
Is Circulating DNA and Tumor Cells in Myeloma the Way Forward?


Abstract
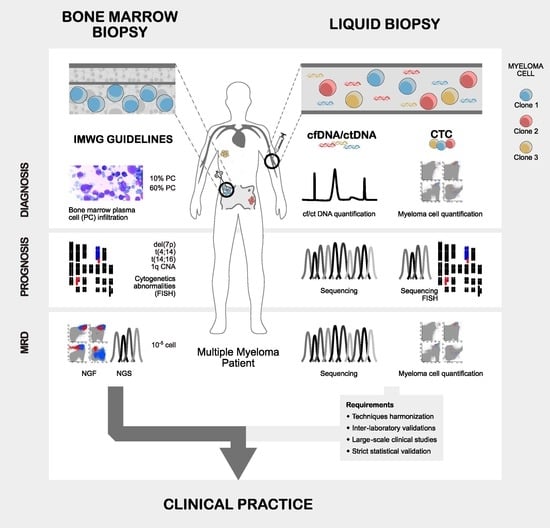
:
1. Introduction
2. Direct vs. Indirect Assessment of Myeloma Plasma Cells
2.1. Myeloma Plasma Cells Direct Assessment through Bone Marrow Biopsies
2.2. Myeloma Indirect Assessment through Circulating Tumoral DNA
2.3. Myeloma Indirect Assessment through Circulating Tumor Plasma Cells
2.4. Statistical Validation of Biomarkers for Multiple Myeloma
2.4.1. Validation of Diagnostic Biomarkers
2.4.2. Validation of Prognostic Biomarkers
2.4.3. Validation of Biomarkers for Depth of Response
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer Globocan: Multiple Myeloma. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/35-Multiple-myeloma-fact-sheet.pdf (accessed on 28 October 2021).
- International Agency for Research on Cancer Globocan: Europe. Available online: https://gco.iarc.fr/today/data/factsheets/populations/908-europe-fact-sheets.pdf (accessed on 29 October 2021).
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Larson, D.; Kyle, R.A. Diagnosis of Smoldering Multiple Myeloma. N. Engl. J. Med. 2011, 365, 474–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshman, A.; Vincent Rajkumar, S.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk Stratification of Smoldering Multiple Myeloma Incorporating Revised IMWG Diagnostic Criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. A Long-Term Study of Prognosis in Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.; Caetano, J.; Ferreira, B.; Barahona, F.; Carneiro, E.A.; João, C. The Immune Microenvironment in Multiple Myeloma: Friend or Foe? Cancers 2021, 13, 625. [Google Scholar] [CrossRef]
- Lopes, R.; Ferreira, B.V.; Caetano, J.; Barahona, F.; Carneiro, E.A.; João, C. Boosting Immunity against Multiple Myeloma. Cancers 2021, 13, 1221. [Google Scholar] [CrossRef]
- Waldeyer, W. Ueber Bindegewebszellen. Arch. Mikrosk. Anat. 1875, 11, 176–194. [Google Scholar] [CrossRef]
- Marschalkó, T. Ueber Die Sogenannten Plasmazellen, Ein Beitrag Zur Kenntniss Der Herkunft Der Entzündlichen Infiltrationszellen. Arch. Für Dermatol. Syph. 1895, 30, 3–52. [Google Scholar] [CrossRef]
- Wright, J.H. A Case of Multiple Myeloma. Boston Med. Surg. J. 1900, 4, 195–204. [Google Scholar]
- Ramón, Y.; Cajal, S. Manual de Anatomía Patológica General: Seguida de Un Resumen de Microscopia Aplicada á La Histología y Bacteriología Patológicas; Forgotten Books: London, UK, 1913; ISBN 9781332475421. [Google Scholar]
- Rosenthal, N. Value of the Sternal Puncture in the Diagnosis of Multiple Myeloma. J. Mt. Sinai Hosp. 1938, 4, 1001–1019. [Google Scholar]
- Arinkin, M.I. Die Intravitale Untersuchungsmethodik Des Knochenmarks. Folia Haematol. 1929, 38, 192–199. [Google Scholar]
- Diwany, M. Sternal Marrow Puncture in Children. Arch. Dis. Child. 1940, 15, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Committee of the Chronic Leukemia Myeloma Task Force, National Cancer Institute. Proposed Guidelines for Protocol Studies—II Plasma Cell Myeloma. Cancer Chemother. Rep. 1973, 4, 141. [Google Scholar]
- Durie, B.G.M.; Salmon, S.E. A Clinical Staging System for Multiple Myeloma Correlation of Measured Myeloma Cell Mass with Presenting Clinical Features, Response to Treatment, and Survival. Cancer 1975, 36, 842–854. [Google Scholar] [CrossRef]
- Merlini, G.; Waldenstrom, J.G.; Jayakar, S.D. A New Improved Clinical Staging System for Multiple Myeloma Based on Analysis of 123 Treated Patients. Blood 1980, 55, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Greipp, P. Smoldering Multiple Myeloma. Br. J. Haematol. 1980, 302, 1347–1349. [Google Scholar] [CrossRef]
