1. Introduction
The preparation of polymers carrying active functional groups in their structure is an important direction in the chemistry of macromolecular compounds. Such polymers can be used to attach non-polymeric molecules to them, such as drugs and other biologically active compounds [
1], dyes [
2], and catalytic agents [
3].
A key group of polymers for biomedical applications is microbial polyhydroxyalkanoates (PHAs), biodegradable biocompatible polyesters of hydroxy derivatives of fatty acids obtained by biotechnological methods [
4]. They usually have a high molecular weight (hundreds of thousands of Da), but for some applications it is necessary to obtain oligomeric PHAs with chemical functionality. Such oligomers can be used in controlled drug delivery systems [
5]; controlled pesticide release systems for agriculture [
6]; to obtain copolymers with oligomers of a different nature, such as polyurethanes [
7]; and graft copolymers with hyaluronan [
8]. In most cases, PHA oligomers for these purposes are obtained by transesterification or thermal degradation, less often by reduction. These methods provide the formation of terminal hydroxyl and (or) carboxyl groups, as well as double bonds (in the form of crotonic acid residues). The development of additional functionalization methods can allow the introduction of new end groups, and thus expand the possibilities of the subsequent attachment of biologically active substances or macromolecules.
Thionyl chloride, an acid chloride of sulfurous acid, is widely used in organic synthesis as a chlorinating agent, for example, in the preparation of carboxylic acid chlorides and alkyl chlorides. The ability of thionyl chloride to interact with alcohols and carboxylic acids with the formation of alkyl chlorides and acyl chlorides, respectively, is well known [
9]. For esters, however, only special cases of interaction with SOCl
2 are known: these are, in particular, reactions of the formation of acyl chlorides from tert-butyl esters (in this case, it is noted that benzyl, methyl, ethyl, and isopropyl esters are essentially unreactive under these conditions) [
10].
At the same time, the reaction to obtain 4-chlorobutanoyl chloride from gamma-butyrolactone in the presence of zinc chloride is known [
11], as is the ability of a mixture of ethyl acetate with thionyl chloride to react exothermically when placed in a zinc container [
12]. This suggests the ability of zinc compounds to catalyze this reaction. However, it is not considered as universal for esters; its mechanism has also not been studied, which may be due to its relatively low rate under usual conditions.
The aim of this study was to investigate the possibility of the reaction of poly-3-hydroxybutyrate with thionyl chloride in chloroform in the presence of zinc chloride. An additional goal was to obtain functionalized PHB oligomers, potentially suitable for the subsequent attachment of biologically active compounds.
2. Materials and Methods
2.1. Materials
Poly-3-hydroxybutyrate (PHB) of microbial biosynthesis (weight-average molecular weight, 840 kDa; polydispersity, 2.69) was obtained at the Siberian Federal University [
13]. The reagents and/or solvents used were thionyl chloride for synthesis (Merck, Darmstadt, Germany), extra-pure chloroform (Ekos-1, Staraya Kupavna, Russia), extra-pure hexane (Component-Reaktiv, Moscow, Russia), and zinc chloride (98.8%, Bochemie a.s., Bohumín, Czech Republic).
2.2. Degradation of PHB with Thionyl Chloride and the Isolation of Oligomer Samples
Here, 2.15 g of PHB (0.025 mol in terms of the –O–CH(CH3)–CH2–CO– monomer fragment, taking into account its molecular weight of 86 g/mol) was dissolved in 100 mL of chloroform at 50 °C under stirring by a magnetic stirrer. After complete dissolution, at a temperature of 50 °C and under strong stirring (750 rpm), 340 mg (0.0025 mol) of finely crystalline zinc chloride was added, followed by 7.26 mL (0.1 mol) of thionyl chloride. Continued heating and stirring under reflux were performed in the nitrogen atmosphere. Samples were periodically taken by precipitating the polymer with hexane at a polymer solution-to-hexane volume ratio of 2:5, followed by washing and purification on a Schott filter. The experiment was carried out for 96 h.
