
Clinical Implementation of MicroRNAs in Cancer Immunology

Department of Gynecology, University Medical Center Hamburg-Eppendorf, 20246 Hamburg, Germany
Int. J. Transl. Med. 2024, 4(1), 53-71; https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm4010003
Submission received: 12 December 2023
/
Revised: 2 January 2024
/
Accepted: 3 January 2024
/
Published: 5 January 2024
Abstract
:MicroRNAs (miRNAs), or small non-coding RNAs, modulate the expression of mRNAs and, consequently, a variety of signal transduction pathways. Due to their dysregulation in cancer, they exert oncogenic pressure and have an impact on the immune system with their protective functions. These immunosuppressive characteristics of miRNAs in cancer promote cancer progression and metastasis, causing the dysregulation of immune cells and the immune escape of tumor cells. In contrast, there are also tumor suppressor miRNAs that are able to activate the immune system. Therefore, studies on the altered expression of miRNAs that consider both the oncogenic and tumor-suppressive aspects of miRNAs have become an important research field for advancing immunotherapeutic interventions using miRNAs or their inhibitors as therapeutics. In the current review, their potential in the immunomodulation of immune cells and their use as immune stimulatory molecules to elicit specific cytotoxic responses against the tumor are discussed.
1. Introduction
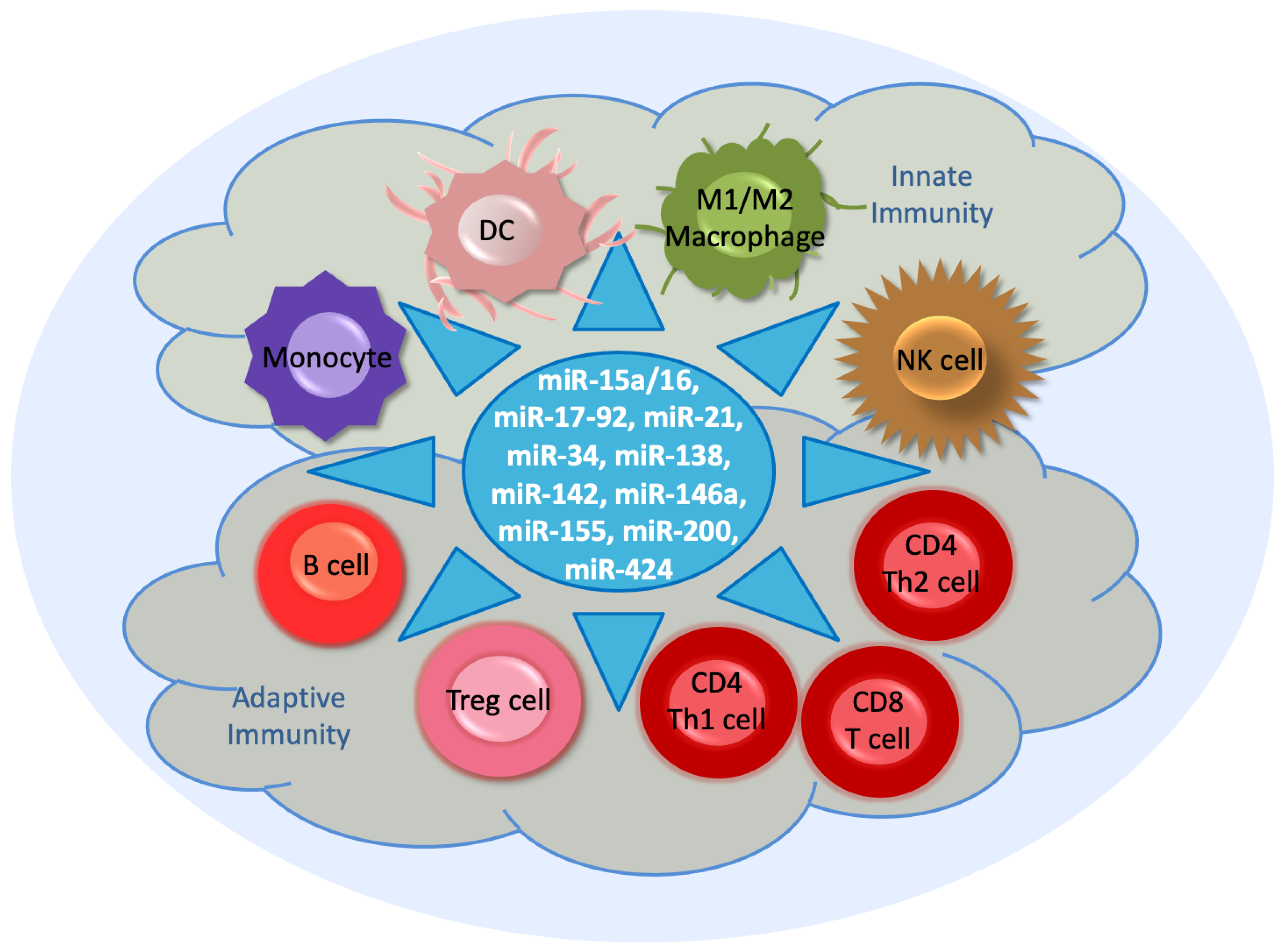
The immune system, which functions in host defense, is an interactive complex network consisting of lymphoid cells, humoral factors, and cytokines [1] (Figure 1). There are two arms of the immune system. One is the innate immune response, which occurs at a very early stage of tumor progression. It consists of phagocytic cells and specific proteins that recognize conserved features of microorganisms through the Toll-like receptor (TLR) on antigen-presenting cells (APCs), such as monocyte-derived dendritic cells (DCs), along with their presentation of histocompatibility complex (MHC) class I molecules [2]. In the innate immunity, natural killer (NK) cells, a type of cytotoxic lymphocytes, serve as the main effector cells toward cancer. They provide a rapid response to intracellular pathogens [3].
Myeloid cells, which comprise macrophages, DCs, monocytes, and granulocytes, represent a major component of the tumor microenvironment. They are derived from hematopoietic stem cells in the bone marrow. Their terminal differentiation is achieved in response to specific colony-stimulating factors (CSFs) with subsequent release into the circulation. During tumorigenesis, myeloid cells are promptly recruited in local tissues or the tumor microenvironment via diverse chemokines. Here, they are activated for phagocytosis and to secret inflammatory cytokines [4].
DCs initiate the adaptive or acquired immune response and determine tolerance. This arm of the immune system mainly involves interactions between B and T cells, which occur by the major MHC-II on B cells and the T cell receptor on antigen-specific T cells [1]. Activated CD4 T helper (Th1 and Th2) cells play a critical role in the activation of mature, antigen-presenting DCs. CD4 T cells provoke a long-lasting, effective immune response from cytotoxic CD8 T cells. An effective, secondary expansion of CD8 T cells occurs by the simultaneous interaction of DCs with CD4 and CD8 T cells [5].
Additionally, regulatory T (Treg) cells are involved in the immune system because they contribute to maintain immune system homeostasis and tolerance. Treg cells inhibit the activation and differentiation of CD4 and CD8 T cells, thereby inducing responses to autologous and tumor-expressed antigens [5]. Macrophages are a diverse group of myeloid immune cells, and they eliminate pathogens through phagocytosis. Based on macrophage polarization, macrophages are classified as M1 and M2 macrophages. M1 macrophages are typically activated by interferon-γ (IFNγ) or lipopolysaccharide. They produce proinflammatory cytokines and phagocytize microbes and initiate an immune response. In addition, they generate nitric oxide or reactive oxygen intermediates to protect against bacteria and viruses. M2 macrophages are activated by interleukins, including IL-4, IL-10, or IL-13. They are associated with wound healing and tissue repair [6] (Figure 1).
Within the tumor microenvironment, immune cells incline to target and kill cancer cells. However, the cancer cells may eventually escape from immune surveillance and even inhibit the cytotoxic function of the immune cells. The circumvention of immune response by tumor cells occurs through diverse mechanisms, such as the expression of PD-1 (programmed death-ligand 1), the secretion of immunosuppressive cytokines (e.g., IL-10 and TGF-β), the induction of distinct regulatory lymphoid or myeloid cells, or the loss of immunogenic tumor antigens presented in the context of the MHC complex [7].
In this respect, myeloid-derived suppressor cells (MDSCs), with their immunosuppressive characteristics, promote tumor immune escape. They represent a heterogenic subpopulation of myeloid cells that differ from their normal, healthy counterparts and originate from hematopoietic stem cells as a result of an altered myelopoiesis in response to pathogenic stimuli. They are, thus, pathologically activated myeloid cells, and they are not present in healthy individuals, but they appear in cancer. Since they tremendously expand during tumor progression, they play critical roles in tumor development, metastasis, and treatment resistance, and thus, they are eligible as potential therapeutic targets for cancer therapy. Although MDSCs are implicated in the suppression of different immune cells, their main targets are T cells. This inhibition occurs, among others, through the increased expression of PD-L1 in MDSCs to interact with PD-1 on T cells [8].
