
ACT Up TIL Now: The Evolution of Tumor-Infiltrating Lymphocytes in Adoptive Cell Therapy for the Treatment of Solid Tumors



Abstract
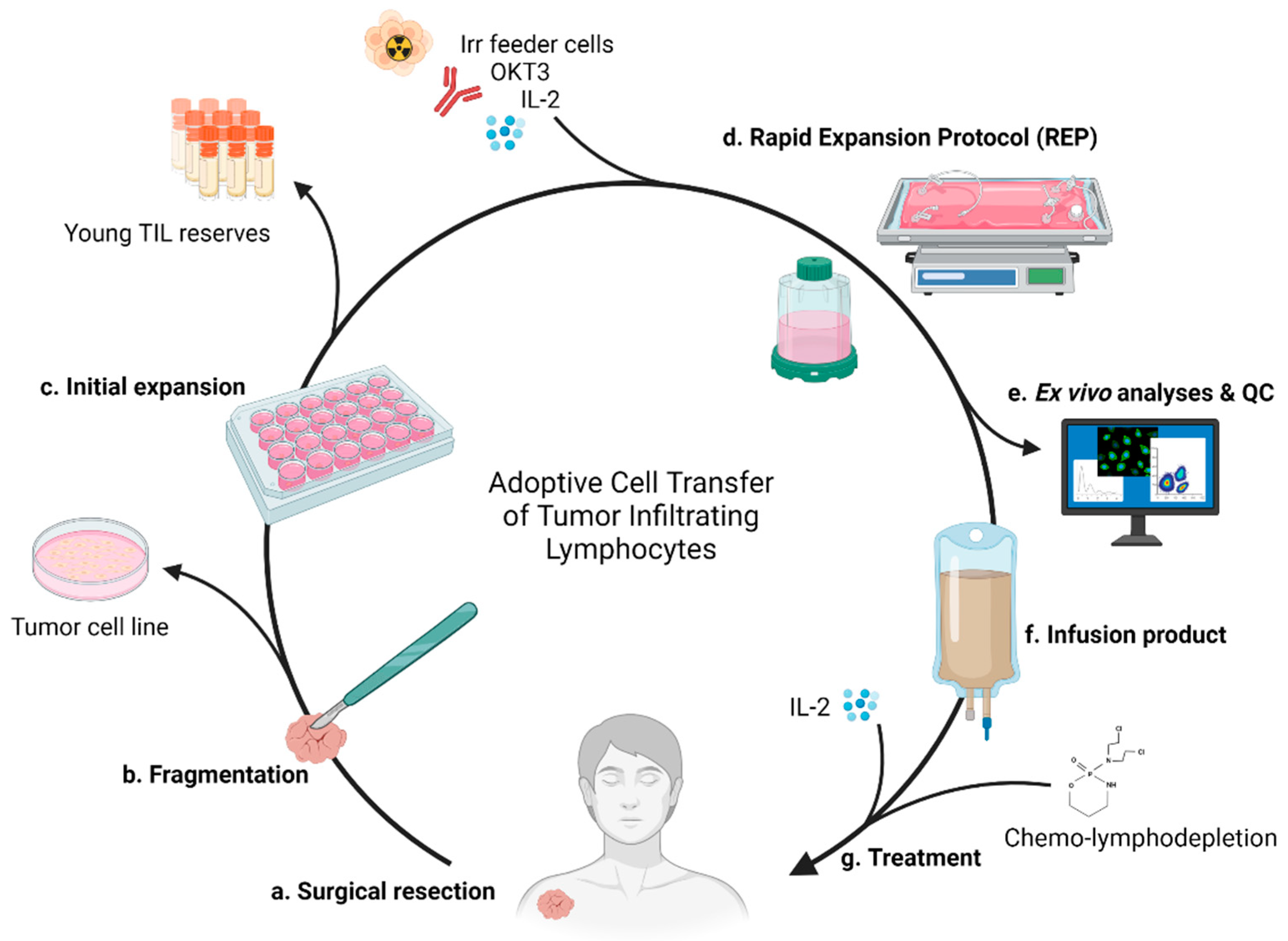
:1. Introduction
2. History
2.1. Early Experiments
2.2. Optimizing Methods: The Rapid Expansion Protocol
2.3. Discovering Correlates of Response and Optimizations to Therapy
2.4. Current Standard of Care and Therapeutic Process
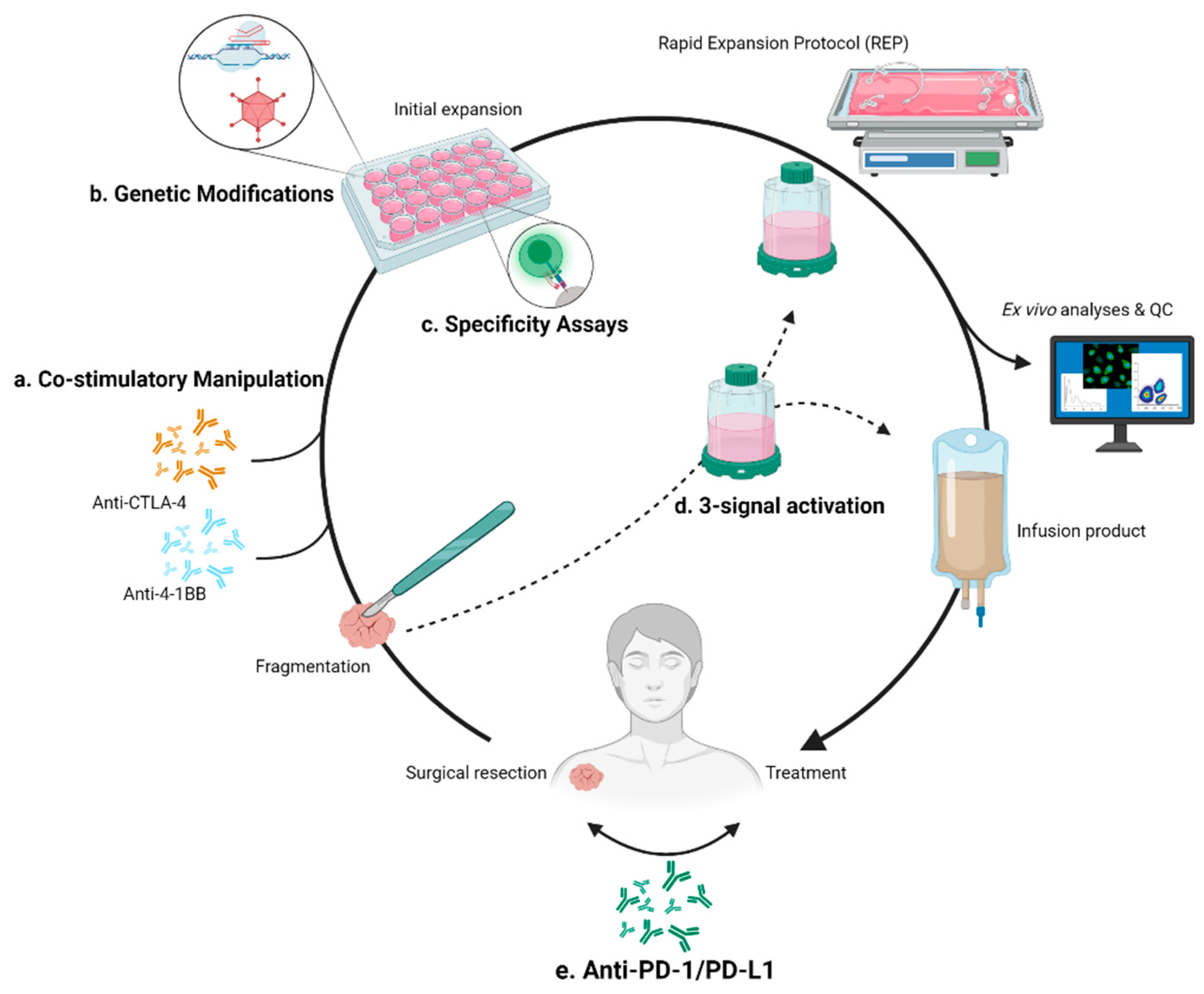
3. Treatment Complications and Resolutions
3.1. Surgery
3.2. Expansion Complications
3.2.1. Expansion Resolutions Targeting 4-1BB
3.2.2. Expansion Resolutions Targeting CTLA-4
3.3. Severe Adverse Events
3.4. The Effect of CPI-Resistance on TIL Therapy
4. TIL-Based ACT in Non-Melanoma Solid Tumors
4.1. Checkpoint Inhibition Combination
4.2. Specific Targeting
5. Genetic Modification of TILs
5.1. IL-2
5.2. IL-12
5.3. CXCR2
5.4. Gene Knockouts
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, W. The Checkpoint Immunotherapy Revolution: What Started as a Trickle Has Become a Flood, despite Some Daunting Adverse Effects, New Drugs, Indications, and Combinations Continue to Emerge. Pharm. Ther. 2016, 41, 185–191. [Google Scholar]
- Haslam, A.; Prasad, V. Estimation of the Percentage of Us Patients with Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. Available online: http://0-www-Jstor-Org.brum.beds.ac.uk/Stable/2890840 (accessed on 16 March 2021). [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of Adoptive T Cell Therapy in Cancer. Semin. Immunopathol. 2019, 41, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Maus, M.V. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically Engineered T Cells for Cancer Immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Fu, Q.; Chen, N.; Ge, C.; Li, R.; Li, Z.; Zeng, B.; Li, C.; Wang, Y.; Xue, Y.; Song, X.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Melanoma: A Systematic Review and Meta-Analysis. Oncoimmunology 2019, 8, e1593806. [Google Scholar] [CrossRef] [Green Version]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 3360. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- MacCarty, W.C. Factors Which Influence Longevity in Cancer. A Study of 293 Cases. Ann. Surg. 1922, 76, 9–12. [Google Scholar] [PubMed]
- Elston, C.W.; Bagshawe, K.D. Cellular Reaction in Trophoblastic Tumours. Br. J. Cancer 1973, 28, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, M.; Angelini, L.; Bertolotti, A.; Colizza, S. Host Resistance in Relation to Survival in Breast Cancer. Br. Med. J. 1974, 4, 268–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mule, J.J.; Shu, S.; Schwarz, S.L.; Rosenberg, S.A. Adoptive Immunotherapy of Established Pulmonary Metastases with LAK Cells and Recombinant Interleukin-2. Science 1984, 225, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.; Mochizuki, D.; Gillis, S. T-Cell Growth Factors: Interleukin 2. Immunol. Today 1980, 1, 113–117. [Google Scholar] [CrossRef]
- Ruscetti, F.W.; Gallo, R.C. Human T-Lymphocyte Growth Factor: Regulation of Growth and Function of T-Lymphocytes. Blood 1981, 57, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, B.E.A.; Viazumder, A.; Zhang, H.U.A.Z.; Rosenberg, S.A. Lymphokine-Activated Killer Cell Phenomenon, Lysis of Natural Killer-Resistant Fresh Solid Tumor Cells by Interleukin 2-Activated Autologous Human Peripheral Blood Lymphocytes. J. Exp. Med. 1982, 155, 1823–1841. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Grimm, E.A.; Mcgrogan, M.; Doyle, M.; Kawasaki, E.; Koths, K.; Mark, D.F. Biological Activity of Recombinant Human Interleukin-2 Produced in Escherichia Coli. Science 1984, 223, 1412–1415. [Google Scholar] [CrossRef]