2.3. Molecular Weight Analysis
The molecular weight and molar mass distribution of the PHB oligomers were analyzed by gel permeation chromatography using an Agilent 1260 Infinity chromatograph (Agilent Technologies, Santa Clara, CA, USA) equipped with an isocratic pump, an autosampler, and a refractometric detector. The Agilent PLgel Mixed-C column was used. Sample preparation was carried out by dissolving approximately 10 mg of the polymer in 2 mL of chloroform and filtering the resulting solution (Millex-FH PTFE Filter Unit, pore diameter 0.45 μm). Chloroform was used as an eluent and the flow rate and temperature were 1.0 mL/min and 40°C, respectively. The sample volume was 50 µL and the polymer concentration in the solution was 5 mg/mL. The calibration curve was obtained with a set of Agilent EasiVial PS-H narrowly dispersed polystyrene standards. Weight-average (Mw) and number-average (Mn) molecular weight and polydispersity (D) were calculated using Agilent ChemStation for LC Systems, Rev. B.04.03, with ChemStation GPC Data Analysis Software, Rev. B.01.02.
2.4. Gas Chromatomass Spectrometry
Chloroform (1 mL), methanol (0.85 mL), and concentrated sulfuric acid (0.15 mL) were added to a polymer sample (approximately 10 mg); the mixture was kept in a tightly closed 4 mL vial for 4 h at a temperature of 60 °C. After cooling, 1 mL of distilled water was added and the lower (chloroform) layer containing methyl esters of monomeric acids was collected. Methyl esters were analyzed using an Agilent 6890 N gas chromatograph with an Agilent 5975 C inert XL EI/CI MSD mass spectrometer, in electron impact ionization mode (70 eV), using an Agilent VF-200 ms column (length, 60 m; inner diameter, 250 μm; thickness sorbent layer, 0.10 µm; the composition of the sorbent layer was polytrifluoropropylmethylsiloxane). Samples were injected using an Agilent 7683 B autosampler. Helium was used as the carrier gas at a flow rate of 1.2 mL min−1, in splitless mode. The temperatures of the input device, interface, quadrupole, and ion source were set to 220 °C, 230 °C, 150 °C, and 230 °C, respectively. The following temperature regime of the oven was used: 80 °C for 3 min, then heating to 220 °C at 10 °C min−1. A range of mass/charge ratios from 10 to 500 was analyzed. MSD Chemstation software version E.01.01.335 was used.