- Kyle, R.A.; Child, J.A.; Anderson, K.; Barlogie, B.; Bataille, R.; Bensinger, W.; Bladé, J.; Boccadoro, M.; Dalton, W.; Dimopoulos, M.; et al. Criteria for the Classification of Monoclonal Gammopathies, Multiple Myeloma and Related Disorders: A Report of the International Myeloma Working Group. N. Engl. J. Med. 2003, 121, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Moon, S.Y.; Lee, J.H.; Park, H.K.; Kong, S.Y.; Bang, S.M.; Lee, J.H.; Yoon, S.S.; Lee, D.S. Discrepancies between the Percentage of Plasma Cells in Bone Marrow Aspiration and BM Biopsy: Impact on the Revised IMWG Diagnostic Criteria of Multiple Myeloma. Blood Cancer J. 2017, 7, e530. [Google Scholar] [CrossRef]
- Kastritis, E.; Terpos, E.; Moulopoulos, L.; Spyropoulou-Vlachou, M.; Kanellias, N.; Eleftherakis-Papaiakovou, E.; Gkotzamanidou, M.; Migkou, M.; Gavriatopoulou, M.; Roussou, M.; et al. Extensive Bone Marrow Infiltration and Abnormal Free Light Chain Ratio Identifies Patients with Asymptomatic Myeloma at High Risk for Progression to Symptomatic Disease. Leukemia 2013, 27, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating Tumour DNA Sequence Analysis as an Alternative to Multiple Myeloma Bone Marrow Aspirates. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating Tumour DNA Analysis Demonstrates Spatial Mutational Heterogeneity That Coincides with Disease Relapse in Myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Long, X.; Xu, Q.; Lou, Y.; Li, C.; Gu, J.; Cai, H.; Wang, D.; Xu, J.; Li, T.; Zhou, X.; et al. The Utility of Non-Invasive Liquid Biopsy for Mutational Analysis and Minimal Residual Disease Assessment in Extramedullary Multiple Myeloma; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2020; Volume 189, pp. e45–e48. [Google Scholar]
- Deshpande, S.; Tytarenko, R.G.; Wang, Y.; Boyle, E.M.; Ashby, C.; Schinke, C.D.; Thanendrarajan, S.; Zangari, M.; Zhan, F.; Davies, F.E.; et al. Monitoring Treatment Response and Disease Progression in Myeloma with Circulating Cell-Free DNA. Eur. J. Haematol. 2021, 106, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Sanoja-Flores, L.; Flores-Montero, J.; Garcés, J.J.; Paiva, B.; Puig, N.; García-Mateo, A.; García-Sánchez, O.; Corral-Mateos, A.; Burgos, L.; Blanco, E.; et al. Next Generation Flow for Minimally-Invasive Blood Characterization of MGUS and Multiple Myeloma at Diagnosis Based on Circulating Tumor Plasma Cells (CTPC). Blood Cancer J. 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolli, N.; Sgherza, N.; Curci, P.; Rizzi, R.; Strafella, V.; Delia, M.; Gagliardi, V.P.; Neri, A.; Baldini, L.; Albano, F.; et al. What Is New in the Treatment of Smoldering Multiple Myeloma? J. Clin. Med. 2021, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.; Morgan, G.; van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group Molecular Classification of Multiple Myeloma: Spotlight Review. Leukemia 2009, 23, 2210. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- D’Agostino, M.; Lahuerta, J.-J.; Wester, R.; Waage, A.; Bertsch, U.; Zamagni, E.; Mateos, M.-V.; Larocca, A.; Dall’Olio, D.; van de Donk, N.W.C.J.; et al. A New Risk Stratification Model (R2-ISS) in Newly Diagnosed Multiple Myeloma: Analysis of Mature Data from 7077 Patients Collected By European Myeloma Network within Harmony Big Data Platform. Blood 2020, 136, 34–37. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2020 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [Green Version]
- Greipp, P.R.; Miguel, J.S.; Dune, B.G.M.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Harousseau, J.L.; Kyle, R.A.; et al. International Staging System for Multiple Myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Rustad, E.H.; Coward, E.; Skytøen, E.R.; Misund, K.; Holien, T.; Standal, T.; Børset, M.; Beisvag, V.; Myklebost, O.; Meza-Zepeda, L.A.; et al. Monitoring Multiple Myeloma by Quantification of Recurrent Mutations in Serum. Haematologica 2017, 102, 1266–1272. [Google Scholar] [CrossRef] [Green Version]