- Lotze, M.T.; Grimm, E.A.; Mazumder, A.; Strausser, J.L.; Rosenberg, S.A. Lysis of Fresh and Cultured Autologous Tumor by Human Lymphocytes Cultured in T-Cell Growth Factor. Cancer Res. 1981, 41, 4420–4425. [Google Scholar]
- Lafreniere, R.; Rosenstein, M.S.; Rosenberg, S.A. Optimal Methods for Generating Expanded Lymphokine Activated Killer Cells Capable of Reducing Established Murine Tumors in vivo. J. Immunol. Methods 1986, 94, 37–49. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T.; et al. Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients with Metastatic Cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Muul, L.M.; Solomon, D.; Rosenberg, S.A. Expansion of Human Tumor Infiltrating Lymphocytes for Use in Immunotherapy Trials. J. Immunol. Methods 1987, 102, 127–141. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.J.; Schwarz, S. In Vitro Growth of Murine T Cells. V. The Isolation and Growth of Lymphoid Cells Infiltrating Syngeneic Solid Tumors. J. Immunol. 1979, 51, 1852–1854. [Google Scholar]
- Muul, L.M.; Spiess, P.J.; Director, E.P.; Rosenberg, S.A. Identification of Specific Cytolytic Immune Responses against Autologous Tumor in Humans Bearing Malignant Melanoma. J. Immunol. 1987, 138, 989–995. [Google Scholar]
- Kradin, R.L.; Boyle, L.A.; Preffer, F.I.; Callahan, R.J.; Barlai-Kovach, M.; Strauss, H.W.; Dubinett, S.; Kurnick, J.T. Tumor-Derived Interleukin-2-Dependent Lymphocytes in Adoptive Immunotherapy of Lung Cancer. Cancer Immunol. Immunother. 1987, 24, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Solomon, D.; Avis, F.P.; Chang, A.E.; Freerksen, D.L.; Linehan, W.M.; Lotze, M.T.; Robertson, C.N.; Seipp, C.A.; Simon, P.; et al. Immunotherapy of Patients with Advanced Cancer Using Tumor-Infiltrating Lymphocytes and Recombinant Interleukin-2: A Pilot Study. J. Clin. Oncol. 1988, 6, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. NEJM 1988, 299, 230–234. [Google Scholar] [CrossRef]
- Cheever, M.A.; Thompson, J.A.; Kern, D.E.; Greenberg, P.D. Interleukin-2 Administered In Vivo Induces the Growth and Augments the Function of Cultured T Cells in vivo. J. Biol. Response Mod. 1984, 3, 462–467. [Google Scholar]
- Marabondo, S.; Kaufman, H.L. High-Dose Interleukin-2 (IL-2) for the Treatment of Melanoma: Safety Considerations and Future Directions. Expert Opin. Drug Saf. 2017, 16, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Riddell, S.R.; Greenberg, P.D. High Efficient Transduction of T Lymphocytes Using Rapid Expansion Methods (“REM”). U.S. Patent 6,040,177, 21 March 2000. [Google Scholar]
- Flyer, D.C.; Clary, K.W. Modified Rapid Expansion Methods (“Modified-REM”) for In Vitro Propagation of T Lymphocytes. U.S. Patent 6,316,257 B1, 13 November 2001. [Google Scholar]
- Clevers, H.; Alarcon, B.; Wileman, T.; Terhorst, C. The T Cell Receptor/CD3 Complex: A Dynamic Protein Ensemble. Annu. Rev. Immunol. 1988, 6, 629–662. [Google Scholar] [CrossRef] [PubMed]
- Londei, M.; Grubeck-Loebenstein, B.; De Berardinis, P.; Greenall, C.; Feldmann, M. Efficient Propagation and Cloning of Human T Cells in the Absence of Antigen by Means of OKT3, Interleukin 2, and Antigen-Presenting Cells. Scand. J. Immunol. 1988, 27, 35–46. [Google Scholar] [CrossRef]
- Riddell, S.R.; Greenberg, P.D. The Use of Anti-CD3 and Anti-CD28 Monoclonal Antibodies to Clone and Expand Human Antigen-Specific T Cells. J. Immunol. Methods 1990, 128, 189–201. [Google Scholar] [CrossRef]
- Minguet, S.; Swamy, M.; Alarcón, B.; Luescher, I.F.; Schamel, W.W.A. Full Activation of the T Cell Receptor Requires Both Clustering and Conformational Changes at CD3. Immunity 2007, 26, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Crossland, K.D.; Lee, V.K.; Chen, W.; Riddell, S.R.; Greenberg, P.D.; Cheever, M.A. T Cells from Tumor-Immune Mice Nonspecifically Expanded in Vitro with Anti-CD3 plus IL-2 Retain Specific Function in Vitro and Can Eradicate Disseminated Leukemia in Vivo. J. Immunol. 1991, 146, 4414–4420. [Google Scholar] [PubMed]