The ability of tumor cells to escape immunosurveillance and, thus, the attack from immune cells is currently considered as an essential hallmark of cancer, contributing to tumor progression and metastasis. Therefore, research has focused on immunotherapies, which are used to activate or boost the activation of the immune system to attack cancer cells. There are diverse therapy methods, such as checkpoint inhibitors, namely the blockade of PD-1/PD-L1, lymphocyte-promoting cytokines, engineered T cell receptor (TCR) T cells, agonistic antibodies against co-stimulatory receptors, and cancer vaccines [9]. In particular, the clinical efficiency of immune checkpoint blockades in several cancer types has been acknowledged by the awarding of the Nobel Prize in Physiology or Medicine in 2018 to two pioneers who deal with checkpoint inhibitor research, namely Tasuku Honjo and James Allison. In 1992, Dr. Honjo was the first to identify the programmed cell death-1 gene (PDCD1) [10] and its importance in regulating immune responses for cancer treatment [11], while Dr. Allison was the first to determine the anti-tumor efficacy of the cytotoxic T lymphocyte antigen-4 blockade for the treatment of melanoma [12]. So far, numerous immune checkpoint blockade-based therapies have been developed and approved by the Food and Drug Administration (FDA) for the treatment of a variety of cancer types [13]. Particularly, the investigations of PD-1/PD-L1 have provoked a lot of research interest. PD-L1 is highly expressed on APC under physiological conditions, and its presence prevents autoimmune reaction and provides self-tolerance. PD-1 is located on lymphocytes and blocks the lymphocyte action pathway by binding to PD-L1. This binding inhibits cytokine and proteolytic enzyme secretion mediated by cytotoxic T cells and prevents the destruction of cells that express PD-L1 [10,11].
Despite the advances of these immunotherapies, only a subset of patients usually experience positive clinical response, and many patients suffer from severe toxicity [9]. In this respect, novel approaches to manage cancer immunotherapy in a more controlled manner to reduce toxicities are required. Since microRNAs (miRNAs) participate in the regulation of immune cells, they may provide potential cancer immunotherapeutic approaches by using them as activators of the immune system or as targets. MiRNAs are small ca. 21-nucleotide-long non-coding RNAs (ncRNAs) involved in the posttranscriptional regulation of mRNA expression [14,15]. They are important regulators of immune responses, and they control interactions between immune and cancer cells in the tumor microenvironment. Increasing interest has focused on understanding the role of miRNAs in the regulation of anticancer immune responses to improve the efficacy of different cancer therapeutics [16]. Currently, numerous miRNAs are known to regulate the immune system in various cancer types. For example, more than 50 miRNAs are involved in the immune checkpoint: the PD-1/PD-L1 pathway [17].
In this review, an overview of the different pathways of an arbitrary selection of ten miRNAs in the immunoregulation in different cancer types and their potential roles as therapeutic or target molecules to activate the immune system are described.
2. Biogenesis and Function of miRNAs
MiRNAs are usually transcribed by RNA polymerase II to form primary miRNA (pri-miRNA) transcripts [18,19]. Likewise, similar to protein-coding mRNAs, the pri-miRNA undergoes typical splicing, capping, and polyadenylating processes [20]. The pri-miRNA is processed by the enzyme Drosha to form the 70–100-nucleotide-long stem-loop precursor RNA (pre-miRNA) in the nucleus [21]. Then, the pre-miRNA is exported from the nucleus into the cytoplasm, and its hairpin-like structure is further processed by Dicer, resulting in a duplex miRNA consisting of a single-stranded miRNA derived from the 5′ (5p) and 3′ (3p) regions of the pre-miRNA. One of these 21–23-nucleotide-long strands is loaded into the RNA-induced silencing complex (RISC) containing Argonaute protein to mediate target-specific gene silencing, while the other strand is usually degraded [22,23]. Argonaute proteins are nucleic acid-guided endonucleases that can cleave targets that are complementary to DNA or RNA guides, leading to translational silencing, directly or indirectly, by recruiting additional silencing proteins [24].
Besides this miRNA biogenesis, there is a number of alternative routes to generate miRNAs, including one in which mirtrons bypass the Drosha processing step, with the pre-miRNA being produced by a splicing reaction [25].
The modulation of gene expression by miRNAs occurs via the base-pairing of the miRNA sequence to its corresponding target mRNA, either in the 3’ untranslated region (UTR) or within the coding sequence of the mRNA. This pairing leads to either the translational repression or cleavage of the mRNA, resulting in reduced levels of the target protein and changes in the signaling pathways.
In most cases, miRNAs act as inhibitors of mRNA translation. However, in contrast to this mechanism, miRNAs can also activate translation [14]. The interaction of miRNAs with their mRNA targets is a dynamic process which depends on the subcellular location of miRNAs, the abundancy of miRNAs and target mRNAs, and the affinity of miRNA–mRNA interactions.
miRNAs are secreted into extracellular fluids, e.g., the circulating blood [26,27], and transported to target cells via extracellular vesicles, such as exosomes, or they bind to proteins, including Argonautes [28]. They are involved in a variety of biological processes and are therefore critical for normal development. The aberrant expression of miRNAs is associated with numerous benign and malignant diseases [29]. They may serve as potential biomarkers and signaling molecules for a variety of diseases [30].
However, the functions of miRNAs may also be modulated or abrogated by their interplay with long non-coding RNAs (lncRNAs) [31,32]. LncRNAs, with more than 200 nucleotides in size, are much longer than miRNAs, but they have similar characteristics. Like miRNAs, they are classified as tumor suppressor and oncogenic lncRNAs, are aberrantly expressed in diverse cancer types, and contribute to cancer development, progression, recurrence, and therapeutic resistance via direct or indirect interactions with their mRNAs. The indirect pathway involves the lncRNA–miRNA–mRNA axis. Conservatively, lncRNAs interact with miRNAs as “sponges” or competing endogenous RNAs (ceRNAs), diminishing the repression of mRNAs by miRNAs [33]. In addition to the interaction with miRNAs, lncRNAs can act as miRNA precursors [34]. A known example is the processing of the H19 lncRNA to generate miR-675 [35,36]. MiRNAs are also able of degrading lncRNAs by binding to them in the RISC complex. Finally, lncRNAs can also compete for the same mRNA target, masking the binding site for miRNAs [33].
To study the complex, regulatory network of ncRNAs, they can be screened by using the Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) programs [37].
3. Signaling Pathways
Mitogen-activated protein kinase (MAPK)/MEK/extracellular-signal-regulated kinase (ERK) signaling plays a crucial role in multiple cellular processes, including cell proliferation and survival. Upon the dimerization of receptor tyrosine kinases, the activation of RAS, which occurs via the GTP exchange, triggers the phosphorylation of downstream kinases MEK1/2, which then, in turn, activates ERK1/2. This signaling induces a cascade of substrates, such as cyclin D and CDK4/6, to promote the G1/S phase transition [38]. The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, which is involved in cell growth, apoptosis regulation, and glucose metabolism, is usually activated in many cancer types. Signaling from RAS results in the active catalysis of PI3K, leading to the activation of AKT, which, in turn, promotes multiple downstream signaling cascades, including mTOR. The phosphate and tensin homolog (PTEN) is a negative regulator of this pathway [39]. The inhibitors of both pathways have been shown to modulate the tumor microenvironment, increase immunogenicity, and thus potentially facilitate increased sensitivity to immunotherapy and enhance the response to immune checkpoint inhibitors [40].
The dysregulation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling is also associated with various cancer types. JAKs are associated with cytokine receptors and the mediate tyrosine phosphorylation of receptors, and they can recruit one or more members of the STAT family. Tyrosine-phosphorylated STATs dimerize and are then transported into the nucleus to regulate specific genes [41]. The signaling cross-talk of JAK/STAT involves the TGFβ signaling pathway. In T cells, the transforming growth factor (TGF)-β blocks IL-12-mediated JAK/STAT activation, resulting in decreased T cell proliferation and diminished IFNγ production [42]. Moreover, JAK inhibitors inhibit both the JAK/STAT and PI3K/mTOR pathways, suggesting an interconnection between them. In addition, PI3K negatively regulates STAT activity [43]. The cross-talk between the JAK/STAT signaling pathway components and the NF-κB signaling pathway components is versatile. NF-κB can induce the expression of a variety of inflammatory mediators and is a core transcription factor in various immune responses [44].