2.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
The 1H NMR spectra were recorded at 25 °C using a Bruker Avance III 600 (Bruker, Bremen, Germany) spectrometer at 600 MHz operating frequency in the Krasnoyarsk Regional Center of Research Equipment of Federal Research Center “Krasnoyarsk Science Center SB RAS”.
For the study, approximately 10 mg of a dry sample was dissolved in 1 mL of CDCl3 and incubated until the polymer was completely dissolved and dispersed (12 h). For 1H NMR and COSY spectra, a fivefold dilution of the resulting solution was used. 1H NMR spectra were obtained by accumulating 64 scans with a relaxation delay of 8 s. After the Fourier transform, spectra with a sweep of 24 ppm were obtained. For recording double-quantum-filtered COSY spectra, 128 experiments with 8 scans each were performed, with a delay of 5 s. CHCl3 as an internal standard was used (δCH 7.27).
3. Results
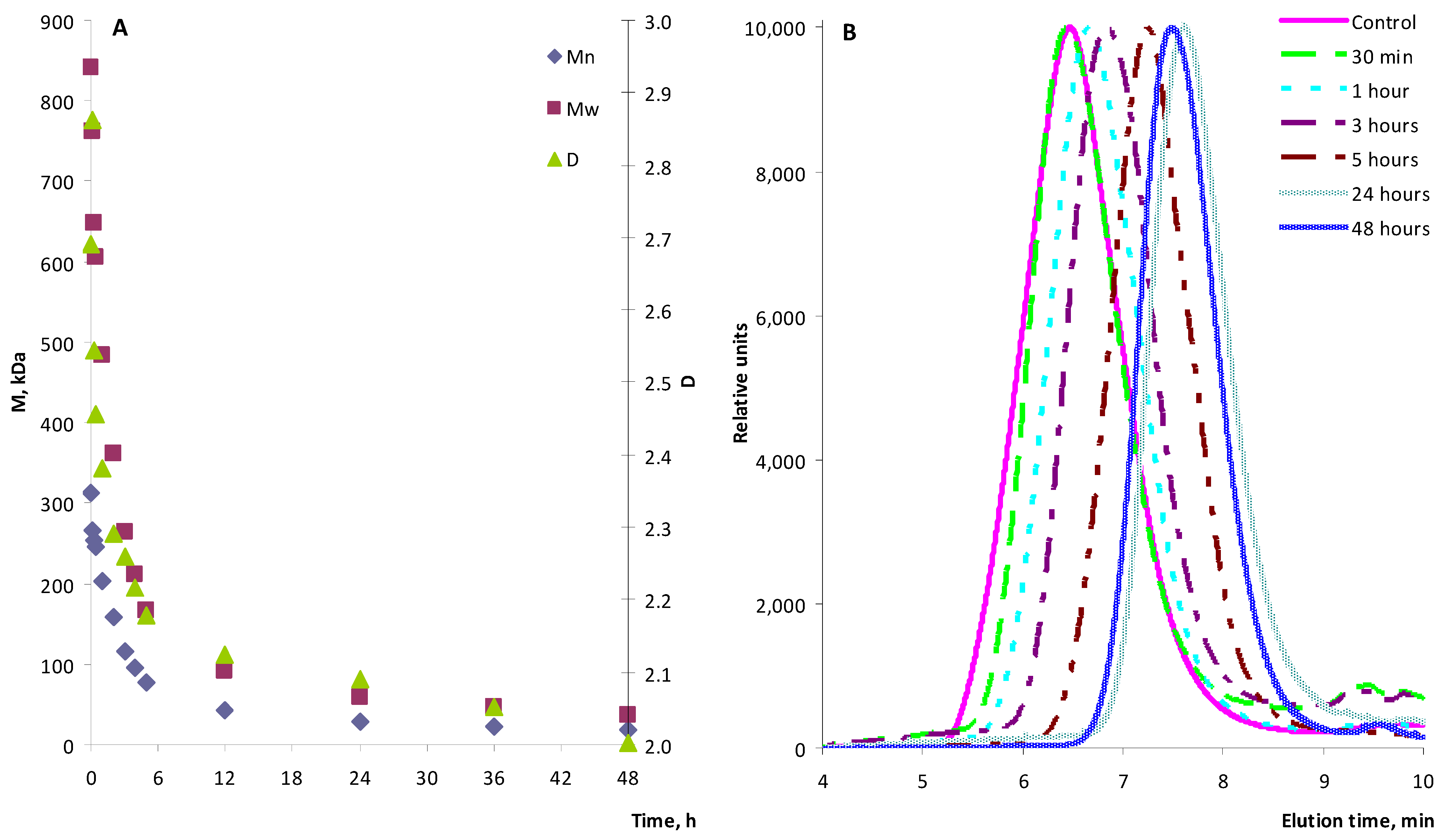
During the reaction, a relatively rapid decrease in the molecular weight of the polymer was noted in the first hour of the experiment; the M
w values decreased during this time from 840 kDa to 483 kDa (