- Gerber, M.; Guichard, M.; Pioch, Y.; Dubois, J.B. The Influence of Interleukin-2, Feeder Cells, and Timing of Irradiation on the Radiosensitivity of Human T Lymphocytes Assessed by the Colony-Forming Assay. Radiat. Res. 1989, 120, 164–176. [Google Scholar] [CrossRef]
- Cerottini, J.; Maria, A.N.I.; Mingari, C. Direct Demonstration of the Clonogenic Potential of Every Human Peripheral Blood T Cell. J. Exp. Med. 1983, 157, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, J.M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of Viral Immunity in Immunodeficient Humans by the Adoptive Transfer of T Cell Clones. Science 1992, 257, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Llames, S.; García-Pérez, E.; Meana, Á.; Larcher, F.; Del Río, M. Feeder Layer Cell Actions and Applications. Tissue Eng. Part B Rev. 2015, 21, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Aebersold, P.; Hyatt, C.; Johnson, S.; Hines, K.; Korcak, L.; Sanders, M.; Lotz, M.; Topalian, S.; Yang, J.; Rosenberg, S.A. Lysis of Autologous Melanoma Cells by Tumor Infiltrating Lymphocytes: Association with Clinical Response. J. Natl. Cancer Inst. 1991, 83, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Hom, S.S.; Dadmarz, R.; White, D.E.; Yannelli, J.R.; Steinberg, S.M.; Rosenberg, S.A.; Topalian, S.L. In Vitro Predictors of Therapeutic Response in Melanoma Patients Receiving Tumor-Infiltrating Lymphocytes and Interleukin-2. J. Clin. Oncol. 1994. [Google Scholar] [CrossRef]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally Cultured Tumor-Infiltrating Lymphocytes Display Optimal Characteristics for Adoptive Cell Therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Schallmach, E.; Kubi, A.; Shalmon, B.; Hardan, I.; Catane, R.; et al. Minimally Cultured or Selected Autologous Tumor-Infiltrating Lymphocytes after a Lympho-Depleting Chemotherapy Regimen in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and Large-Scale Expansion of Minimally Cultured Young Tumor Infiltrating Lymphocytes for Adoptive Transfer Therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shen, X.; Huang, J.; Hodes, R.J.; Rosenberg, S.A.; Robbins, P.F. Telomere Length of Transferred Lymphocytes Correlates with In Vivo Persistence and Tumor Regression in Melanoma Patients Receiving Cell Transfer Therapy. J. Immunol. 2005, 175, 7046–7052. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H. Structure and Function of Telomeres. Nature 1991, 354, 737–740. [Google Scholar] [CrossRef]
- Hodes, R.J.; Hathcock, K.S.; Weng, N.P. Telomeres in T and B Cells. Nat. Rev. Immunol. 2002, 2, 699–706. [Google Scholar] [CrossRef]
- Dudley, M.E.; Gross, C.A.; Somerville, R.P.T.; Hong, Y.; Schaub, N.P.; Rosati, S.F.; White, D.E.; Nathan, D.; Restifo, N.P.; Steinberg, S.M.; et al. Randomized Selection Design Trial Evaluating CD8+-Enriched versus Unselected Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy for Patients with Melanoma. J. Clin. Oncol. 2013, 31, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E.; et al. CD8+ Enriched “Young” Tumor Infiltrating Lymphocytes Can Mediate Regression of Metastatic Melanoma. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donia, M.; Junker, N.; Ellebaek, E.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Characterization and Comparison of “standard” and “Young” Tumour-Infiltrating Lymphocytes for Adoptive Cell Therapy at a Danish Translational Research Institution. Scand. J. Immunol. 2012, 75, 157–167. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical Responses in a Phase II Study Using Adoptive Transfer of Short-Term Cultured Tumor Infiltration Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific Lymphocyte Subsets Predict Response to Adoptive Cell Therapy Using Expanded Autologous Tumor-Infiltrating Lymphocytes in Metastatic Melanoma Patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes in Patients with Metastatic Ovarian Cancer: A Pilot Study. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A Phase I Study of Nonmyeloablative Chemotherapy and Adoptive Transfer of Autologous Tumor Antigen-Specific T Lymphocytes in Patients with Metastatic Melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, M.E.; Wunderlich, J.; Nishimura, M.I.; Yu, D.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Sherry, R.; et al. Adoptive Transfer of Cloned Melanoma-Reactive T Lymphocytes for the Treatment of Patients with Metastatic Melanoma. J. Immunother. 2001, 24, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T Cell Therapy Using Antigen-Specific CD8+ T Cell Clones for the Treatment of Patients with Metastatic Melanoma: In Vivo Persistence, Migration, and Antitumor Effect of Transferred T Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.-C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T Cell Immunity Against a Tumor/Self-Antigen Is Augmented by CD4+ T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Lowery, F.J.; Copeland, A.R.; Bahadiroglu, E.; Mukherjee, R.; Jia, L.; Anibal, J.T.; Sachs, A.; Adebola, S.O.; Gurusamy, D.; et al. Stem-like CD8 T Cells Mediate Response of Adoptive Cell Immunotherapy Against Human Cancer. Science 2020, 370, 1328–1334. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.P.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive Cell Therapy for Patients with Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive Cell Transfer: A Clinical Path to Effective Cancer Immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients with Refractory Metastatic Melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients after Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Larsen, S.M.; Met, Ö.; Svane, I.M. Simplified Protocol for Clinical-Grade Tumor-Infiltrating Lymphocyte Manufacturing with Use of the Wave Bioreactor. Cytotherapy 2014, 16, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.F.; Brenner, L.J.; Gerdemann, U.; Ngo, M.C.; Sili, U.; Liu, H.; Wilson, J.; Dotti, G.; Heslop, H.E.; Leen, A.M.; et al. Accelerated Production of Antigen-Specific T Cells for Preclinical and Clinical Applications Using Gas-Permeable Rapid Expansion Cultureware (G-Rex). J. Immunother. 2010, 33, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Hollyman, D.; Stefanski, J.; Przybylowski, M.; Bartido, S.; Borquez-Ojeda, O.; Taylor, C.; Yeh, R.; Capacio, V.; Olszewska, M.; Hosey, J.; et al. Manufacturing Validation of Biologically Functional T Cells Targeted to CD19 Antigen for Autologous Adoptive Cell Therapy. J. Immunother. 2009, 32, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Forget, M.A.; Malu, S.; Liu, H.; Toth, C.; Maiti, S.; Kale, C.; Haymaker, C.; Bernatchez, C.; Huls, H.; Wang, E.; et al. Activation and Propagation of Tumor-Infiltrating Lymphocytes on Clinical-Grade Designer Artificial Antigen-Presenting Cells for Adoptive Immunotherapy of Melanoma. J. Immunother. 2014, 37, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Ullenhag, G.J.; Sadeghi, A.M.; Carlsson, B.; Ahlström, H.; Mosavi, F.; Wagenius, G.; Tötterman, T.H. Adoptive T-Cell Therapy for Malignant Melanoma Patients with TILs Obtained by Ultrasound-Guided Needle Biopsy. Cancer Immunol. Immunother. 