4. Immunoregulation by miRNAs
MiRNAs regulate diverse processes, such as differentiation activation and the effector functions of cells of innate and adaptive immunity, and they have essential effects on cancer progression. Variable miRNA expression levels have been detected in cancer cells and the tumor environment, and their up- and downregulation determines the extent of their impact on tumor progression [45]. In the following paragraphs, ten miRNAs (miR-15a/16, miR-17-92, miR-21, miR-34, miR-138, miR-142, miR-146a, miR-155, miR-200, and miR-424) that, according to the author, are important in the immune system, are introduced. Of course, there are other miRNAs that have a similar relevance, as shown in Table 1, but for a detailed description of their features, the selection was limited.
4.1. MiR-15a/16
Since their discovery in chronic lymphocytic leukemia, miR-15a and miR-16 have been described to act as tumor suppressors or potential oncomiRs in different cancer types. MiR-15a/16 is a member of a miRNA cluster that exhibits immune modulation [47]. For example, these miRNAs are downregulated in cancer-associated fibroblasts of prostate cancer patients. By binding to associated targets, they promote tumor expansion and invasiveness by their activity on stromal and cancer cells [48].
MiR-15a/16-deficient mice have high levels of CD4 T cells, which produce TGF-β1 and IL-10 and inhibit the IFN-γ production of CD8 T cells, which promote tumor evasion via the secretion of immune-suppressive molecules. MiR-16 was reported to modulate T cell functions via targeting the natural killer cell receptor, NKG2D [49].
In addition, miR-16 inversely correlates with PD-L1, PD-1, CD80, and cytotoxic-T-lymphocyte-associated antigen 4 (CTLA-4) expression. Mechanistic investigations revealed that miR-195 and miR-16 inhibit the PD-L1 immune checkpoint [50]. The downregulation of PD-L1 by miR-16 provokes macrophage polarization that shifts from M2 to M1 status, functionally activating CD4 T cells [51].
4.2. MiR-17-92 Cluster
The miR-17-92 cluster comprising miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a was first studied in malignant B cell lymphoma. It is involved in the development of the immune system and in the immune cells of both the innate and adaptive systems, including B cells; the subsets of T cells, such as Th1, Th2, T follicular helper cells, and Treg cells; as well as monocytes/macrophages; NK cells; and DCs [52]. The Th2-skewing tumor microenvironment was reported to induce the downregulation of miR-17-92 expression in T cells and, hence, decrease the persistence of tumor-specific T cells [53].
MiR-20a und miR-17-5p of this cluster, together with miR-106a, regulate monocyte differentiation and maturation by inhibiting the expression of the transcription factor, acute myeloid leukaemia-1 (AML1), to control the MAPK, TGF-β, and JAK-STAT signaling pathways. During monocytopoiesis, these three miRNAs are downregulated, which causes increased levels of AML1, which then, in turn, promotes the transcription of the M-CSF receptor (central regulator of monocytic–macrophage differentiation and maturation) and inhibits the expression of these miRNAs [54].
The miR-17–92 cluster is overexpressed in Th1 cells. Here, miR-17 and miR-19b control Th1 responses by promoting proliferation, reducing activation-induced cell death, enhancing IFN-γ production, and suppressing Treg cell differentiation. MiR-17 and miR-19b exert their regulatory function by binding TGFβ receptor II and cAMP response element binding protein 1 (CREB1) and PTEN, respectively [53,55].
The loss of miR-17-92 in CD4 T cells leads to tumor immune evasion by modulating the biological characteristics of Treg cells by acting on target genes, such as Forkhead box P3 (FOXP3) and CTLA-4 [56].
4.3. MiR-21
Mir-21 is one of the most abundant miRNAs in mammalian cells. The levels of miR-21 are elevated in diverse cancer types and promote cell proliferation, metastasis, and drug resistance. In the cells of the tumor immune infiltrate, and in particular, in macrophages, the expression of miR-21 seems to be responsible for promoting tumor growth [57]. These features indicate that targeting miR-21 combined with conventional chemotherapeutic agents could enhance their therapeutic efficacy and overcome drug resistance and cancer recurrence [58].
MiR-21 has been identified as the key regulator of PDCD4 by binding to its 3’-UTR to promote tumor proliferation, migration, and invasion in colon, breast, and bladder carcinomas [59]. MiR-21 also regulates CD8 T cell proliferation by targeting dual specificity phosphatase 10 (DUSP10) and B cell CLL/lymphoma 6 (BCL-6) [60]. As a classic oncogene, miR-21 attenuates classical macrophage activation (M1 polarization) by targeting the key adaptor molecules in the TLR/NF-κB pathway [61].
Additionally, the RNA-editing enzyme ADAR1 enhances Treg cell function via the modulation of the miR-21b/Foxp3 axis [62].
4.4. MiR-34
MiR-34 plays a crucial role in repressing tumor progression by participating in the epithelial–mesenchymal transition (EMT) via EMT transcription factors and p53 [63]. EMT is a process that leads to cancer invasion and metastasis, and inhibits polarized epithelial cells, making them lose their adhesion property to obtain mesenchymal cell phenotypes [64].
In head and neck cancers, miR-34a acts as a tumor suppressor and targets the proto-oncogene MET. In the tumor microenvironment, it provokes a decreased number of immunosuppressive PDL1-expressing tumor-associated macrophages. Higher levels of miR-34a are significantly associated with a higher frequency of Th1 cells and CD8 naïve T cells [65]. Moreover, tumor immune evasion is regulated by the p53/miR-34/PDL1 axis. P53 mutations are associated with lower levels of miR-34a. In lung cancer, p53 regulates PDL1 via miR-34, which directly binds to the PDL1 3’UTR. In addition, miR-34a delivery reduced the number of radiation-induced macrophages and T-reg cells [66].
MiR-34a and miR-34c act as repressors of the UL16 binding protein 2 (ULBP2), the stress-induced ligand of the natural killer cell receptor NKG2D. Both miRNAs directly target the 3’UTR of ULBP2 mRNA. Treatment with a small molecule inhibitor decreases the ULBP2 levels in a p53-dependent manner, which is caused by a p53-mediated increase in the cellular miR-34 levels [67].
NF-κB-driven miR-34a disrupts the equilibrium Treg/Th17 cell balance by directly targeting Foxp3 [68].
4.5. MiR-138
MiR-138 acts as a tumor suppressor by targeting numerous target genes, which are involved in cell proliferation, apoptosis, invasion, and migration and can sensitize tumors to chemotherapies [69].
MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. It suppresses the expressions of CTLA-4, PD-1, and FoxP3 in transfected human CD4 T cells. In vivo, the miR-138 treatment of immune-competent mice demonstrates marked tumor glioma regression and an associated decrease in intratumoral FoxP3 Treg cells, CTLA-4, and PD-1 expression [70]. Furthermore, in lung cancer, miR-138-5p is also found and targets PD-L1 to reduce the expression of Ki67 in tumor cells and decrease the tolerance effect on DCs [71]. In breast cancer, miRNA-138-5p also restrains T cell exhaustion via repressing PD-L-1, resulting in the disruption of the PD-L-1/PD-1 interaction and the modulation of effector cytokines in T cells [72].
The delivery of miR-138-5p from breast cancer cells to tumor-associated macrophages via exosomes that are able to shuttle with their cell-specific cell cargo between cells [28] downregulates the expression of the epigenetic factor lysine demethylase 6B (KDM6B) and inhibits M1 polarization, whereas it stimulates M2 polarization [73].
4.6. MiR-142
The miR-142 locus is preferentially expressed in hematopoietic cells. The inactivation of this locus in mice highlights its importance through the deeply disturbed immune system [74].
In an in vitro model of lung cancer, the overexpression of miR-142-5p suppresses PTEN protein expression and induces PI3K, p-Akt, and PD-L1 protein expressions. The repression of PD-L1 and the inhibition of PTEN reduce the cancer effects of CD4 T cells following miR-142-5p downregulation. Thus, miR-142-5p regulates CD4 T cells in lung cancer through PD-L1 expression via the PTEN pathway [75]. In addition, MiR-142-5p overexpression on tumor cells inhibits the expression of PD-L1 on tumor cells, resulting in increases in CD4 and CD8 T lymphocytes, a decrease in PD-1 T lymphocytes, and increases in IFN-γ and TNF-α [76].
MiR-142-3p downregulation promotes macrophage differentiation and determines the acquisition of their immunosuppressive function in tumors. It downregulates gp130, the common subunit of the IL-6 cytokine receptor family, by binding to its 3’UTR, and it represses C/EBPβ LAP* by binding to its 5’ mRNA coding sequence, impairing macrophage differentiation both in vitro and in vivo. Mice that constitutively express miR-142-3p in the bone marrow show a marked increase in survival following immunotherapy with tumor-specific T lymphocytes [77].