Figure 1). Subsequently, the decline continued at a slower rate; by 5 h of the experiment, M
w decreased to 167 kDa, and by 96 h (the end of the experiment), to 16.7 kDa. The initial and final values of M
n were 312 kDa and 8.7 kDa, respectively. The polydispersity also gradually decreased from 2.69 at the beginning of the experiment to 1.92 at the end. Complete dissolution of zinc chloride in the reaction medium did not occur; its suspension was noted until the end of the experiment.
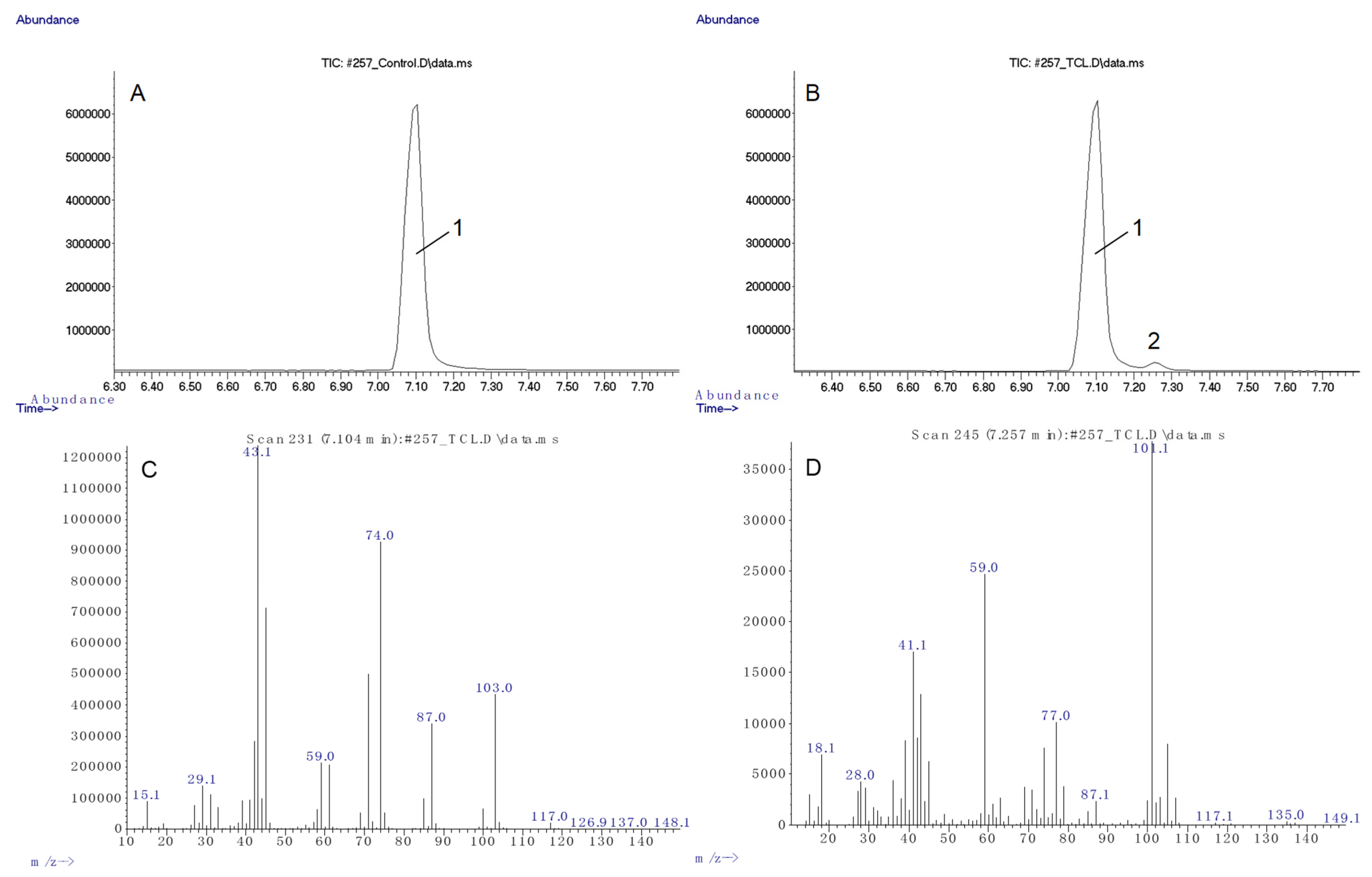
Polyester samples were first subjected to cleavage with methanol to methyl esters of the corresponding hydroxy acids, followed by their identification using gas chromatography with mass spectrometry. For the starting PHB, a single peak was recorded (
Figure 2A), corresponding to 3-hydroxybutyric acid methyl ester (the mass spectrum is shown in
Figure 2C). Chromatograms of samples subjected to degradation in thionyl chloride contained an additional minor peak (
Figure 2B), the mass spectrum of which (
Figure 2D) corresponded to methyl 3-chlorobutyrate, which indicated the presence of 3-chlorobutyric acid residues in the polymer, apparently formed at the site of polymer chain breaks by thionyl chloride.
