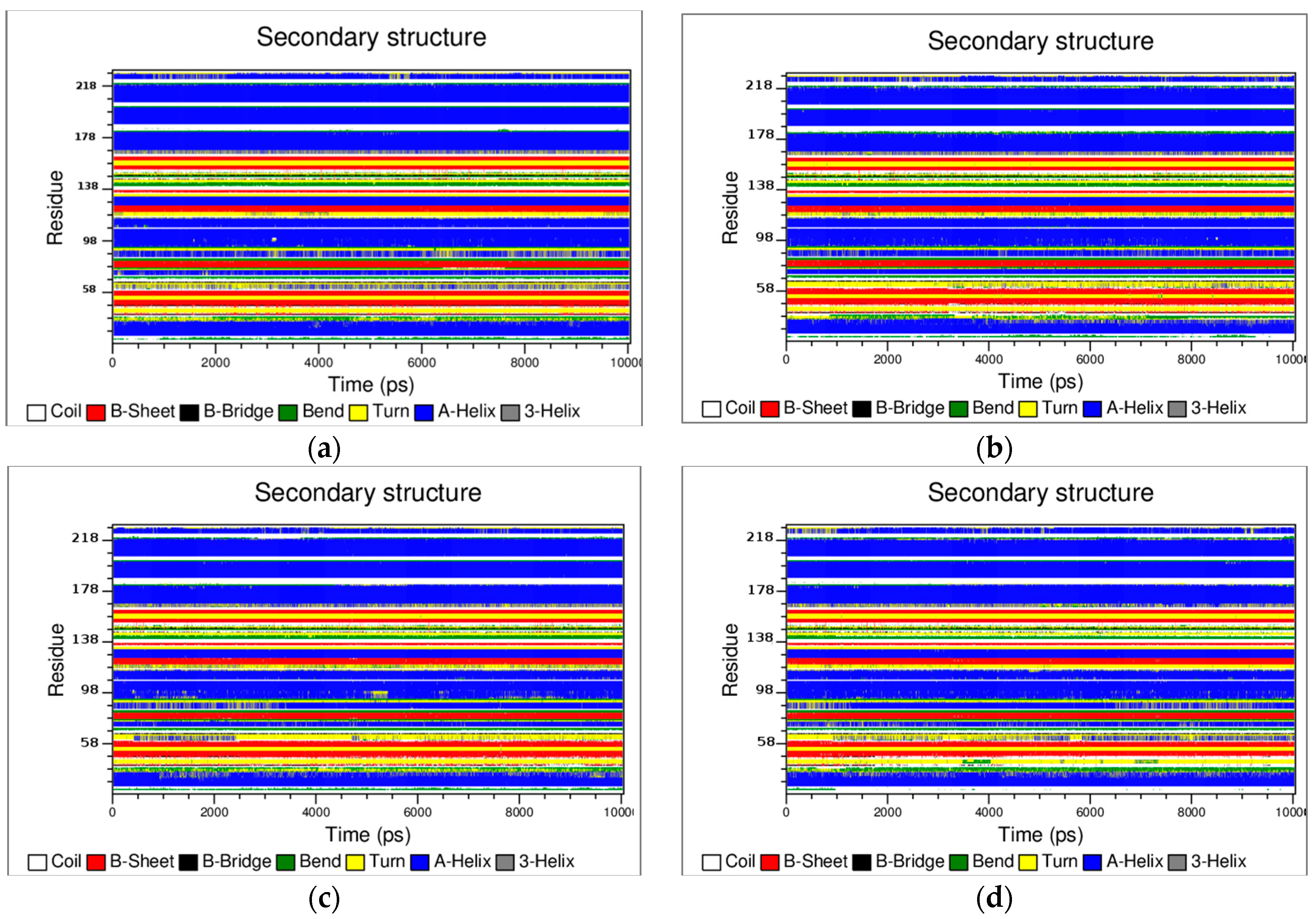
3.2. The Structural Flexibility
3.3. Hydrogen Bond Lifetime
3.3.1. The Psychrophile Enzyme VsEndA
3.3.2. The Mesophile Enzyme VcEndA
4. Conclusions
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
References
- D’Amico, S.; Collins, T.; Marx, J.C.; Feller, G.; Gerday, C. Psychrophilic microorganisms: Challenges for life. EMBO Rep. 2006, 7, 385–389. [Google Scholar] [CrossRef]
- Muga, A.; Moro, F. Thermal adaptation of heat shock proteins. Curr. Protein Pept. Sci. 2008, 9, 552–566. [Google Scholar] [CrossRef]
- Somero, G.N. Adaptation of enzymes to temperature: Searching for basic strategies. Comp. Biochem. Physiol. B 2004, 139, 321–333. [Google Scholar] [CrossRef]
- Trivedi, S.; Gehlot, H.S.; Rao, S.R. Protein thermostability in Archaea and Eubacteria. Genet. Mol. Res. 2006, 5, 816–827. [Google Scholar]
- Daniel, R.M.; Danson, M.J.; Eisenthal, R.; Lee, C.K.; Peterson, M.E. The effect of temperature on enzyme activity: New insights and their implications. Extremophiles 2008, 12, 51–59. [Google Scholar] [CrossRef]
- Schiraldi, C.; De Rosa, M. The production of biocatalysts and biomolecules from extremophiles. Trends Biotechnol. 2002, 20, 515–521. [Google Scholar] [CrossRef]
- Chiuri, R.; Maiorano, G.; Rizzello, A.; del Mercato, L.L.; Cingolani, R.; Maffia, M.; Pompa, P.P. Exploring local flexibility/rigidity in psychrophilic and mesophilic carbonicanhydrases. Biophys. J. 2009, 96, 1586–1596. [Google Scholar] [CrossRef]
- Li, W.F.; Zhou, X.X.; Lu, P. Structural features of thermozymes. Biotechnol. Adv. 2005, 23, 271–281. [Google Scholar] [CrossRef]
- Vieille, C.; Zeikus, G.J. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001, 65, 1–43. [Google Scholar] [CrossRef]
- Sterner, R.; Liebl, W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 39–106. [Google Scholar] [CrossRef]
- Del Vecchio, P.; Elias, M.; Merone, L.; Graziano, G.; Dupuy, J.; Mandrich, L.; Manco, G. Structural determinants of the high thermal stability of SsoPox from the hyperthermophilic archaeon Sulfolobus Solfataricus. Extremophiles 2009, 13, 461–470. [Google Scholar] [CrossRef]
- Feller, G. Protein stability and enzyme activity at extreme biological temperatures. J. Phys. Condens. Matter 2010, 22, 323101. [Google Scholar] [CrossRef]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef]
- Liang, Z.X.; Tsigos, I.; Lee, T.; Bouriotis, V.; Resing, K.A.; Ahn, N.G.; Klinman, J.P. Evidence for increased local flexibility in psychrophilic alcohol dehydrogenase relative to its thermophilic homologue. Biochemistry 2004, 43, 14676–14683. [Google Scholar] [CrossRef]