4.7. MiR-146a
MiR-146a acts as a tumor suppressor miRNA in some cancer types, whereas it acts as an oncogenic miRNA in others. Genetic and epigenetic events responsible for its repression cause the overexpression of its target genes, leading to increased cell proliferation, invasion, metastasis, and cell survival [79].
MiR-146a is upregulated by T cell receptor-mediated activation. It attenuates classical macrophage activation (M1 polarization) by the TLR/NF-κB pathway, and targets IL1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6). The downregulation of both of these targets leads to reduced TLR signaling and attenuated pro-inflammatory cytokine responses controlled by NF-κB [80].
In Treg cells, miR-146a is highly expressed and binds to the transcription factor STAT1, which is essential for the differentiation of Th1 effector cells. This targeting prevents the acquisition of Th1-like properties by Treg cells and enhances the effective suppressor function of Treg cells. On the other hand, increased STAT1 activation in Treg cells subjected to a selective removal of the suppressor of cytokine signaling 1 (SOCS1), a key negative regulator of SAT1 phosphorylation downstream of the IFNγ receptor, correlates with analogous Th1-mediated pathology [81].
4.8. MiR-155
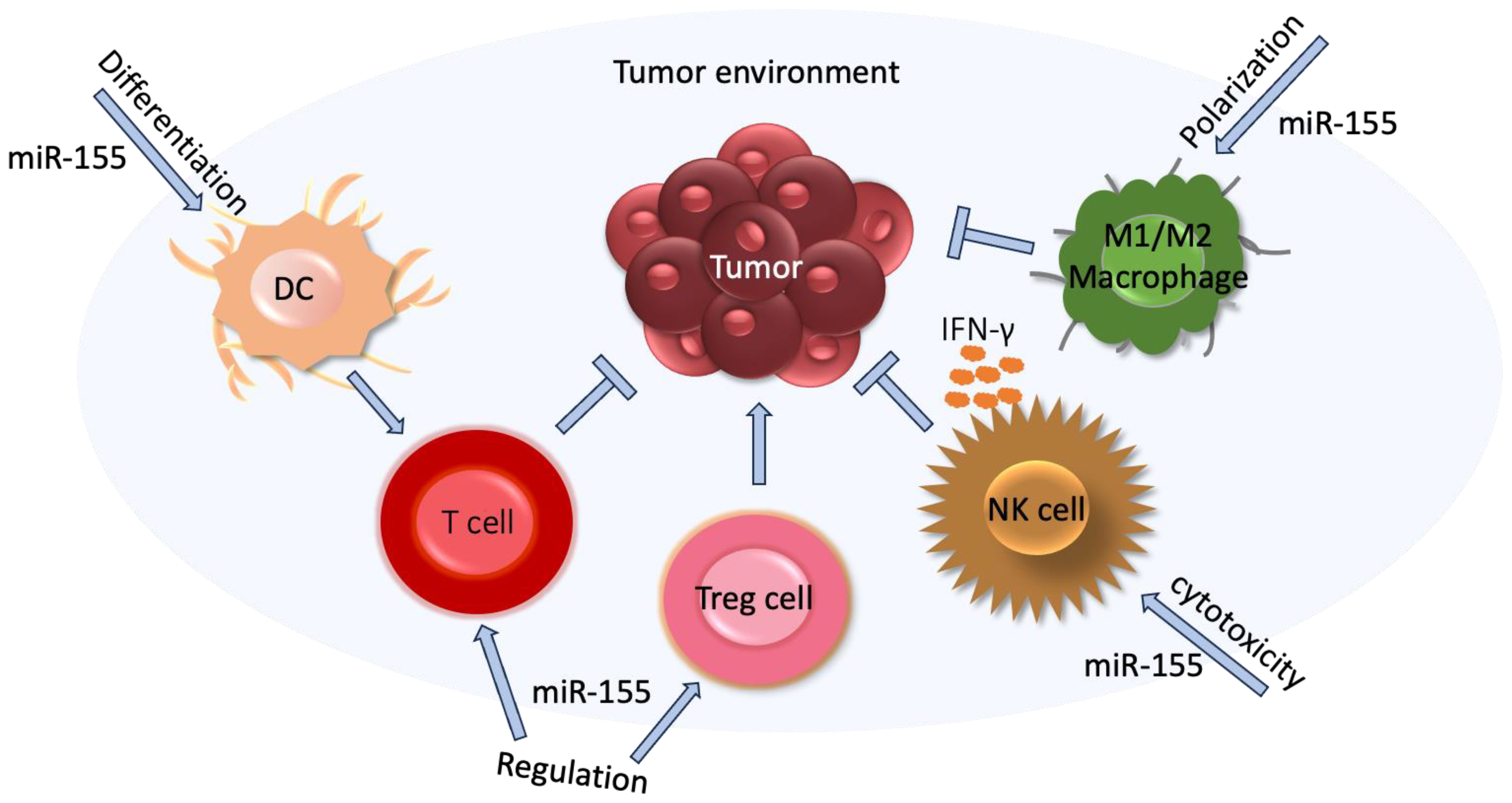
MiR-155 is an oncogenic miRNA. So far, nearly 200 mRNA targets of miR-155 have been identified from the literature and involved in numerous pathways, such as apoptosis, differentiation, angiogenesis, proliferation, EMT, and in the immune system [82,83]. Figure 2 presents an example of the impact of miR-155 on the tumor and tumor environment.
MiR-155 plays an important role in monocytic differentiation, a process of the maturation of emigrating monocytes into different classes of tissue macrophages that promote tumor progression, angiogenesis, and metastasis through regulating T cell function [84].
MiR-155 also plays a role in the regulation of effector functions of NK cells, increasing the synthesis of IFN-γ in activated NK cells by targeting hematopoietic cell-specific 5′ inositol phosphatase 32 (SHIP-1), a potent-negative regulator of NK-cell effector functions [85]
Moreover, miR-155 can regulate the polarization of CD4 T cells. The silencing of miR-155 promotes the differentiation of CD4 T cell towards Th2 cells by increasing the levels of its target c-Maf, a potent transactivator of the IL-4 promoter, leading to a higher production of Th2 cytokines IL-4, IL-5, and IL-10 [86]. In addition, miR-155 also plays a role in the regulation of the subpopulation of T cells, namely Treg cells which are characterized by the high expression of the transcription factor FOXP3, strong immunosuppressive activity and regulating homeostasis in health and disease. The binding of Foxp3 to the promoter region of the B cell integration cluster (BIC) gene results in processing miR-155 so that the deficiency of miR-155 impairs the development of Treg cells and reduces the numbers of thymic and splenic Treg cells [87].
Furthermore, miR-155 is upregulated by T cell receptor-mediated activation and controls the proliferation of CD8 T cells by targeting SOCS1, a negative regulator of IL-2 receptor signaling (which inhibits proinflammatory response), and the anti-proliferative effect of type I IFN signaling [88,89]. Low levels of miR-155, which are inversely associated with increased levels of SOCS1, decrease STAT5 signaling downstream of the IL-2 receptor. Consequently, they reduce Foxp3 synthesis and diminish the proliferative potential of Treg cells [90]. By binding to SOCS1, miR-155 can also activate macrophages [91].
4.9. MiR-200 Family
MiR-200 is a family of five molecules: miR-200a, miR-200b, miR-429, miR-200c, and miR-141 [92]. As suppressors of EMT and metastasis, they inhibit the expression of EMT-inducing transcription factors zinc finger E-box binding homeobox 1 and 2 genes (ZEB1 and ZEB2), leading to an increased expression of the cell–cell adhesion molecule, E-cadherin [64].
In lung cancer, PD-L1 expression was reported to be regulated by the miR-200/ZEB1 axis. PD-L1 expression is upregulated in non-metastatic epithelial cells during constitutive ZEB1 expression, while stable, inducible, or transient miR-200 expression (or ZEB1 silencing) suppresses PD-L1 expression in mesenchymal lung cancer cells [93]. The association of low expression levels of miR-200 with increased PD-L1 expression and the consequent suppression of CD8 T cell infiltration cause a strong proliferation and metastasis of tumor cells [94].
The miR-200 family is exclusively expressed in pancreatic adenocarcinoma cells. Interactions between cancer cells and the fibrotic tumor-associated stroma within the microenvironment modulate the miRNA expression and contributes to tumor progression [95].