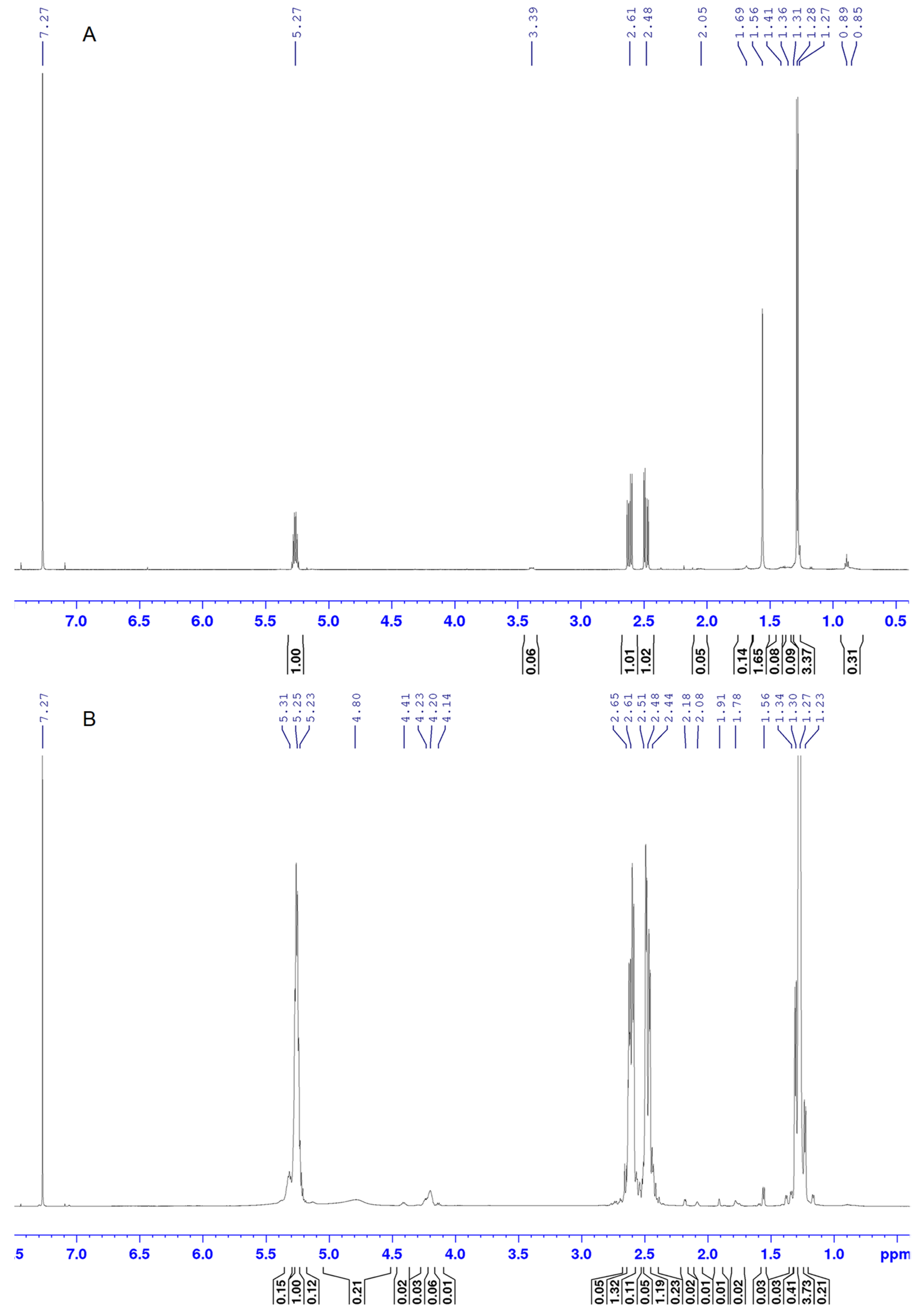
The
1H NMR spectrum of the control PHB sample (
Figure 3A) shows peaks typical of this polymer (δ
Me, 1.28; δ
CH2a, 2.48; δ
CH, 5.27; δ
CH2b, 2.61). There are also peaks corresponding to minor inclusions of monomers of 3-hydroxyvalerate (δ
Me, 0.85) and longer-chain 3-hydroxy acids (δ
Me, 0.89).
The
1H NMR spectrum of the polymer sample after degradation in thionyl chloride (
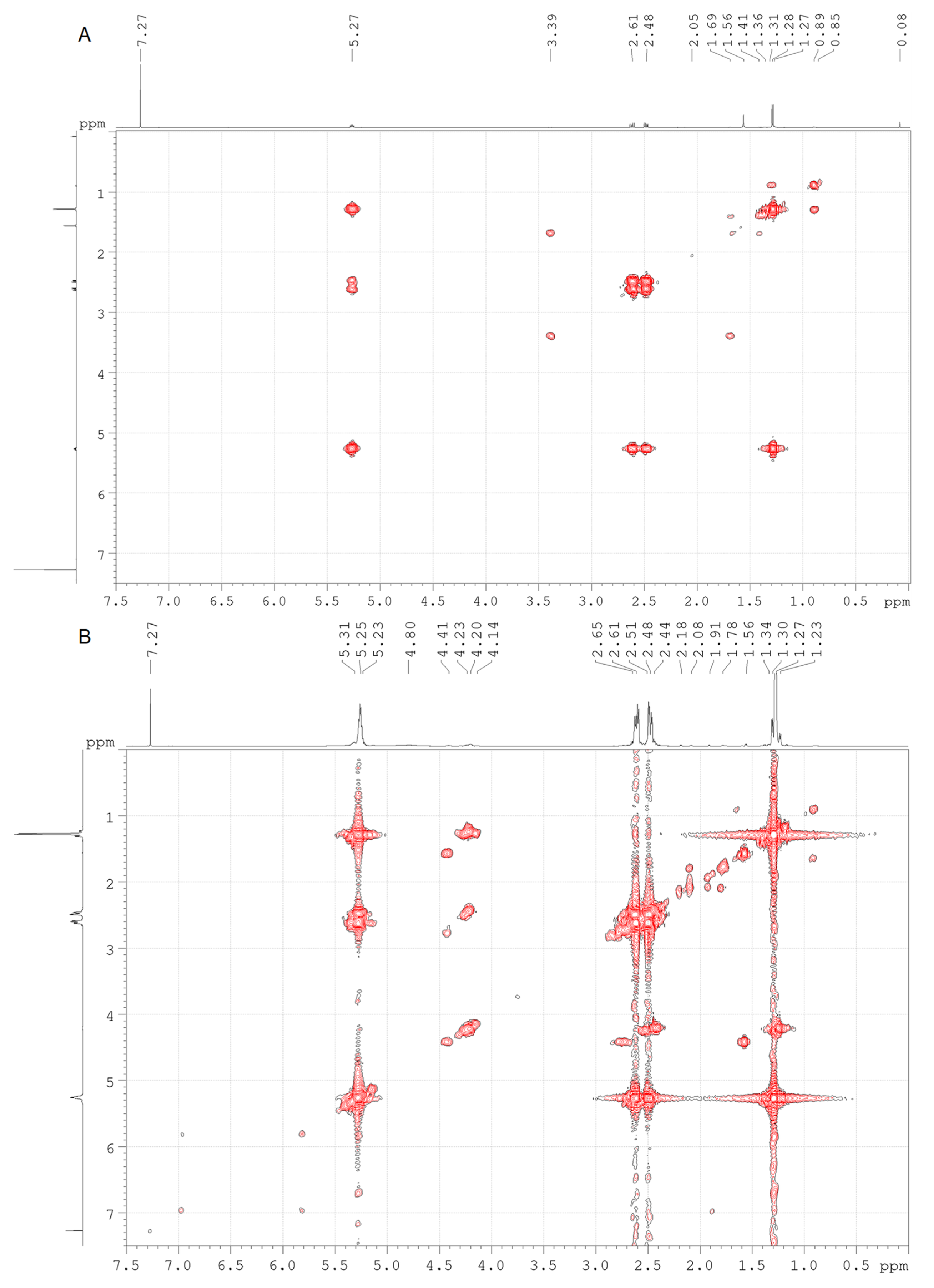
Figure 3A) is represented by broadened peaks, with the appearance of multiplets in the region of 4.10–4.50 ppm. Using the obtained COSY spectra (
Figure 4), the multiplets in this region were assigned to acid chlorides of the head monomers of 3-hydroxybutyric acid (4.14, 4.20, and 4.22 ppm), as well as to the tail monomers of 3-chlorobutyric acid (4.41 ppm) (
Table 1).
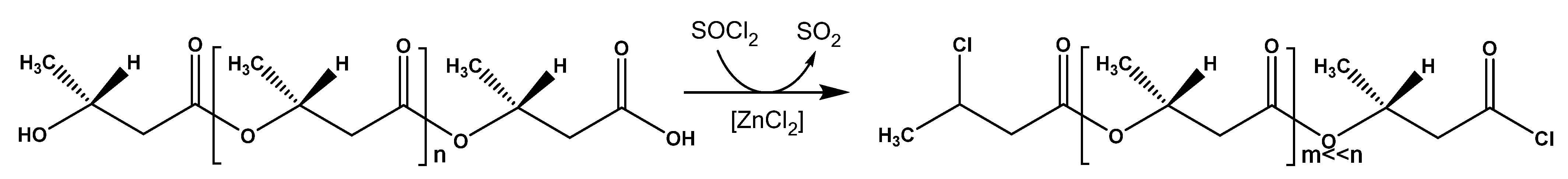
The data obtained allowed us to assert that the PHB degradation reaction proceeded along the path of cleavage of ester bonds with the formation of residues of 3-chlorobutyric and acid chloride groups at the ends of the resulting oligomeric fragments (
Figure 5).