2012, 61, 725–732. [Google Scholar] [CrossRef]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 Are Members of a Tumor Necrosis Factor (TNF)-Nerve Growth Factor Receptor Subfamily That Bind TNF Receptor-Associated Factors and Activate Nuclear Factor ΚB. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J. CD137 Accurately Identifies and Enriches for Naturally Occurring Tumor-Reactive T Cells in Tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacon, J.A.; Sarnaik, A.A.; Chen, J.Q.; Creasy, C.; Kale, C.; Robinson, J.; Weber, J.; Hwu, P.; Pilon-Thomas, S.; Radvanyi, L. Manipulating the Tumor Microenvironment Ex Vivo for Enhanced Expansion of Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy. Clin. Cancer Res. 2015, 21, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harao, M.; Forget, M.A.; Roszik, J.; Gao, H.; Babiera, G.V.; Krishnamurthy, S.; Chacon, J.A.; Li, S.; Mittendorf, E.A.; Desnyder, S.M.; et al. 4-1BB-Enhanced Expansion of CD8 TIL from Triple-Negative Breast Cancer Unveils Mutation-Specific CD8 T Cells. Cancer Immunol. Res. 2017, 5, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavera, R.J.; Forget, M.A.; Kim, Y.U.; Sakellariou-Thompson, D.; Creasy, C.A.; Bhatta, A.; Fulbright, O.J.; Ramachandran, R.; Thorsen, S.T.; Flores, E.; et al. Utilizing T-Cell Activation Signals 1, 2, and 3 for Tumor-Infiltrating Lymphocytes (TIL) Expansion: The Advantage over the Sole Use of Interleukin-2 in Cutaneous and Uveal Melanoma. J. Immunother. 2018, 41, 399–405. [Google Scholar] [CrossRef]
- Nielsen, M.; Krarup-Hansen, A.; Hovgaard, D.; Petersen, M.M.; Loya, A.C.; Westergaard, M.C.W.; Svane, I.M.; Junker, N. In Vitro 4-1BB Stimulation Promotes Expansion of CD8+ Tumor-Infiltrating Lymphocytes from Various Sarcoma Subtypes. Cancer Immunol. Immunother. 2020, 69, 2179–2191. [Google Scholar] [CrossRef]
- Shin, S.M.; Kim, Y.H.; Choi, B.K.; Kwon, P.M.; Lee, H.-W.; Kwon, B.S. 4-1BB Triggers IL-13 Production from T Cells to Limit the Polarized, Th1-Mediated Inflammation. J. Leukoc. Biol. 2007, 81, 1455–1465. [Google Scholar] [CrossRef]
- Elpek, K.G.; Yolcu, E.S.; Franke, D.D.H.; Lacelle, C.; Schabowsky, R.-H.; Shirwan, H. Ex Vivo Expansion of CD4 + CD25 + FoxP3 + T Regulatory Cells Based on Synergy between IL-2 and 4-1BB Signaling. J. Immunol. 2007, 179, 7295–7304. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory Monoclonal Antibodies for Cancer Therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A Molecular Perspective of CTLA-4 Function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef] [Green Version]
- Friese, C.; Harbst, K.; Borch, T.H.; Westergaard, M.C.W.; Pedersen, M.; Kverneland, A.; Jönsson, G.; Donia, M.; Svane, I.M.; Met, Ö. CTLA-4 Blockade Boosts the Expansion of Tumor-Reactive CD8+ Tumor-Infiltrating Lymphocytes in Ovarian Cancer. Sci. Rep. 2020, 10, 3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated Il2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C. Toxicities Associated with Adoptive T-Cell Transfer for Cancer. Cancer J. 2015, 21, 506–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma: Intent-to-Treat Analysis and Efficacy after Failure to Prior Immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [Green Version]
- Andersen, R.; Borch, T.H.; Draghi, A.; Gokuldass, A.; Rana, A.H.M.; Pedersen, M.; Nielsen, M.; Kongsted, P.; Kjeldsen, J.W.; Westergaard, C.W.M.; et al. T Cells Isolated from Patients with Checkpoint Inhibitor-Resistant Melanoma Are Functional and Can Mediate Tumor Regression. Ann. Oncol. 2018, 29, 1575–1581. [Google Scholar] [CrossRef]