4.10. MiR-424
So far, the downregulation of miR-424 has been reported in a number of cancer types. Cancer cells can also secrete miR-424 into the extracellular matrix via exosomes, which affects immune cells in the tumor environment. In contrast, its upregulation has also been described in other cancer types. The expression of this miRNA is regulated by the methylation status of its promoter. Additionally, there are some lncRNAs that act as molecular sponges for miR-424, thus binding to it and inhibiting its action [96].
In chemoresistant ovarian cancer, miR-424 regulates the PD-L1/PD-1 and CD80/CTLA-4 pathways. MiR-424 inversely correlates with PD-L1, PD-1, CD80, and CTLA-4 expression. Analyses have revealed that miR-424 inhibits PD-L1 and CD80 expression through its binding to their 3’-UTRs. The blockage of the PD-L1 immune checkpoint by miR-424 may reverse chemoresistance. This synergistic effect of chemotherapy and immunotherapy is accompanied with the proliferation of functional cytotoxic CD8 T cells and the inhibition of myeloid-derived suppressive cells and Treg cells [97].
As a tumor suppressor, miR-424 also regulates monocytic differentiation by controlling the MAPK, TGF-β, and JAK-STAT signaling pathways [84].
5. MiRNAs in Immunotherapies
In the tumor microenvironment, infiltrated Treg cells restrict protective immune surveillance, impede effective antitumor immune responses, and contribute to the formation of an immunosuppressive microenvironment [5]. The selective depletion of tumor-infiltrating Treg cells and the elicitation of effective T cell responses offer a potential approach for anti-tumor immunity. MiRNAs regulate both cancer and immune cells and, consequently, have the ability to potentially improve antitumor responses [56]. To date, the ability of nanoparticles or exosomes to deliver miRNAs have shown a great therapeutic potential to treat a variety of cancer types.
Presently, a variety of therapeutic approaches using miRNAs are being developed and are available [45,98].
5.1. MiR-15a/16
The efficacy of the miR-16 mimetic has been evaluated in a phase I clinical trial in patients with malignant pleural mesothelioma (NCT02369198). In this study, van Zandwijk et al. [99] engineered targomiRs, which are minicells loaded with miR-16-based mimic miRNA and targeted to EGF receptors that are designed to counteract the loss of the miR-15 and miR-16 family miRNAs. However, the acceptable safety profile and early signs of activity of targomiRs in the patients inspire additional studies on targomiRs in combination with chemotherapy or immune checkpoint inhibitors.
5.2. MiR-17-92 Cluster
T cells from tumor-bearing mice and glioma patients have lower levels of miR-17-92 than cells from their non-tumor-bearing counterparts. A disruption of the IL-4 signaling through either the IL-4 neutralizing antibody or the knockout of STAT6 reverses the miR-17-92 cluster suppression in Th2 cells. Okada et al. indicated that the Th2 cell-skewing tumor microenvironment can induce the downregulation of miR-17-92 expression in T cells, thereby diminishing the persistence of tumor-specific T cells and tumor destruction. Thus, the genetic engineering of T cells that express miR-17-92 may represent a promising approach for cancer immunotherapy [53].
5.3. MiR-21
In melanoma, miR-21-3p upregulation contributes to IFNγ-driven ferroptosis [100], a cell death, which is driven by iron-dependent phospholipid peroxidation and synergizes with the anti-PD-1 antibody. Guo et al. constructed miR-21-3p-loaded gold nanoparticles. In a mouse model, their systemic delivery increased the immunotherapy efficacy of the anti-PD-1 antibody without prominent side effects. Activating transcription factor 3 (ATF3) was found to promote miR-21-3p transcription in IFN-γ-driven ferroptosis [101].
Xi et al. [102] showed that in the presence of tumor cells, the genetic deficiency of miR-21 promotes the polarization of macrophages toward an M1-like phenotype in vivo and in vitro, and consequently, it confers host mice with enhanced anti-tumor immunity. By downregulating JAK2 and STAT1, miR-21 inhibits the IFNγ-induced STAT1 signaling pathway, which is required for macrophage M1 polarization. Moreover, miR-21 depletion in macrophages and PD-1 antibody treatment offers superior anti-tumor activity than either agent alone.
5.4. MiR-34
A miR-34a liposomal mimic, MRX34, is the first human miRNA-based drug that has been evaluated in clinical trials in patients with various cancer types [103]. Regrettably, this trial was terminated due to serious immune-mediated adverse events in cancer patients, suggesting the effect of this miRNA on several immune system regulatory genes. Nevertheless, miR-34a continues to be considered as a therapeutic target in cancer. For example, Meng et al. [104] showed that synthesized miR-34a simultaneously decreased the expression of PD-L1 as well as the checkpoint molecule, B7-H3, which inhibits tumor antigen-specific immune responses, and it demonstrated superior antitumor activity in colorectal cancer models in vitro and in vivo.
5.5. MiR-138
Song et al. [71] showed that miR-138-5p treatment decreases the proliferation of tumor cells and increases the number of tumor-infiltrated DCs. MiR-138-5p not only downregulates the expression of cyclin D3 (CCND3), CCD20, Ki67, and MCM in lung cancer cells, but also regulates the maturation of DCs in a mouse model and improves DCs’ capability to enhance T cells to kill tumor cells. Thus, miR-138-5p inhibits tumor growth and activates the immune system by downregulating PD-1/PD-L1.
In glioma models, Wei et al. [70] showed that miR-138 inhibits CTLA-4 and PD-1, leading to the significant regression of subcutaneous tumors.
5.6. MiR-142
As suggested by Khani et al. [105], the administration of exosomes containing miR-142 together with the miRNA Let-7i may be effective in immunotherapy against solid tumors. The administration of these exosomes elicits cytotoxic T cells with increasing IFNγ and Granzyme B production. Notably, their intramuscular injection has a significant effect on DC maturation and T cell activation along with tumor shrinkage.
5.7. MiR-146a
Recombinant human granulocyte/macrophage colony stimulating factor (GM-CSF) displays antitumor immunologic activity in prostate cancer and other cancer types. Triozzi et al. [106] observed a decrease in the serum levels of miR-146a in patients treated with GM-CSF. More DC activation and fewer myeloid-derived suppressors and Treg cells are found when administered at lower doses intermittently and continuously compared with when administered at higher doses daily and cyclically. Thus, the serum levels of miR-146a are potentially useful biomarkers of these effects.
5.8. MiR-155
MiR-155 may be an immunological clinical target for hematological malignancies. The miR-155 inhibitor, cobomarsen, has been examined and implemented well in the clinical trials NCT02580552, NCT03837457, and NCT03713320 using patients with hematologic malignancies in whom miR-155 is overexpressed, such as patients with cutaneous T cell lymphoma, mycosis fungoides, chronic lymphocytic leukemia, diffuse large B cell lymphoma, ABC subtype, and adult T cell leukemia/lymphoma [107].
5.9. MiR-200 Family
In a mouse model of colorectal cancer, the miR-200 members, which are inhibitors of PD-L1 [93], HIF-1α [108], and the VEGF pathway, have been analyzed [109]. In a study, Nguyen et al. [110] combined PD-L1 inhibition therapy and BRAF-targeted therapy using nanoparticles targeted with the C-X-C motif chemokine receptor 4 (CXCR4). This G-protein coupled receptor is usually upregulated on tumor cells [111]. The delivery of dabrafenib (Dab), a BRAF inhibitor, and miR-200c, a downregulator of PD-L1 expression by these nanoparticles, had significant efficacy on tumor cells. Both immunogenic cell death (ICD) and the inhibition of PD-L1 expression, induced by treatment with CXCR-4-targeted nanoparticles, improved the DC maturation in the lymph node, improved CD8 T cell activation in the spleen, and increased effector T cell infiltration into the tumor, whereas the immunosuppressive factors like PD-L1 expression and Treg cells were significantly reduced. These findings promote the immune responses against the tumor [110].
5.10. MiR-424
MiR-424 is a direct inhibitor of PD-L1 and CD80. In experiments in animal models of ovarian cancer carried out by Xu et al. [97], the restoration of miR-424 expression increased tumor sensitivity to chemotherapy. Thus, the administration of miR-424 leads to tumor regression and decreased tumor cell chemoresistance due to the activation of the T cell immune response.
6. Conclusions
In the present review article, a short overview on the role of ten miRNAs in the immune system and their potential clinical applications in immunotherapies was described. These ten miRNAs were arbitrarily selected due to their frequent descriptions and increasing profiles in the literature. In vivo experiments have repeatedly shown that these selected miRNAs affect significant tumor regression. However, there are numerous further studies on other miRNAs that are also promising as immunotherapeutic agents that were not mentioned because of the elaborateness of the article [11,12,61].