- Biancon, G.; Gimondi, S.; Vendramin, A.; Carniti, C.; Corradini, P. Noninvasive Molecular Monitoring in Multiple Myeloma Patients Using Cell-Free Tumor DNA: A Pilot Study. J. Mol. Diagn. 2018, 20, 859–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-Exome Sequencing of Cell-Free DNA and Circulating Tumor Cells in Multiple Myeloma. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rajkumar, S.V.; Kyle, R.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Lust, J.A.; Gertz, M.A.; Greipp, P.R.; Witzig, T.E. Prognostic Value of Circulating Plasma Cells in Monoclonal Gammopathy of Undetermined Significance. J. Clin. Oncol. 2005, 23, 5668–5674. [Google Scholar] [CrossRef]
- Peceliunas, V.; Janiulioniene, A.; Matuzeviciene, R.; Zvirblis, T.; Griskevicius, L. Circulating Plasma Cells Predict the Outcome of Relapsed or Refractory Multiple Myeloma. Leuk. Lymphoma 2012, 53, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Kyle, R.A.; Larson, D.R.; Witzig, T.E.; Kumar, S.; Dispenzieri, A.; Morice, W.G.; Rajkumar, S.V. High Levels of Peripheral Blood Circulating Plasma Cells as a Specific Risk Factor for Progression of Smoldering Multiple Myeloma. Leukemia 2013, 27, 680–685. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Rajkumar, S.; Gupta, V.; Morice, W.G.; Timm, M.M.; Singh, P.P.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Kapoor, P.; et al. Quantification of Clonal Circulating Plasma Cells in Newly Diagnosed Multiple Myeloma: Implications for Redefining High-Risk Myeloma. Leukemia 2014, 28, 2060–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, R.; Muchtar, E.; Kumar, S.K.; Jevremovic, D.; Buadi, F.K.; Dingli, D.; Dispenzieri, A.; Hayman, S.R.; Hogan, W.J.; Kapoor, P.; et al. Risk Stratification in Myeloma by Detection of Circulating Plasma Cells Prior to Autologous Stem Cell Transplantation in the Novel Agent Era. Blood Cancer J. 2016, 6, e512. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Huhn, S.; Weinhold, N.; Nickel, J.; Pritsch, M.; Hielscher, T.; Hummel, M.; Bertsch, U.; Huegle-Doerr, B.; Vogel, M.; Angermund, R.; et al. Circulating Tumor Cells as a Biomarker for Response to Therapy in Multiple Myeloma Patients Treated within the GMMG-MM5 Trial. Bone Marrow Transplant. 2017, 52, 1194–1198. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Rajkumar, S.; Dispenzieri, A.; Dingli, D.; Timm, M.M.; Morice, W.G.; Lacy, M.Q.; Buadi, F.K.; Go, R.S.; Leung, N.; et al. Quantification of Circulating Clonal Plasma Cells via Multiparametric Flow Cytometry Identifies Patients with Smoldering Multiple Myeloma at High Risk of Progression. Leukemia 2017, 31, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, R.; Muchtar, E.; Kumar, S.K.; Jevremovic, D.; Buadi, F.K.; Dingli, D.; Dispenzieri, A.; Hayman, S.R.; Hogan, W.J.; Kapoor, P.; et al. Serial Measurements of Circulating Plasma Cells before and after Induction Therapy Have an Independent Prognostic Impact in Patients with Multiple Myeloma Undergoing Upfront Autologous Transplantation. Haematologica 2017, 102, 1439–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, A.J.; Stevenson, P.A.; Libby, E.N.; Becker, P.S.; Coffey, D.G.; Green, D.J.; Hyun, T.S.; Fromm, J.R.; Gopal, A.K.; Holmberg, L.A. Circulating Plasma Cells at the Time of Collection of Autologous PBSC for Transplant in Multiple Myeloma Patients Is a Negative Prognostic Factor Even in the Age of Post-Transplant Maintenance Therapy. Biol. Blood Marrow Transplant. 2018, 24, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcés, J.J.; Bretones, G.; Burgos, L.; Valdes-Mas, R.; Puig, N.; Cedena, M.T.; Alignani, D.; Rodriguez, I.; Puente, D.Á.; Álvarez, M.G.; et al. Circulating Tumor Cells for Comprehensive and Multiregional Non-Invasive Genetic Characterization of Multiple Myeloma. Leukemia 2020, 34, 3007–3018. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A Large Meta-Analysis Establishes the Role of MRD Negativity in Long-Term Survival Outcomes in Patients with Multiple Myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal Residual Disease Negativity Using Deep Sequencing Is a Major Prognostic Factor in Multiple Myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef] [Green Version]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.J.; Vidriales, M.B.; García-Sanz, R.; et al. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [Green Version]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring Multiple Myeloma by Next-Generation Sequencing of V(D)J Rearrangements from Circulating Myeloma Cells and Cell-Free Myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef]