- Borch, T.H.; Andersen, R.; Ellebaek, E.; Met, Ö.; Donia, M.; Marie Svane, I. Future Role for Adoptive T-Cell Therapy in Checkpoint Inhibitor-Resistant Metastatic Melanoma. J. Immunother. Cancer 2020, 8, e000668. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Hong, J.J.; Rosenberg, S.A.; Dudley, M.E.; Yang, J.C.; White, D.E.; Butman, J.A.; Sherry, R.M. Successful Treatment of Melanoma Brain Metastases with Adoptive Cell Therapy. Clin. Cancer Res. 2010, 16, 4892–4898. [Google Scholar] [CrossRef] [Green Version]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Establishment of Adoptive Cell Therapy with Tumor Infiltrating Lymphocytes for Non-small Cell Lung Cancer Patients. Cancer Immunol. Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef]
- Ma, Y.; Ou, J.; Lin, T.; Chen, L.; Wang, J.; Qiao, D.; Lai, S.; Duan, C.; Chen, Y.; Chang, R.; et al. Phenotypic Analysis of Tumor-infiltrating Lymphocytes from non-small Cell Lung Cancer and Their Potential Application for Adoptive Cell Therapy. Immunopharmacol. Immunotoxicol. 2020, 42, 319–329. [Google Scholar] [CrossRef]
- Jiang, S.S.; Tang, Y.; Zhang, Y.; Weng, D.; Zhou, Z.; Pan, K.; Pan, Q.; Wang, Q.; Liu, Q.; He, J.; et al. A Phase I Clinical Trial Utilizing Autologous Tumor-infiltrating Lymphocytes in Patients with Primary Hepatocellular Carcinoma. Oncotarget 2015, 6, 41339–41349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of Tumor-infiltrating Lymphocytes (TIL) from Human Pancreatic Tumors. J. Immunother. Cancer. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating Lymphocytes (TILs) from Patients with Glioma. Oncoimmunology 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodmer, S.; Strommer, K.; Frei, K.; Siepl, C.; Tribolet, N.; Heid, I.; Fontana, A. Immunosupression and Transforming Growth Factor-beta in Glioblastoma. Preferential Production of Transforming Growth Factor-beta 2. J. Immunol. 1989, 143, 3222–3229. [Google Scholar] [PubMed]
- Quattrocchi, K.B.; Miller, C.H.; Cush, S.; Bernard, S.A.; Dull, S.T.; Smith, M.; Gudeman, S.; Varia, M.A. Pilot Study of Local Autologous Tumor Infiltrating Lymphocytes for the Treatment of Recurrent Malignant Gliomas. J. Neurooncol. 1999, 45, 141–157. [Google Scholar] [CrossRef]
- Westergaard, M.C.W.; Andersen, R.; Chong, C.; Kjeldsen, J.W.; Pedersen, M.; Friese, C.; Hasselager, T.; Lajer, H.; Coukos, G.; Bassani-Sternberg, M.; et al. Tumour-Reactive T Cell Subsets in the Microenvironment of Ovarian Cancer. Br. J. Cancer 2019, 120, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ Tumor-Infiltrating Lymphocytes and a High CD8+/Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Kverneland, A.H.; Pedersen, M.; Wulff Westergaard, M.C.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive Cell Therapy in Combination with Checkpoint Inhibitors in Ovarian Cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Heeke, C.; Kristensen, N.P.; Tvingsholm, S.A.; Borch, A.; Draghi, A.; Crowther, M.D.; Carri, I.; Munk, K.K.; Holm, J.S.; Bjerregaard, A.; et al. Engraftment of Neoantigen-Reactive CD8+ T Cells Affects the Clinical Outcome in Patients Receiving Adoptive Transfer of Tumor-Infiltrating Lymphocytes. J. Clin. Invest. 2021. In submission. [Google Scholar]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.; Gros, A.; Turcotte, S.; Paul, F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Hansen, U.K.; Ramskov, S.; Bjerregaard, A.M.; Borch, A.; Andersen, R.; Draghi, A.; Donia, M.; Bentzen, A.K.; Marquard, A.M.; Szallasi, Z.; et al. Tumor-Infiltrating T Cells From Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived From Point and Frameshift Mutations. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Kast, F.; Klein, C.; Umaña, P.; Gros, A.; Gasser, S. Advances in Identification and Selection of Personalized Neoantigen/T-Cell Pairs for Autologous Adoptive T Cell Therapies. Oncoimmunology 2021, 10, 1869389. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.V.; Leonard, M.T.; Jeppson, J.D.; Swift, M.; Li, G.; Wong, S.; Peng, S.; Zaretsky, J.M.; Heath, J.R.; Ribas, A.; et al. T Cell Antigen Discovery via Signaling and Antigen-Presenting Bifunctional Receptors. Nat. Methods 2019, 16, 191–198. [Google Scholar] [CrossRef]