4. Discussion
The simplest method for obtaining oligomers is the thermal degradation of PHA at a relatively low temperature, which leads to the gradual degradation of polyester molecules with the formation of unsaturated (in the case of PHB, crotonic, or trans-2-butenoic) acid residues and carboxyl groups at the corresponding ends [
14]. The mechanism of thermal degradation of PHB in the melt has been studied and is proposed as a process of random primary breakage of the polymer chain in the β-elimination reaction (E
i), which proceeds with the elimination of cis-β-CH and a six-membered cyclic transition [
15]. In this case, at the site of the chain break, an unsaturated acid residue is formed. Kawalec et al. [
16] proposed the thermal cleavage of PHA in the presence of carboxylic acid salts according to the E1cB mechanism, which proceeded through α-deprotonation with a carboxylate anion to form the same products. In general, the studied methods of oligomerization lead to the formation of mainly carboxyl, hydroxyl, and alkene residues in the polymer chain. The reaction studied here makes it possible to obtain oligomers containing chloroalkyl and acid chloride groups at opposite ends.
The interaction mechanism of thionyl chloride with carboxylic acids to form acyl chlorides has been thoroughly studied [
17]. Thionyl chloride is electrophilic at the sulfur atom, and is attacked by the nucleophilic oxygen atom of the carbonyl group to form an unstable intermediate. At subsequent stages, the chlorine anion migrates to the positively charged carbonyl carbon, SO
2 is eliminated, and the released second chlorine anion interacts with the hydrogen of the hydroxyl group to form HCl.
The interaction mechanism of thionyl chloride with alcohols is more complex and largely depends on the environment [
18,
19]. It has been established that the reaction proceeds with the formation of ionic alkyl chlorosulfites as an intermediate, followed by their decomposition into sulfur dioxide and alkyl chloride. Depending on the solvent used, such reactions can proceed with the reversal or retention of the optical configuration of the alkyl chloride compared with the original alcohol. In the case of a configuration inversion (for example, in pure thionyl chloride, in kerosene or toluene), the replacement of the chlorosulfite group is assumed as a result of attack by a chloride ion from the rear. In the second case (characteristic, for example, of tetrahydrofuran or dioxane, which are themselves nucleophilic agents), a two-stage process occurs. At the first stage, the solvent replaces chlorosulfite through the mechanism of conventional bimolecular nucleophilic substitution by S
N2. In the second stage, the chloride ion replaces dioxane in the oxonium cation. Double reversal is tantamount to saving the configuration at the asymmetric carbon atom. In addition, in the presence of tertiary bases, the reaction is supposed to proceed according to the S
N2 mechanism in chlorosulfite under the action of the chloride ion as a nucleophile. In this case, the source of the chloride ion is the tertiary amine hydrochloride formed in situ.
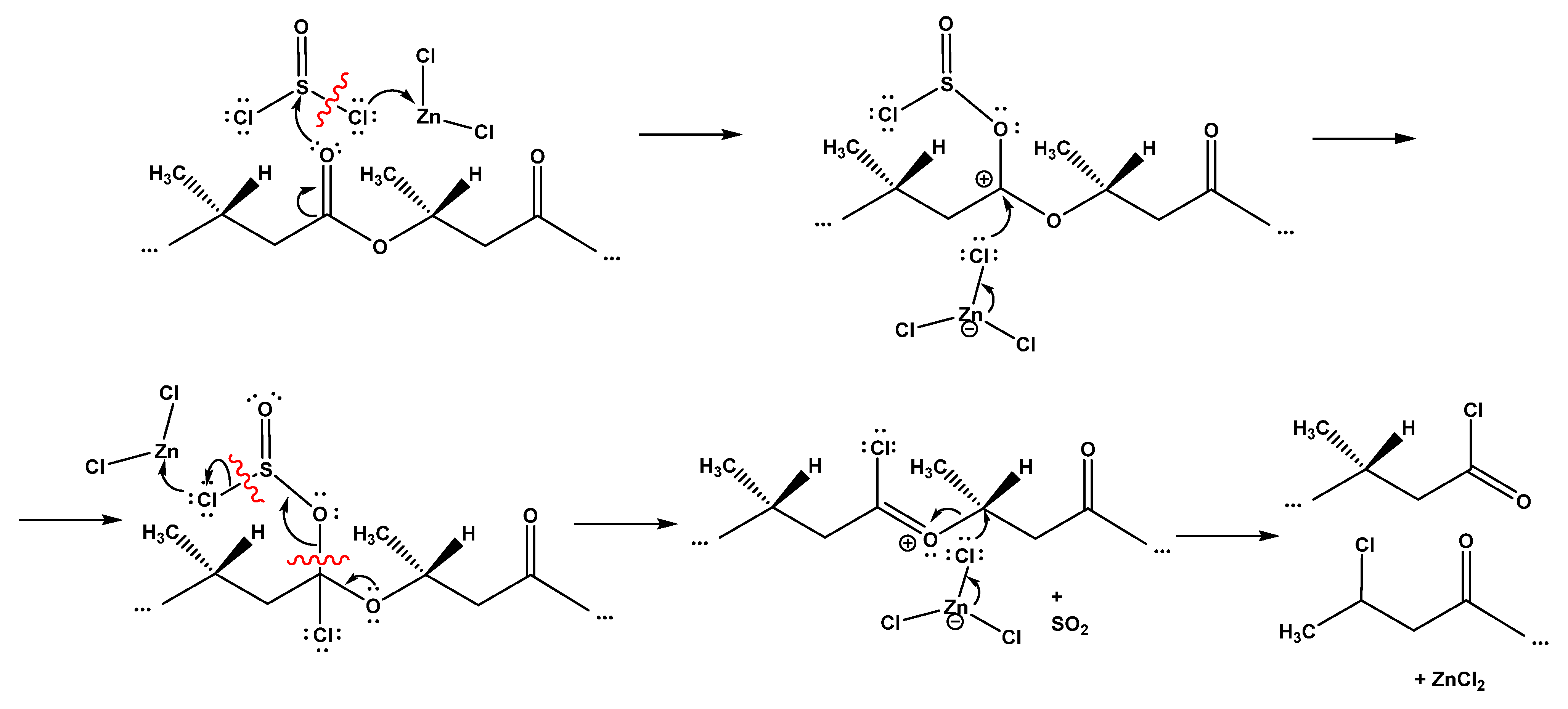