- Georlette, D.; Damien, B.; Blaise, V.; Depiereux, E.; Uversky, V.N.; Gerday, C.; Feller, G. Structural and functional adaptations to extreme temperatures inpsychrophilic, mesophilic, and thermophilic DNA ligases. J. Biol. Chem. 2003, 278, 37015–37023. [Google Scholar] [CrossRef]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef]
- Altermark, B.; Niiranen, L.; Willassen, N.P.; Smalas, A.O.; Moe, E. Comparative studies of endonuclease I from cold-adapted Vibrio salmonicida and mesophilic Vibrio cholerae. FEBS J. 2007, 274, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Riccardi, L.; Villa, C.; Fantucci, P.; De Gioia, L. Flexibility and enzymatic cold adaptation: A comparative molecular dynamics investigation of the elastase family. Biochim. Biophys. Acta 2006, 1764, 1397–1406. [Google Scholar] [CrossRef]
- Spiwok, V.; Lipovova, P.; Skalova, T.; Duskova, J.; Dohnalek, J.; Hasek, J.; Kralova, B. Cold-active enzymes studied by comparative molecular dynamics simulation. J. Mol. Model. 2007, 13, 485–497. [Google Scholar] [CrossRef]
- Kundu, S.; Roy, D. Comparative structural studies of psychrophilic and mesophilic proteinhomologues by molecular dynamics simulation. J. Mol. Graph. Model. 2009, 27, 871–880. [Google Scholar] [CrossRef]
- Michetti, D.; Brandsdal, B.O.; Bon, D.; Isaksen, G.V.; Tiberti, M.; Papaleo, E. A comparative study of cold- and warm-adapted Endonucleases A using sequence analyses and molecular dynamics simulations. PLoS ONE 2017, 12, e0169586. [Google Scholar] [CrossRef]
- Benrezkallah, D.; Dauchez, M.; Krallafa, A.M. Molecular dynamics of the salt dependence of a cold-adapted enzyme: Endonuclease I. J. Biomol. Struct. Dyn. 2015, 33, 2511–2521. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Karplus, M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, J.A.D.; Feig, M.; Brooks, C.L., III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Šali, A.; Blundel, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald potential. J. Chem. Phys. 1995, 103, 8577–8592. [Google Scholar] [CrossRef]
- Luzar, A.; Chandler, D. Hydrogen-bond kinetics in liquid water. Nature 1996, 397, 55–57. [Google Scholar] [CrossRef]
- Luzar, A. Resolving the hydrogen bond dynamics conundrum. J. Chem. Phys. 2000, 113, 10663–10675. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tarek, M.; Tobias, D.J. Role of Protein-Water Hydrogen Bond Dynamics in the Protein Dynamical Transition. Phys. Rev. Lett. 2002, 88, 138101. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | 276 K | 300 K | 318 K | 326 K |