- Kisielow, J.; Obermair, F.J.; Kopf, M. Deciphering CD4+ T Cell Specificity Using Novel MHC–TCR Chimeric Receptors. Nat. Immunol. 2019, 20, 652–662. [Google Scholar] [CrossRef]
- Rao, A.A.; Madejska, A.A.; Pfeil, J.; Paten, B.; Salama, S.R.; Haussler, D. ProTECT—Prediction of T-Cell Epitopes for Cancer Therapy. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Yannelli, J.; Kriegler, M.; Anderson, W.F.; Perez, C.; Chiang, Y.; Schwarz, S.; Cowherd, R.; Delgado, C.; Mule, J.; et al. Functional and Molecular Characterization of Tumor-Infiltrating Lymphocytes Transduced with Tumor Necrosis Factor-α CDNA for the Gene Therapy of Cancer in Humans. J. Immunol. 1993, 150, 4104–4115. [Google Scholar] [PubMed]
- Hwu, P.; Rosenberg, S.A. The Use of Gene-Modified Tumor-Infiltrating Lymphocytes for Cancer Therapy. Ann. N. Y. Acad. Sci. 1994, 716, 188–203. [Google Scholar] [CrossRef]
- Weinstein-Marom, H.; Gross, G.; Levi, M.; Brayer, H.; Schachter, J.; Itzhaki, O.; Besser, M.J. Genetic Modification of Tumor-Infiltrating Lymphocytes via Retroviral Transduction. Front. Immunol. 2020, 11, 584148. [Google Scholar] [CrossRef]
- Heemskerk, B.; Liu, K.E.; Dudley, M.E.; Johnson, L.A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Robbins, P.F.; Morgan, R.A.; Rosenberg, S.A. Adoptive Cell Therapy for Patients with Melanoma, Using Tumor- Infiltrating Lymphocytes Genetically Engineered to Secrete Interleukin-2. Hum. Gene Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Kuchroo, V.K. IL-12 Family Cytokines: Immunological Playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunda, B.M.J.; Luistro, L.; Warrier, R.R.; Wright, R.B.; Hubbard, B.R.; Murphy, M.; Wolf, S.F.; Gately, M.K. Antitumor and Antimetastatic Activity of Interleukin 12 against Murine Tumors. J. Exp. Med. 1993, 178, 1223–1230. [Google Scholar] [CrossRef]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12 Associated Toxicity and Interferon-γ Production. Blood 1997, 90, 2541–2548. [Google Scholar] [CrossRef]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-Infiltrating Lymphocytes Genetically Engineered with an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Haghnegahdar, H.; Du, J.; Wang, D.Z.; Strieter, R.M.; Burdick, M.D.; Nanney, L.B.; Cardwell, N.; Luan, J.; Shattuck-Brandt, R.; Richmond, A. The Tumorigenic and Angiogenic Effects of MGSA/GRO Proteins in Melanoma. J. Leukoc. Biol. 2000, 67, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kruizinga, R.C.; Bestebroer, J.; Berghuis, P.; de Haas, C.J.C.; Links, T.P.; de Vries, E.G.E.; Walenkamp, A.M.E. Role of Chemokines and Their Receptors in Cancer. Curr. Pharm. Des. 2009, 15, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting Migration of T Cells to Chemokine Secreted from Tumors by Genetic Modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef]
- Peng, W.; Ye, Y.; Rabinovich, B.A.; Liu, C.; Lou, Y.; Zhang, M.; Whittington, M.; Yang, Y.; Overwijk, W.W.; Lizée, G.; et al. Transduction of Tumor-Specific T Cells with CXCR2 Chemokine Receptor Improves Migration to Tumor and Antitumor Immune Responses. Clin. Cancer Res. 2010, 16, 5458–5468. [Google Scholar] [CrossRef] [Green Version]
- Idorn, M.; Straten, P.T.; Svane, I.M.; Met, Ö. Transfection of Tumor-Infiltrating T Cells with MRNA Encoding CXCR2. Methods Mol. Biol. 2016, 1428, 261–276. [Google Scholar] [CrossRef]