- Mazzotti, Ć.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.L.; Leleu, X.; Hulin, C.; Manier, S.; Hebraud, B.; Roussel, M.; et al. Myeloma MRD by Deep Sequencing from Circulating Tumor DNA Does Not Correlate with Results Obtained in the Bone Marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Vrabel, D.; Sedlarikova, L.; Besse, L.; Rihova, L.; Bezdekova, R.; Almasi, M.; Kubaczkova, V.; Brožová, L.; Jarkovsky, J.; Plonkova, H.; et al. Dynamics of Tumor-specific CfDNA in Response to Therapy in Multiple Myeloma Patients. Eur. J. Haematol. 2020, 104, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Puig, N.; Cedena, M.T.; Rosiñol, L.; Cordón, L.; Vidriales, M.B.; Burgos, L.; Flores-Montero, J.; Sanoja-Flores, L.; Lopez-Anglada, L.; et al. Measurable Residual Disease by Next-Generation Flow Cytometry in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les Acides Nucléiques Du Plasma Sanguin Chez l’homme. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic Acid (DNA) and Antibodies to DNA in the Serum of Patients with Systemic Lupus Erythematosus. J. Clin. Investig. 1966, 45, 1732–1740. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble Normal and Mutated DNA Sequences from Single-Copy Genes in Human Blood. CEBP 1994, 3, 67–71. [Google Scholar]
- Deligezer, U.; Erten, N.; Akisik, E.E.; Dalay, N. Circulating Fragmented Nucleosomal DNA and Caspase-3 MRNA in Patients with Lymphoma and Myeloma. Exp. Mol. Pathol. 2006, 80, 72–76. [Google Scholar] [CrossRef]
- Hocking, J.; Mithraprabhu, S.; Kalff, A.; Spencer, A. Liquid Biopsies for Liquid Tumors: Emerging Potential of Circulating Free Nucleic Acid Evaluation for the Management of Hematologic Malignancies. Cancer Biol. Med. 2016, 13, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Chiu, R.W.K.; Poon, L.L.M.; Lau, T.K.; Leung, T.N.; Wong, E.M.C.; Lo, Y.M.D. Effects of Blood-Processing Protocols on Fetal and Total DNA Quantification in Maternal Plasma. Clin. Chem. 2001, 47, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Raje, N.S.; Seifer, C.; Kloeber, J.; Isenhart, R.; Ha, G.; Yee, A.J.; O’Donnell, E.K.; Tai, Y.T.; Richardson, P.G.; et al. Genomic Discovery and Clonal Tracking in Multiple Myeloma by Cell-Free DNA Sequencing. Leukemia 2018, 32, 1838–1841. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Hocking, J.; Ramachandran, M.; Choi, K.; Klarica, D.; Khong, T.; Reynolds, J.; Spencer, A. DNA-Repair Gene Mutations Are Highly Prevalent in Circulating Tumour Dna from Multiple Myeloma Patients. Cancers 2019, 11, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithraprabhu, S.; Morley, R.; Khong, T.; Kalff, A.; Bergin, K.; Hocking, J.; Savvidou, I.; Bowen, K.M.; Ramachandran, M.; Choi, K.; et al. Monitoring Tumour Burden and Therapeutic Response through Analysis of Circulating Tumour DNA and Extracellular RNA in Multiple Myeloma Patients. Leukemia 2019, 33, 2022–2033. [Google Scholar] [CrossRef]
- Dennis Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.Z.; Magnus Hjelm, N. Rapid Clearance of Fetal DNA from Maternal Plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov Identifier: NCT04122092 Evaluation of Ultrasensitive Chromosomal Aneuploidy Detection for Detecting Minimal Residual Disease in Multiple Myeloma (EUCADD). Available online: https://clinicaltrials.gov/ct2/show/NCT04122092 (accessed on 30 December 2021).