|---|---|---|---|---|
| Tyr38_BB_Wat | 24.504 | 4.664 | −66.291 | 5.167 |
| Tyr38_SC_Wat | 5.136 | 4.480 | 2.312 | 2.503 |
| Tyr38_BB_P | 87.630 | 357.496 | 382.376 | 42.699 |
| Tyr38_SC_P | none | 38.040 | none | none |
| Asn52_BB_Wat | 3.938 | 2.636 | 8.453 | 2.644 |
| Asn52_SC_Wat | 2.568 | 1.832 | 1.300 | 0.989 |
| Asn52_BB_P | none | 97.19 | none | none |
| Asn52_SC_P | none | 130.661 | 9.132 | −93.1 |
| Lys96_BB_Wat | 1.675 | 1.488 | 1.046 | 0.918 |
| Lys96_SC_Wat | 5.849 | 3.092 | 2.069 | 1.665 |
| Lys96_BB_P | 50.979 | 59.337 | 90.849 | 19.532 |
| Lys96_SC_P | 31.262 | 19.126 | 17.477 | 21.534 |
| Lys182_BB_Wat | 13.875 | 121.496 | −16.277 | 2.157 |
| Lys182_SC_Wat | 4.317 | 2.903 | 1.743 | 1.677 |
| Lys182_BB_P | 199.917 | 226.472 | 79.354 | 37.415 |
| Lys182_SC_P | −11.894 | 15.507 | 797.646 | 76.648 |
| Tyr225_BB_Wat | 20.737 | 7.027 | 3.549 | 3.436 |
| Tyr225_SC_Wat | 2.590 | 1.851 | 1.542 | 1.368 |
| Tyr225_BB_P | 854.846 | 401.057 | 775.720 | 159.523 |
| Tyr225_SC_P | 198.984 | 103.911 | 510.568 | 2.001 |
| Temperature | 276 K | 300 K | 318 K | 326 K |
|---|---|---|---|---|
| Lys53_BB_Wat | 7.786 | 16.241 | 7.691 | 5.408 |
| Lys53_SC_Wat | 4.362 | 3.471 | 1.906 | 1.878 |
| Lys53_BB_P | none | −2748.665 | 37.428 | 25.194 |
| Lys53_SC_P | 79.268 | 101.087 | 80.117 | 96.125 |
| Gln96_BB_Wat | 2.043 | 1.637 | 0.936 | 0.937 |
| Gln96_SC_Wat | 1.846 | 1.293 | 0.821 | 0.559 |
| Gln96_BB_P | 144.256 | 504.247 | 30.880 | 96.679 |
| Gln96_SC_P | 143.457 | 71.147 | 37.072 | 22.270 |
| Gly140_Wat | 8.630 | 9.920 | 3.771 | 1.400 |
| Gly140_P | none | None | −93.416 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benrezkallah, D.; Sediki, H.; Krallafa, A.M. The Structural Flexibility of Cold- and Warm-Adapted Enzymes (Endonucleases I) by Molecular Dynamics Simulation. Chem. Proc. 2021, 3, 128. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08387
Benrezkallah D, Sediki H, Krallafa AM. The Structural Flexibility of Cold- and Warm-Adapted Enzymes (Endonucleases I) by Molecular Dynamics Simulation. Chemistry Proceedings. 2021; 3(1):128. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08387
Chicago/Turabian StyleBenrezkallah, Djamila, Hayat Sediki, and Abdelghani Mohammed Krallafa. 2021. "The Structural Flexibility of Cold- and Warm-Adapted Enzymes (Endonucleases I) by Molecular Dynamics Simulation" Chemistry Proceedings 3, no. 1: 128. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08387