- Forget, M.A.; Tavera, R.J.; Haymaker, C.; Ramachandran, R.; Malu, S.; Zhang, M.; Wardell, S.; Fulbright, O.J.; Toth, C.L.; Gonzalez, A.M.; et al. A Novel Method to Generate and Expand Clinical-Grade, Genetically Modified, Tumor-Infiltrating Lymphocytes. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Beane, J.D.; Lee, G.; Zheng, Z.; Mendel, M.; Abate-Daga, D.; Bharathan, M.; Black, M.; Gandhi, N.; Yu, Z.; Chandran, S.; et al. Clinical Scale Zinc Finger Nuclease-Mediated Gene Editing of PD-1 in Tumor Infiltrating Lymphocytes for the Treatment of Metastatic Melanoma. Mol. Ther. 2015, 23, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Ritthipichai, K.; Machin, M.; Juillerat, A.; Poirot, L.; Fardis, M.; Chartier, C. Genetic Modification of Iovance’s TIL through TALEN-Mediated Knockout of PD-1 as a Strategy to Empower TIL Therapy for Cancer. Ann. Oncol. 2020, 31, S720. [Google Scholar] [CrossRef]
- Chamberlain, C.A.; Bennett, E.P.; Kverneland, A.; Jensen, K.M.; Svane, I.M.; Donia, M.; Met, Ö. Highly Efficient CRISPR/Cas9-mediated Knockout of PD-1 for Tumor-infiltrating Lymphocyte-based Adoptive T cell Therapy. 2021; Manuscript in preparation. [Google Scholar]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish Actively Silences TCR Signaling in CD8+ T Cells to Maintain Tumor Tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal Checkpoint Regulates T Cell Neoantigen Reactivity and Susceptibility to PD1 Blockade. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A Fast and Versatile Algorithm That Searches for Potential off-Target Sites of Cas9 RNA-Guided Endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for SgRNA Design, off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Trial ID | Phase | Cancer Type(s) | CPI(s) Used | Estimated Completion Date | Sponsor |
|---|---|---|---|---|---|
| NCT03296137 | I/II | All metastatic cancers | Ipilimumab Nivolumab | September 2020 | Herlev Hospital CCIT-DK |
| NCT02027935 | II | Metastatic melanoma | Ipilimumab | January 2023 | MDACC |
| Cutaneous melanoma | |||||
| NCT03638375 | I/II | Metastatic melanoma | Nivolumab | May 2023 | Leiden University Medical Center |
| NCT03645928 | II | Metastatic melanoma, Squamous cell carcinoma of the head and neck, Non-small cell lung cancer | Ipilimumab Nivolumab Or Pembrolizumab | December 2024 | Iovance Biotherapeutics, Inc |
| NCT01993719 | II | Metastatic melanoma | Pembrolizumab | September 2029 | NCI |
| NCT01174121 | II | Metastatic colorectal cancer, Progressive glioblastoma, Metastatic pancreatic cancer, metastatic ovarian cancer, Metastatic breast carcinoma | Pembrolizumab | December 2024 | NCI |
| NCT03215810 | I | Non-small cell lung cancer, Metastatic non-small cell lung cancer, Squamous cell carcinoma, Advanced non-small cell lung cancer, Adenosquamous carcinoma, Adenocarcinomas | Nivolumab | December 2023 | H. Lee Moffitt Cancer Center and Research Institute |
| NCT03108495 | II | Cervical carcinoma | Pembrolizumab | December 2026 | Iovance Biotherapeutics, Inc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hulen, T.M.; Chamberlain, C.A.; Svane, I.M.; Met, Ö. ACT Up TIL Now: The Evolution of Tumor-Infiltrating Lymphocytes in Adoptive Cell Therapy for the Treatment of Solid Tumors. Immuno 2021, 1, 194-211. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1030012
Hulen TM, Chamberlain CA, Svane IM, Met Ö. ACT Up TIL Now: The Evolution of Tumor-Infiltrating Lymphocytes in Adoptive Cell Therapy for the Treatment of Solid Tumors. Immuno. 2021; 1(3):194-211. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1030012
Chicago/Turabian StyleHulen, Thomas Morgan, Christopher Aled Chamberlain, Inge Marie Svane, and Özcan Met. 2021. "ACT Up TIL Now: The Evolution of Tumor-Infiltrating Lymphocytes in Adoptive Cell Therapy for the Treatment of Solid Tumors" Immuno 1, no. 3: 194-211. https://0-doi-org.brum.beds.ac.uk/10.3390/immuno1030012