In the case of esters, the proposed mechanism is closer to the interaction of thionyl chloride with carboxylic acids, but nucleophilicity of the chloride ions is probably not sufficient for an analogous reaction to take place. In this case, zinc chloride, playing the role of a Lewis acid, can enhance the nucleophilicity of chlorine, ensuring that the reaction proceeds to the irreversible stage of SO
2 elimination (
Figure 6). At the last stage, zinc chloride (in the form of ZnCl
3−) can act as a chlorine donor to the chlorinated ester intermediate, providing final breakage of the bond between the oxygen of the acyl chloride and the carbon near the former alcohol group. The obtained results do not suggest the spatial configuration of the substituents at the carbon atom previously associated with the “alcohol” oxygen atom; an inversion of this configuration is specified as a result of the described reaction, but more research is needed to confirm this assumption. Therefore, in
Figure 6, the configuration of the substituents at this atom is not indicated.
Although this reaction proceeds at a relatively low rate, which limits its use for the usual preparation of the corresponding acid chlorides and chloroalkanes, it can be used, in particular, for the preparation of functionalized oligomers of aliphatic polyesters, because cleavage of only part of the ester groups is sufficient in this case.
Methods for obtaining functionalized oligomers using chemical reagents (alcohols, reducing agents, etc.) are generally used in addition to thermal degradation. For example, PHB-diols of various molecular weights were obtained through transesterification of the polymer with 1,4-butanediol by varying the alcoholysis time [
20]. The ordinary hydrolysis of PHA under acidic [
21] and alkaline [
22] conditions is also possible. Obtaining low-molecular-weight polyester diols by reduction with sodium borohydride can also be performed [
23]. In general, these methods make it possible to obtain either diol oligomers containing alcohol groups at both ends or oligomers with a carboxyl group at the O-terminus and a hydroxyl group at the C-terminus. The use of the proposed reaction with thionyl chloride makes it possible to obtain oligomers with chloroalkyl and acid chloride groups, which expands the possibilities of their subsequent modification.
5. Conclusions
Thus, the present study shows the possibility of interaction of esters (by the example of 3-hydroxybutyric acid polyesters) with thionyl chloride in the presence of a zinc chloride catalyst with the formation of acid chloride and chloroalkyl groups at the rupture site of the ester bond, as well as the applicability of this reaction to obtain PHB oligomers with the corresponding groups. Such oligomers can be used for the attachment of biologically active substances or macromolecules. Acyl chloride groups can also interact, e.g., with hydroxyl or amine groups, obtaining different graft copolymers.
In addition, the very reaction of interaction of esters with thionyl chloride makes it relatively easy to obtain corresponding acyl chlorides, which can serve as precursors for a wide range of derivatives (aldehydes, ketones, anhydrides, amides, new esters, etc.).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}