An important feature of miRNAs is their ability to bind to and affect the expression of several genes simultaneously and thus influence several signaling pathways. This broad impact indicates the possibility of using miRNAs in the future as an alternative to an immunotherapy or combined treatment. MiRNA-targeted therapies include the downregulation of oncogenic miRNAs by inhibitors and the upregulation of tumor-suppressive miRNAs by miRNA mimics. Likewise, lncRNAs may even be used as antagonists for oncogenic miRNAs. Restoring and blocking miRNA function may be carried out by the delivery of synthetic or viral vectors encoded for miRNA mimics, and by the antisense-mediated inhibition of oncogenic miRNAs, respectively. Moreover, the efficiency of synthetic miRNA mimics and inhibitors is limited by their potential degradation by RNases [112]. To increase their stability, they can be modified by either RNA phosphodiester or ribose sugar backbones [113]. Moreover, the specific interaction between lncRNA and miRNA should also be considered because it can change the efficiency of the synthetic miRNAs. Thus, it is important to define the lncRNA–miRNA–mRNA axis to determine the ability of miRNAs to target mRNAs [114]. Additionally, to inhibit the spreading of oncogenic miRNAs by exosomes, the expansion of exosomes can be inhibited by blocking their release by cancer cells, their migration and their uptake by other cells, as detailed and reviewed by Rashed et al. [115] (Figure 3).
However, the ability of miRNAs to inhibit several genes can also lead to adverse effects. Serious immune-mediated adverse effects were observed using miR-34 as a therapeutic agent in a clinical trial, suggesting the effect of this miRNA on several immune system regulatory genes, leading to the termination of this trial. To overcome such side effects in cancer patients, it is important to collect further data of the spectrum of miRNA targets in a particular cancer type. This includes additional studies on the miRNA/mRNA interaction landscapes and the search for an optimal miRNA mimic structure to allow for an increase in the efficiency and selectivity of interaction. The efficacy is also dependent on a transport system directed to the tumor cells. The ability of nanoparticles or exosomes, which are vesicles that are able to carry miRNAs and shuttle from cell to cell, to deliver miRNAs to the recipient cells have been reported to enhance miRNA effects by the site-directed delivery and eventually to reduce their side effects [110,116,117,118]. In addition to using miRNAs in clinical practice, techniques have to broaden to provide more specific binding to their mRNA targets.
Thus, to achieve their clinical application, several challenges have to be overcome. To achieve this, long-term monitoring platforms, in vivo systems, and multicenter studies are required.
In conclusion, future detailed studies will show whether the concept of using miRNAs will enter the clinic.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
A review article that summarizes the data from PubMed (https://pubmed.ncbi.nim.gov).
Conflicts of Interest
The author declares no conflict of interest.
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken-Ysla, L.; Tyurina, Y.Y.; Kagan, V.E.; Gabrilovich, D.I. Functional states of myeloid cells in cancer. Cancer Cell 2023, 41, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 459–477. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1979) 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, G.; Cava, C.; Castiglioni, I. Micrornas: New biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef] [PubMed]
- Kipkeeva, F.; Muzaffarova, T.; Korotaeva, A.; Mansorunov, D.; Apanovich, P.; Nikulin, M.; Malikhova, O.; Stilidi, I.; Karpukhin, A. The Features of Immune Checkpoint Gene Regulation by microRNA in Cancer. Int. J. Mol. Sci. 2022, 23, 9324. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Bortolin-Cavaille, M.L.; Dance, M.; Weber, M.; Cavaille, J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009, 37, 3464–3473. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science (1979) 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Slezak-Prochazka, I.; Durmus, S.; Kroesen, B.J.; van den Berg, A. MicroRNAs, macrocontrol: Regulation of miRNA processing. RNA 2010, 16, 1087–1095. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P. MicroRNA Shuttle from Cell-To-Cell by Exosomes and Its Impact in Cancer. Noncoding RNA 2019, 5, 28. [Google Scholar] [CrossRef]
- Schwarzenbach, H. CNAPS and General Medicine: Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring. An Introduction; Gahan, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 5, pp. 143–163. [Google Scholar]
- Berindan-Neagoe, I.; Monroig, P.d.C.; Pasculli, B.; Calin, G.A. MicroRNAome genome: A treasure for cancer diagnosis and therapy. CA Cancer J. Clin. 2014, 64, 311–336. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P.B. Interplay between LncRNAs and microRNAs in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 8095. [Google Scholar] [CrossRef]
- Müller, V.; Oliveira-Ferrer, L.; Steinbach, B.; Pantel, K.; Schwarzenbach, H. Interplay of lncRNA H19/miR-675 and lncRNA NEAT1/miR-204 in breast cancer. Mol. Oncol. 2019, 13, 1137–1149. [Google Scholar] [CrossRef]
- Venkatesh, J.; Wasson, M.C.D.; Brown, J.M.; Fernando, W.; Marcato, P. LncRNA-miRNA axes in breast cancer: Novel points of interaction for strategic attack. Cancer Lett. 2021, 509, 81–88. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Schwarzenbach, H. Biological and Clinical Relevance of H19 in Colorectal Cancer Patients. EBioMedicine 2016, 13, 9–10. [Google Scholar] [CrossRef]
- Vennin, C.; Spruyt, N.; Dahmani, F.; Julien, S.; Bertucci, F.; Finetti, P.; Chassat, T.; Bourette, R.P.; Le Bourhis, X.; Adriaenssens, E. H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget 2015, 6, 29209–29223. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Bright, J.J.; Sriram, S. TGF-β Inhibits IL-12-Induced Activation of Jak-STAT Pathway in T Lymphocytes. J. Immunol. 1998, 161, 1772–1777. [Google Scholar] [CrossRef]
- Tasian, S.K.; Doral, M.Y.; Borowitz, M.J.; Wood, B.L.; Chen, I.M.; Harvey, R.C.; Gastier-Foster, J.M.; Willman, C.L.; Hunger, S.P.; Mullighan, C.G.; et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012, 120, 833–842. [Google Scholar] [CrossRef]
- Karin, M.; Baud, V. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar]
- Xing, Y.; Wang, Z.; Lu, Z.; Xia, J.; Xie, Z.; Jiao, M.; Liu, R.; Chu, Y. MicroRNAs: Immune modulators in cancer immunotherapy. Immunother. Adv. 2021, 1, ltab006. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The role of cancer-derived microRNAs in cancer immune escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef]
- Huang, E.; Liu, R.; Chu, Y. MiRNA-15a/16: As tumor suppressors and more. Future Oncol. 2015, 11, 2351–2363. [Google Scholar] [CrossRef]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Saccá, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D.; et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef]
- Sullivan, R.P.; Leong, J.W.; Schneider, S.E.; Keppel, C.R.; Germino, E.; French, A.R.; Fehniger, T.A. MicroRNA-Deficient Murine NK Cells Exhibit Impaired Development and Survival but Enhanced IFN-γ Production In Vitro and In Vivo. Blood 2011, 118, 357. [Google Scholar] [CrossRef]
- Tao, Z.; Xu, S.; Ruan, H.; Wang, T.; Song, W.; Qian, L.; Chen, K. MiR-195/-16 Family Enhances Radiotherapy via T Cell Activation in the Tumor Microenvironment by Blocking the PD-L1 Immune Checkpoint. Cell. Physiol. Biochem. 2018, 48, 801–814. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Shen, Y.; Miao, J.; Liu, H.; Li, G.; Wang, Z. MiR-16 regulates mouse peritoneal macrophage polarization and affects T-cell activation. J. Cell. Mol. Med. 2016, 20, 1898–1907. [Google Scholar] [CrossRef]
- Kuo, G.; Wu, C.Y.; Yang, H.Y. MiR-17-92 cluster and immunity. J. Formos. Med. Assoc. 2019, 118, 2–6. [Google Scholar] [CrossRef]