- Ashworth, T. A Case of Cancer in Which Cells Similar to Those in the Tumours Were Seen in the Blood after Death. Australas. Medical J. 1869, 14, 146–149. [Google Scholar]
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Ghobrial, I.M. Myeloma as a Model for the Process of Metastasis: Implications for Therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Sanoja-Flores, L.; Flores-Montero, J.; Pérez-Andrés, M.; Puig, N.; Orfao, A. Detection of Circulating Tumor Plasma Cells in Monoclonal Gammopathies: Methods, Pathogenic Role, and Clinical Implications. Cancers 2020, 12, 1499. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Witzig, T.E.; Dingli, D.; Tracz, M.J.; Gertz, M.A.; Lacy, M.Q.; Lust, J.A.; Dispenzieri, A.; Greipp, P.R.; Kyle, R.A.; et al. Circulating Plasma Cells Detected by Flow Cytometry as a Predictor of Survival in 302 Patients with Newly Diagnosed Multiple Myeloma. Blood 2005, 106, 2276–2279. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, W.I.; Jevremovic, D.; Nandakumar, B.; Dispenzieri, A.; Buadi, F.K.; Dingli, D.; Lacy, M.Q.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. Enhancing the R-ISS Classification of Newly Diagnosed Multiple Myeloma by Quantifying Circulating Clonal Plasma Cells. Am. J. Hematol. 2020, 95, 310–315. [Google Scholar] [CrossRef]
- Bae, M.H.; Park, C.J.; Kim, B.H.; Cho, Y.U.; Jang, S.; Lee, D.H.; Seo, E.J.; Yoon, D.H.; Lee, J.H.; Suh, C. Increased Circulating Plasma Cells Detected by Flow Cytometry Predicts Poor Prognosis in Patients with Plasma Cell Myeloma. Cytometry B. Clin. Cytom. 2018, 94, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Pérez-Andrés, M.; Vídriales, M.B.; Almeida, J.; de Las Heras, N.; Mateos, M.; López-Corral, L.; Gutiérrez, N.C.; Blanco, J.; Oriol, A.; et al. Competition between Clonal Plasma Cells and Normal Cells for Potentially Overlapping Bone Marrow Niches Is Associated with a Progressively Altered Cellular Distribution in MGUS vs. Myeloma. Leukemia 2011, 25, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vij, R.; Mazumder, A.; Klinger, M.; O’dea, D.; Paasch, J.; Martin, T.; Weng, L.; Park, J.; Fiala, M.; Faham, M.; et al. Deep Sequencing Reveals Myeloma Cells in Peripheral Blood in Majority of Multiple Myeloma Patients. Clin. Lymphoma Myeloma Leuk. 2014, 14, 131–139.e1. [Google Scholar] [CrossRef]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic Interrogation of Circulating Multiple Myeloma Cells at Single-Cell Resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov Identifier: NCT02627261 Multiple Myeloma Minimal Residual Disease (MMRD). Available online: https://clinicaltrials.gov/ct2/show/NCT02627261 (accessed on 3 November 2021).
- Wu, V.; Moshier, E.; Leng, S.; Barlogie, B.; Cho, H.J.; Jagannath, S.; Madduri, D.; Mazumdar, M.; Parekh, S.; Chari, A. Risk Stratification of Smoldering Multiple Myeloma: Predictive Value of Free Light Chains and Group-Based Trajectory Modeling. Blood Adv. 2018, 2, 1470–1479. [Google Scholar] [CrossRef]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. NEJM 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Eo, S.-H.; Kang, H.J.; Hong, S.-M.; Cho, H. K-Adaptive Partitioning for Survival Data, with an Application to Cancer Staging. J. Stat. Softw. 2013, 10, 1–15. [Google Scholar]
- Rajkumar, S.V.; Fonseca, R.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Therneau, T.M.; Kyle, R.A.; Greipp, P.R.; Gertz, M.A. Methods for Estimation of Bone Marrow Plasma Cell Involvement in Myeloma: Predictive Value for Response and Survival in Patients Undergoing Autologous Stem Cell Transplantation. Am. J. Hematol. 2001, 68, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Hájek, R.; Delforge, M.; Raab, M.S.; Schoen, P.; DeCosta, L.; Spicka, I.; Radocha, J.; Pour, L.; Gonzalez-McQuire, S.; Bouwmeester, W. Development and Validation of a Novel Risk Stratification Algorithm for Relapsed Multiple Myeloma. Br. J. Haematol. 2019, 187, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Bouwmeester, W.; Briggs, A.; van Hout, B.; Hájek, R.; Gonzalez-McQuire, S.; Campioni, M.; DeCosta, L.; Brozova, L. Methodology of a Novel Risk Stratification Algorithm for Patients with Multiple Myeloma in the Relapsed Setting. Oncol.Ther. 2019, 7, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Lee, L. NADA: Nondetects and Data Analysis for Environmental Data. Available online: https://cran.r-project.org/ (accessed on 3 November 2021).
- Roche Molecular Systems, Basel, Switzerland. Cobas EGFR Mutation Test V2. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/cobas-egfr-mutation-test-v2 (accessed on 30 December 2021).