- Sasaki, K.; Kohanbash, G.; Hoji, A.; Ueda, R.; McDonald, H.A.; Reinhart, T.A.; Martinson, J.; Lotze, M.T.; Marincola, F.M.; Wang, E.; et al. MiR-17-92 expression in differentiated T cells—Implications for cancer immunotherapy. J. Transl. Med. 2010, 8, 8–17. [Google Scholar] [CrossRef]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell Biol. 2007, 9, 775–785. [Google Scholar] [CrossRef]
- Jiang, S.; Li, C.; Olive, V.; Lykken, E.; Feng, F.; Sevilla, J.; Wan, Y.; He, L.; Li, Q.J. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 2011, 118, 5487–5497. [Google Scholar] [CrossRef]
- Fayyad-Kazan, H.; Rouas, R.; Fayyad-Kazan, M.; Badran, R.; El Zein, N.; Lewalle, P.; Najar, M.; Hamade, E.; Jebbawi, F.; Merimi, M.; et al. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J. Biol. Chem. 2012, 287, 9910–9922. [Google Scholar] [CrossRef]
- Sahraei, M.; Chaube, B.; Liu, Y.; Sun, J.; Kaplan, A.; Price, N.L.; Ding, W.; Oyaghire, S.; García-Milan, R.; Mehta, S.; et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J. Clin. Investig. 2019, 129, 5518–5536. [Google Scholar] [CrossRef]
- Arghiani, N.; Matin, M.M. MiR-21: A Key Small Molecule with Great Effects in Combination Cancer Therapy. Nucleic Acid Ther. 2021, 31, 271–283. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, N.; Hao, F.; Song, Y.; Wang, Z.; Ni, Y.; Ding, L. The Regulatory Role of Non-coding RNAs on Programmed Cell Death Four in Inflammation and Cancer. Front. Oncol. 2019, 9, 919. [Google Scholar] [CrossRef]
- Chang, C.C.; Zhang, Q.Y.; Liu, Z.; Clynes, R.A.; Suciu-Foca, N.; Vlad, G. Downregulation of Inflammatory MicroRNAs by Ig-like Transcript 3 Is Essential for the Differentiation of Human CD8+ T Suppressor Cells. J. Immunol. 2012, 188, 3042–3052. [Google Scholar] [CrossRef]
- Iurca, I.; Tirpe, A.; Zimta, A.A.; Moldovan, C.; Gulei, D.; Slabý, O.; Condorelli, G.; Berindan-Neagoe, I. Macrophages Interaction and MicroRNA Interplay in the Modulation of Cancer Development and Metastasis. Front. Immunol. 2020, 11, 870. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, X.; Li, M.; Zhao, J.; Wang, X.; Zhu, H. ADAR1 improved Treg cell function through the miR-21b/Foxp3 axis and inhibits the progression of acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Int. Immunopharmacol. 2023, 115, 109620. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2022, 211, 157–182. [Google Scholar] [CrossRef]
- Wu, X.; Cheng, Y.S.L.; Matthen, M.; Yoon, A.; Schwartz, G.K.; Bala, S.; Taylor, A.M.; Momen-Heravi, F. Down-regulation of the tumor suppressor miR-34a contributes to head and neck cancer by up-regulating the MET oncogene and modulating tumor immune evasion. J. Exp. Clin. Cancer Res. 2021, 40, 70. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef]
- Xie, M.; Wang, J.; Gong, W.; Xu, H.; Pan, X.; Chen, Y.; Ru, S.; Wang, H.; Chen, X.; Zhao, Y.; et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J. Autoimmun. 2019, 102, 96–113. [Google Scholar] [CrossRef]
- Sha, H.H.; Wang, D.D.; Chen, D.; Liu, S.W.; Wang, Z.; Yan, D.L.; Dong, S.C.; Feng, J.F. MiR-138: A promising therapeutic target for cancer. Tumor Biol. 2017, 39, 1010428317697575. [Google Scholar] [CrossRef]
- Wei, J.; Nduom, E.K.; Kong, L.Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qia, W.; Zhou, S.; et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016, 18, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, P.; Song, P.; Li, Y.; Zhou, S.; Su, Q.; Li, X.; Yu, Y.; Li, P.; Feng, M.; et al. MicroRNA-138-5p Suppresses Non-small Cell Lung Cancer Cells by Targeting PD-L1/PD-1 to Regulate Tumor Microenvironment. Front. Cell. Dev. Biol. 2020, 8, 540. [Google Scholar]
- Rasoolnezhad, M.; Safaralizadeh, R.; Hosseinpourfeizi, M.A.; Banan-Khojasteh, S.M.; Baradaran, B. MiRNA-138–5p: A strong tumor suppressor targeting PD-L-1 inhibits proliferation and motility of breast cancer cells and induces apoptosis. Eur. J. Pharmacol. 2021, 896, 173933. [Google Scholar] [CrossRef]
- Xun, J.; Du, L.; Gao, R.; Shen, L.; Wang, D.; Kang, L.; Chen, C.; Zhang, Z.; Zhang, Y.; Yue, S.; et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-Associated macrophages through inhibition of KDM6B. Theranostics 2021, 11, 6847–6859. [Google Scholar] [CrossRef]
- Olson, W.J.; Derudder, E. The miR-142 miRNAs: Shaping the naïve immune system. Immunol. Lett. 2023, 261, 37–46. [Google Scholar] [CrossRef]
- Wan, J.; Ling, X.; Peng, B.; Ding, G. MiR-142-5p regulates CD4+ T cells in human non-small cell lung cancer through PD-L1 expression via the PTEN pathway. Oncol. Rep. 2018, 40, 272–282. [Google Scholar] [CrossRef]
- Jia, L.; Xi, Q.; Wang, H.; Zhang, Z.; Liu, H.; Cheng, Y.; Guo, X.; Zhang, J.; Zhang, Q.; Zhang, L.; et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem. Biophys. Res. Commun. 2017, 488, 425–431. [Google Scholar] [CrossRef]
- Sonda, N.; Simonato, F.; Peranzoni, E.; Calì, B.; Bortoluzzi, S.; Bisognin, A.; Wang, E.; Marincola, F.M.; Naldini, L.; Gentner, B.; et al. MiR-142-3p Prevents Macrophage Differentiation during Cancer-Induced Myelopoiesis. Immunity 2013, 38, 1236–1249. [Google Scholar] [CrossRef]
- Xu, S.; Wei, J.; Wang, F.; Kong, L.Y.; Ling, X.Y.; Nduom, E.; Gabrusiewicz, K.; Doucette, T.; Yang, Y.; Yaghi, N.K.; et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J. Natl. Cancer Inst. 2014, 106, dju162. [Google Scholar] [CrossRef]
- Iacona, J.R.; Lutz, C.S. miR-146a-5p: Expression, regulation, and functions in cancer. Wiley Interdiscip. Rev. RNA 2019, 10, e1533. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1236–1243. [Google Scholar] [CrossRef]
- Vigorito, E.; Kohlhaas, S.; Lu, D.; Leyland, R. miR-155: An ancient regulator of the immune system. Immunol. Rev. 2013, 253, 146–157. [Google Scholar] [CrossRef]
- Schmeier, S.; MacPherson, C.R.; Essack, M.; Kaur, M.; Schaefer, U.; Suzuki, H.; Hayashizaki, Y.; Bajic, V.B. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genom. 2009, 10, 595. [Google Scholar] [CrossRef]
- Trotta, R.; Chen, L.; Ciarlariello, D.; Josyula, S.; Mao, C.; Costinean, S.; Yu, L.; Butchar, J.B.; Tridandapani, S.; Croce, C.M.; et al. miR-155 regulates IFN-γ production in natural killer cells. Blood 2012, 119, 3478–3485. [Google Scholar] [CrossRef]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science (1979) 2007, 316, 608–611. [Google Scholar] [CrossRef]
- Kohlhaas, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting Edge: The Foxp3 Target miR-155 Contributes to the Development of Regulatory T Cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector cd8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef]
- Lu, L.F.; Thai, T.H.; Calado, D.P.; Chaudhry, A.; Kubo, M.; Tanaka, K.; Loeb, G.B.; Lee, H.; Yoshimura, A.; Rajewsky, K.; et al. Foxp3-Dependent MicroRNA155 Confers Competitive Fitness to Regulatory T Cells by Targeting SOCS1 Protein. Immunity 2009, 30, 80–91. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Etzrodt, M.; De Palma, M.; Pittet, M.J. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 2013, 34, 350–359. [Google Scholar] [CrossRef]
- Zhang, H.F.; Xu, L.Y.; Li, E.M. A Family of Pleiotropically Acting MicroRNAs in Cancer Progression, miR-200: Potential Cancer Therapeutic Targets. Curr. Pharm. Des. 2014, 20, 1896–1903. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Chen, L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Lin, W.; Diao, L.; Wang, J.; Roybal, J.; et al. Regulation of tumor cell PD-L1 expression by microRNA-200 and control of lung cancer metastasis. J. Clin. Oncol. 2014, 32 (Suppl. S15), 5s. [Google Scholar] [CrossRef]
- Han, S.; Gonzalo, D.H.; Feely, M.; Delitto, D.; Behrns, K.E.; Beveridge, M.; Zhang, D.Y.; Thomas, R.; Trevino, J.G.; Schmittgen, T.D.; et al. The pancreatic tumor microenvironment drives changes in miRNA expression that promote cytokine production and inhibit migration by the tumor associated stroma. Oncotarget 2017, 8, 54054–54067. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Askari, A.; Hussen, B.M.; Taheri, M.; Akbari Dilmaghani, N. Role of miR-424 in the carcinogenesis. Clin. Transl. Oncol. 2024, 26, 16–38. [Google Scholar] [CrossRef]