- Fernández de Larrea, C.; Kyle, R.; Rosiñol, L.; Paiva, B.; Engelhardt, M.; Usmani, S.; Caers, J.; Gonsalves, W.; Schjesvold, F.; Merlini, G.; et al. Primary Plasma Cell Leukemia: Consensus Definition by the International Myeloma Working Group According to Peripheral Blood Plasma Cell Percentage. Blood Cancer J. 2021, 11, 192. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Type of Biopsy | Cohort (Sample) | Method | Findings | Reference |
|---|---|---|---|---|
| PB | NDMM = 389 | Kaplan–Meier curves | Shorter OS for patients with BMPC ≥ 10% vs. <10%. p = 0.01 | Lee at al., 2017 [23] |
| BM | SMM = 651 | Kaplan–Meier curves Log-rank test | Shorter TTP for BMPC ≥ 60% vs. BMPC < 60%, p < 0.001 95% of patients with BM ≥ 60% progressed to active myeloma within 2 years after diagnosis Median TTP = 7 months (95% CI, 1.0 to 12.9) | Rajkumar et al., 2011 [4] |
| SMM = 96 | ROC Multivariable Cox regression model | BMPC cut-off ≥ 60% in patients progressing at 18 months (specificity = 95.5%) BMPC ≥ 60% associated with high-risk progression (p < 0.001; HR = 13.7) | Kastritis et al., 2013 [24] | |
| cfDNA | MM = 53 (64) Other cancers = 56 | QIAamp Circulating Nucleic Acid kit (Qiagen) | MM = 20.1 ng/mL plasma Other cancers = 10.3 ng/mL p < 0.001 | Kis et al., 2017 [25] |
| MM = 37 HD = 21 | QIAamp Circulating Nucleic Acid kit (Qiagen) | MM = 23 ng/mL (5–195) HD = 15 ng/mL (6–32) p = 0.0085 | Mithraprabhu et al., 2017 [26] | |
| EM-MM = 8 (22) MM = 10 (23) | QIAamp Circulating Nucleic Acid kit (Qiagen) | MM = 16.4 ng/mL (4.3–214.7) EMM = 43.6 ng/mL (3.5–313.5) p = 0.009 | Long et al., 2020 [27] | |
| MM = 77 SMM = 25 | QIAamp Circulating Nucleic Acid kit (Qiagen) | MM = 25.2 ng/mL (0.2–467.14) SMM = 12 ng/mL (4.6–39.5) p = 0.0001 cfDNA concentration related to BMPC, R2 = 0.13, p = 0.001 | Deshpande et al., 2021 [28] | |
| CTPC | Paired BM and PB samples MM = 72 SMM = 25 MGUS = 150 HD = 71 | NGF | MM = 1.9 CTPC/μL (0.007–339.9) SMM = 0.16 CTPC/μL (0.005–12.9) MGUS = 0.008 CTPC/μL (˂0.001–9.8) p < 0.05 Correlation between BMPC and the absolute number of CTPC (rho = 0.78; p < 0.001). Cut-off discriminating MGUS and MM—CTPC/μL ≥ 0.058 (p < 0.001). | Sanoja-Flores et al., 2018 [29] |
| Type of Biopsy | Cohort (Sample) | Method | Findings | Reference |
|---|---|---|---|---|
| PB | MM = 10,750 | Cox regression model Survival tree model | ISS model composed by:
| Greipp et al., 2005 [35] |
| BM | MM = 3060 | K-adaptive partitioning Univariable and multivariable Cox regression model | R-ISS model composed by:
| Palumbo et al., 2015 [32] |
| MM = 7077 | Multivariable Cox regression model | R2-ISS model composed by:
| D’Agostino et al., 2020 [33] | |
| cfDNA | Paired BM and blood samples MM = 53 Training = 23 MM Validation = 17 MM | LBSeq WGS | 51 total mutations 49 (96.1%) BM-cfDNA mutations 2 (3.9%) BM-only mutations 5 (9.8%) cfDNA-only mutations >98% specificity | Kis et al., 2017 [25] |
| Paired BM and blood samples MM = 18 | Serum samples = ddPCR (NRAS, KRAS, BRAF) BM samples = WES + PCR | 35 total mutations 34 (97.1%) BM-cfDNA mutations 1 (2.9%) BM-only mutation 0 (0%) cfDNA-only mutation Correlation of mutations levels between BM and cfDNA (r = 0.507, n = 34, p < 0.002) Covariation between M protein level and ctDNA | Rustad et al., 2017 [36] | |
| Paired BM and blood samples MM = 48 [NDMM = 15 RRMM = 33] | OnTarget Mutation detection (OMD) | 128 total mutations 38 (29.7%) BM-cfDNA mutations 59 (46.1%) BM-only mutations 31 (24.2%) cfDNA -only mutations cfDNA-only mutations RRMM > NDMM (27.2% vs. 6.6%, p = 0.25 Chi-squared test) | Mithraprabhu et al., 2017 [26] | |
| MM = 22 (CR) Sequential cfDNA samples | NGS for IgH gene rearrangement MFC | Patients with ≥4.7% (n = 12) of IGH cfDNA had inferior PFS than patients with <4.7% (n = 10) (HR = 3.507, p = 0.04988, log- rank test) | Biancon et al., 2018 [37] | |