- Xu, S.; Tao, Z.; Hai, B.; Liang, H.; Shi, Y.; Wang, T.; Song, W.; Chen, Y.; QuYang, J.; Chen, J.; et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 2016, 7, 11406. [Google Scholar] [CrossRef]
- Cortez, M.A.; Anfossi, S.; Ramapriyan, R.; Menon, H.; Atalar, S.C.; Aliru, M.; Welsh, J.; Calin, G.A. Role of miRNAs in immune responses and immunotherapy in cancer. Genes Chromosomes Cancer 2019, 58, 244–253. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Guo, W.; Wu, Z.; Chen, J.; Guo, S.; You, W.; Wang, S.; Ma, J.; Wang, H.; Wang, X.; Wang, H.; et al. Nanoparticle delivery of miR-21-3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J. Immunother. Cancer 2022, 10, e004381. [Google Scholar] [CrossRef]
- Xi, J.; Huang, Q.; Wang, L.; Ma, X.; Deng, Q.; Kumar, M.; Zhou, Z.; Li, L.; Zeng, Z.; Young, K.H.; et al. MiR-21 depletion in macrophages promotes tumoricidal polarization and enhances PD-1 immunotherapy. Oncogene 2018, 37, 3151–3165. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Chen, Y.; Yang, M.; Zhang, H.; Wang, W. Concomitant inhibition of B7-H3 and PD-L1 expression by a novel and synthetic microRNA delivers potent antitumor activities in colorectal tumor models. Investig. New Drugs 2021, 39, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Khani, A.T.; Sharifzad, F.; Mardpour, S.; Hassan, Z.M.; Ebrahimi, M. Tumor extracellular vesicles loaded with exogenous Let-7i and miR-142 can modulate both immune response and tumor microenvironment to initiate a powerful anti-tumor response. Cancer Lett. 2021, 501, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Achberger, S.; Aldrich, W.; Elson, P.; Garcia, J.; Dreicer, R. Differential immunologic and microRNA effects of 2 dosing regimens of recombinant human granulocyte/macrophage colony stimulating factor. J. Immunother. 2012, 35, 587–594. [Google Scholar] [CrossRef]
- Witten, L.; Slack, F.J. MiR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Byun, Y.; Choi, Y.C.; Jeong, Y.; Lee, G.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, K. MiR-200c downregulates HIF-1α and inhibits migration of lung cancer cells. Cell Mol. Biol. Lett. 2019, 24, 28. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, S.; Wu, H.; Zhang, L.; Dai, X.; Hu, J.; Xue, J.; Liu, T.; Liang, Y.; Wu, G. MiR-200c increases the radiosensitivity of non-small-cell lung cancer cell line A549 by targeting VEGF-VEGFR2 pathway. PLoS ONE 2013, 8, e78344. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Phung, C.D.; Tran, T.H.; Pham, T.T.; Pham, L.M.; Nguyen, T.T.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; et al. Manipulating immune system using nanoparticles for an effective cancer treatment: Combination of targeted therapy and checkpoint blockage miRNA. J. Control. Release 2021, 329, 524–537. [Google Scholar] [CrossRef]
- Buck, A.K.; Serfling, S.E.; Lindner, T.; Hänscheid, H.; Schirbel, A.; Hahner, S.; Fassnacht, M.; Einsele, H.; Werner, R.A. CXCR4-targeted theranostics in oncology. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4133–4144. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From garbage bins to promising therapeutic targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Tenchov, R.; Sasso, J.M.; Wang, X.; Liaw, W.S.; Chen, C.A.; Zhou, Q.A. Exosomes Nature’s Lipid Nanoparticles, a Rising Star in Drug Delivery and Diagnostics. ACS Nano 2022, 16, 17802–17846. [Google Scholar] [CrossRef]
- Xie, J.; Shen, Q.; Huang, K.; Zheng, T.; Cheng, L.; Zhang, Z.; Yu, Y.; Liao, G.; Wang, X.; Li, C. Oriented Assembly of Cell-Mimicking Nanoparticles via a Molecular Affinity Strategy for Targeted Drug Delivery. ACS Nano 2019, 13, 5268–5277. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A Novel Nanoparticle Drug Delivery System: The Anti-inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
Figure 1.
Schema of the innate and adaptive immune system. In the middle, the circle contains ten selected miRNAs, as described in the text. They influence the actions of the immune cells that are involved in the innate and adaptive immune response and surround the circle. DC, monocyte-derived dendritic cells; NK, natural killer; Th, T helper; Treg, regulatory T.
Figure 1.
Schema of the innate and adaptive immune system. In the middle, the circle contains ten selected miRNAs, as described in the text. They influence the actions of the immune cells that are involved in the innate and adaptive immune response and surround the circle. DC, monocyte-derived dendritic cells; NK, natural killer; Th, T helper; Treg, regulatory T.

Figure 2.
The role of miR-155 in cancer immune escape. As described in the text above, miR-155, with its oncogenic potential, regulates immune cells in tumor environment and consequently promotes tumor progression. DC, monocyte-derived dendritic cells; NK, natural killer; Treg, regulatory T.
Figure 2.
The role of miR-155 in cancer immune escape. As described in the text above, miR-155, with its oncogenic potential, regulates immune cells in tumor environment and consequently promotes tumor progression. DC, monocyte-derived dendritic cells; NK, natural killer; Treg, regulatory T.

Figure 3.
MiRNA target therapies. As described in the text and shown in the boxes, there are different possibilities to use miRNAs as therapeutic targets or to deliver tumor-suppressive miRNAs to target cells. As also shown, tumor cells release exosomes and the interaction of an immune cell with tumor cells.
Figure 3.
MiRNA target therapies. As described in the text and shown in the boxes, there are different possibilities to use miRNAs as therapeutic targets or to deliver tumor-suppressive miRNAs to target cells. As also shown, tumor cells release exosomes and the interaction of an immune cell with tumor cells.


{kind=link}
{kind=link}
{kind=link}
Table 1.
A selection of other miRNAs that play roles in immunoregulation, as reviewed in [46].
Table 1.
A selection of other miRNAs that play roles in immunoregulation, as reviewed in [46].
| miRNAs | Targets |
|---|---|
| miR-10a | MDSCs |
| miR-18a | PD-L1 |
| miR-20a | MHC-I chain-related molecules A/B, T cells |
| miR-24 | T cells |
| miR-27a | Fibroblasts |
| miR-29a | MDSCs, MHC-I chain-related molecules B |
| miR-93 | MHC-I chain-related molecules A, PD-L1 |
| miR-106a | MHC-I chain-related molecules A/B, T cells |
| miR-106b | PD-L1 |
| miR-107 | MHC-I chain-related molecules B, MDSCs |
| miR-125b | MHC-I chain-related molecules A, macrophages |
| miR-140 | PD-L1 |
| miR-148a | PD-L1 |
| miR-153 | MHC-I chain-related molecules A |
| miR-181d | Macrophages |
| miR-193a | PD-L1 |
| miR-195 | MHC-I chain-related molecules B, PD-L1 |
| miR-197 | PD-L1 |
| miR-222 | Macrophages |
| miR-302c | MHC-I chain-related molecules A/B |
| miR-340 | PD-L1 |
| miR-373 | MHC-I chain-related molecules A/B |
| miR-375 | PD-L1 |
| miR-383 | PD-L1 |
| miR-497 | PD-L1 |
| miR-519a | MHC-I chain-related molecules A |
| miR-520c | MHC-I chain-related molecules A/B |
| miR-690 | T cells |
| miR-873 | PD-L1 |
| miR-891a | T cells |
| miR-940 | Macrophages |
| miR-1246 | Macrophages |
| miR-1247 | Fibroblasts |
| miR-1908 | T cells |
| miR-3127 | PD-L1 |
| miR-3609 | PD-L1 |
MDSCs, myeloid-derived suppressor cells; MHC-I, major histocompatibility complex class I; PD-L1, programmed death-ligand 1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schwarzenbach, H. Clinical Implementation of MicroRNAs in Cancer Immunology. Int. J. Transl. Med. 2024, 4, 53-71. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm4010003
AMA Style
Schwarzenbach H. Clinical Implementation of MicroRNAs in Cancer Immunology. International Journal of Translational Medicine. 2024; 4(1):53-71. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm4010003
Chicago/Turabian StyleSchwarzenbach, Heidi. 2024. "Clinical Implementation of MicroRNAs in Cancer Immunology" International Journal of Translational Medicine 4, no. 1: 53-71. https://0-doi-org.brum.beds.ac.uk/10.3390/ijtm4010003