| Paired BM and cfDNA samples, MM = 9 Paired BM, cfDNA and CTPC samples, MM = 4 | ULP-WGS WES | 17% of BM-only mutations 12% of cfDNA-only mutations Tumor fraction in cfDNA correlate with disease stage (p < 0.001) and RISS (p = 0.032) 99% of BMPC mutations found in cfDNA or CTPC 94% of cfDNA or CTPC mutations found in BMPC | Manier et al., 2018 [38] | |
| Paired BM and blood samples EM = 8 (22) MM = 10 (23) | NGS and ddPCR | 16 total mutations 12 (66.7%) EM-cfDNA mutations 5 (31.2%) EM-BM mutations ctDNA better represents EM mutations than BM biopsies (ROC = 0.873 vs. 0.621) | Long et al., 2020 [27] | |
| cfDNA MM = 77 ctDNA MM = 17 | Ultra-Low Pass (ULP)-WGS | cfDNA > 25.2 ng/mL is related to shorter PFS (HR = 6.4) e OS (HR = 4.4) High ctDNA level correlates with high-risk GEP70 (p = 0.0027, Spearman r = 0.69) | Deshpande et al., 2021 [28] | |
| CTPC | MGUS = 325 | PC immunofluorescence | MGUS with CTPC (n = 63, 19%) were twice as likely to experience progression to plasma cell disorder (HR = 2.1, p < 0.03) | Kumar et al., 2005 [39] |
| RRMM = 42 | 6-color MFC | Shorter TTP (=51 days) and OS (=308 days) when CTPC have aberrant phentoype compared to other patients (TTP = 258 days, OS = 856 days; TTP = 581 days, OS = 1006 days; p < 0.001 and p = 0.007 for TTP and OS, respectively). | Peceliunas et al., 2012 [40] | |
| SMM = 91 | PC Immunofluorescence | High CTPC increase risk of progression within 2 year (14/91, 15% of patients; risk of progression: 71% versus 24%, respectively, p ≤ 0.001). High CTPC levels reduces OS (49 months versus 148 months; HR = 5.9, p < 0.001) | Bianchi et al., 2013 [41] | |
| NDMM = 157 | 6-color MFC (detection limite of 20/150,000 events) | CTPC ≥ 400/150,000 (n = 37, 24%) associated with adverse cytogenetics, shorter TTNT and OS (14 months and 32 months vs. 26 months and not reached, respectively, p < 0.001). | Gonsalves et al., 2014 [42] | |
| ACST MM = 840 | 6-color MFC | Shorter PFS and OS in patients with CTPC (15.1 months vs. 29.6 months and 41.0 months vs. not reached, respectively, p < 0.001). CTPC is a predictive factor of mortality (HR = 2.5, p = 0.001) and sCR post-transplant (HR = 0.4, p < 0.001). | Chakraborty et al., 2016 [43] | |
| Paired BM and PB samples MM = 29 (8) | FACS WES | 100% of clonal mutations in patient BM were detected in CTPC and that 99% of clonal mutations in CTPC were present in BM MM. | Mishima et al., 2017 [44] | |
| MM = 41 (104 PB; 29 BM) Clinical trial- EudraCT no. 2010-019173-16 | ASO-PCR (Detection limit ≤ 10− 6) | CTPC reduced by 97% after therapy induction and by 86% after ASCT. | Huhn et al., 2017 [45] | |
| SMM = 100 | 6-color MFC (Detection limit 20/150,000 events) | Patients with ≥150 CTPC (n = 9) with higher risk of progression to MM within 2 years (97% specificity and 78% sensitivity). TTP shorter for SMM ≥ 150 CTPC (9 months vs. not reached, p < 0.001). | Gonsalves et al., 2017 [46] | |
| NDMM = 247 | 6-color MFC (detection limit 10/150,000 events) | Less sCR for patients with CTPC (12% (n = 48) vs. 32% (n = 117) p = 0.018. Higher risk of mortality for patients with CTPC (HR = 5.7, p < 0.001) vs. patients without | Chakraborty et al., 2017 [47] | |
| Paired BM and PB samples MM = 72 (Solitary plasmacytoma = 17) SMM = 25 MGUS = 150 | NGF (Detection limit ≥ 106/tube) | R-ISS III patients have higher CTPC counts vs. R-ISS I and II (p = 0.001 and p = 0.004, respectively). Increased PFS and OS in patients with < 0.1 CTPC/μL (94% vs. 40%, p = 0.014; 100% vs. 67%, p = 0.03, respectively). Cut-off ≥ 0.058 CTPC/μL discriminates MGUS vs. MM. | Sanoja-Flores et at., 2018 [29] | |
| ASCT MM = 227 | 7-color NGF | Patients with CTPC (n = 27, 18.8%) have poorer PFS (p = 0.031) and higher risk of progression or death (43%, p = 0.04) when combined with high-risk cytogenetics and ISS. | Cowan et al., 2018 [48] | |
| Paired BM and PB samples MM = 53 (NDMM = 37; RRMM = 16) | NGF WES | First-time sequencing of triple-matched samples. CTPC detected in the PB of all patients (3.5 CTPC/µL, range: 0.115–1248). Detected 537/658 mutations (82%) in CTPC present in BM tumor cells. Detected 48/52 altered genes (92%) in CTPC also present in BM or EM tumor cells. | Garcés et al., 2020 [49] |
| Type of Biopsy | Cohort (Sample) | Method | Findings | Reference |
|---|---|---|---|---|
| BM | Start of maintenance therapy, MM = 224 After maintenance therapy, MM = 183 | Kaplan–Meier curves Log-rank test Cox regression model | PFS and OS significantly extended in MRD negative vs. MRD positive at start and after of maintenance therapy PFS: HR = 0.22/OS: HR = 0.24 OS: HR = 0.18/OS: HR = 0.26 PFS longer in patients with MRD < 10−6 vs. MRD between 10−6 and 10−5 (HR = 1.94) | Perrot et al., 2018 [51] |
| MM = (31) BM | Spearman’s test for paired data Log-rank test | Good correlation between NGF and NGS (rho = 0.62, p = 0.001). MRD negative patients by NGF presented extended PFS (p = 0.01) vs. patients with MRD positive. | Flores-Montero et al., 2017 [52] | |
| cfDNA | Early relapse, MM = 28 Late relapse, MM = 25 | QIAamp Circulating Nucleic Acid kit (Qiagen) | cfDNA level higher in late relapse than in early relapse (p = 0.016) | Kis et al., 2017 [25] |
| MM = 7 | ddPCR | in 3/7 patients ctDNA level coincided with serological changes of relapsein 4/7 patients, ctDNA level anticipated serological changes associated with relapse | Mithraprabhu et al., 2017 [26] | |
| Paired cfDNA and CTPC samples MM = 27 | NGS for IgH gene rearrangement | Association between cfDNA/CTPC levels and response status (p < 0.001). Better clearance of cfDNA than M protein in responder patients. | Oberle et al., 2017 [53] | |
| MM = 22 MRD negative = 6 | NGS for IgH gene rearrangement (5 × 105 reads) MFC | n = 6 patients with negative MRD (<5 cells/105) by MFC Correlation with IGH cfDNA (<10−5, r = 0.5831, p = 0.0044, Pearson’s correlation test). Longer PFS for patients with lower level of IGH cfDNA (p < 0.001) | Biancon et al., 2018 [37] | |
| Paired BM and blood samples MM = 37 MRD negative = 11 | NGS for IgH gene rearrangement | Negative predictive value (Specificity) = 36% (10/28) Positive predictive value (Sensitivity) = 89% (8/9) No quantitative correlation between ctDNA and BM mutations | Mazzotti et al., 2018 [54] | |
| Paired BM and blood samples MM = 12 MRD negative = 6 | ASO-qPCR for IgH gene rearrangement MFC (10−6) | Negative predictive value (Specificity) = 83.3% (5/6) Positive predictive value (Sensitivity) = 66.7% (4/6) More patients with low level ctDNA have reached CR vs. patients with high level of ctDNA. ctDNA better reflect MRD status than M-protein level | Vrabel et al., 2020 [55] | |
| CTPC | MM = 41 (29 BM;104 PB) EudraCT no. 2010-019173-16 | ASO-PCR detection limit: ≤10− 6 | MRD negative = 27 MRD positive = 14 Correlation between BMPC and CTPC in MRD positive (tau = 0.604; p = 0.003). | Huhn et al., 2017 [45] |
| NDMM = 458 PETHEMA/GEM2012MENOS65) | NGF detection limit ≤2.9 × 10−6 | MRD negative = 205 MRD positive = 61 MRD negative: 82% reduction in the risk of progression or death (HR = 0.18; p < 0.001), 88% reduction in the risk of death (HR = 0.12; p < 0.001). | Paiva et al., 2020 [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnault Carneiro, E.; Barahona, F.; Pestana, C.; João, C. Is Circulating DNA and Tumor Cells in Myeloma the Way Forward? Hemato 2022, 3, 63-81. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato3010006
Arnault Carneiro E, Barahona F, Pestana C, João C. Is Circulating DNA and Tumor Cells in Myeloma the Way Forward? Hemato. 2022; 3(1):63-81. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato3010006
Chicago/Turabian StyleArnault Carneiro, Emilie, Filipa Barahona, Carolina Pestana, and Cristina João. 2022. "Is Circulating DNA and Tumor Cells in Myeloma the Way Forward?" Hemato 3, no. 1: 63-81. https://0-doi-org.brum.beds.ac.uk/10.3390/hemato3010006