Modelling and Recognition of Protein Contact Networks by Multiple Kernel Learning and Dissimilarity Representations


Abstract
:1. Introduction
- Feature generation and/or feature engineering, where numerical features are extracted ad-hoc from structured patterns (e.g., using their properties or via measurements) and can be further merged according to different strategies (e.g., in a multi-modal way [11]);
- Ad-hoc dissimilarities in the input space, where custom dissimilarity measures are designed in order to process structured patterns directly in the input domain without moving towards Euclidean (or metric) spaces. Common—possibly parametric—edit distances include the Levenshtein distance [12] for sequence domains and graph edit distances [13] for graphs domains;
- By analysing the kernel weights, one can determine the most suitable representation(s) for the problem at hand;
- The patterns elected as representatives for the dissimilarity space (hence determined as pivotal for tracking the decision boundary amongst the problem-related classes) can give some further insights for the problem at hand.
2. Theoretical Background
- They must well-characterize the decision boundary between patterns in the input space;
3. Proposed Methodology
- The individual receives the full dissimilarity matrices between training data samples, i.e., as in Equation (12);
- Considering the and values in its genetic code, the (multiple) kernel matrix is evaluated by using Equation (14);
- A -SVM is trained using the regularisation term from the genetic code and the kernel matrix from step #3;
- The individual receives the full dissimilarity matrices between training and validation data, each of which is computed by considering all possible -pairs where x belongs to the validation set and y belongs to the training set, i.e., as in Equation (12);
- The ‘reduced’ dissimilarity matrices are projected thanks to , i.e., as in Equation (13);
- The (multiple) kernel matrix between training and validation data is evaluated thanks to and , alike Equation (14);
- The (multiple) kernel matrix from step #7 is fed to the SVM trained on step #4 and the predicted classes on the validation set are returned;
- The fitness function is evaluated.
4. Tests and Results
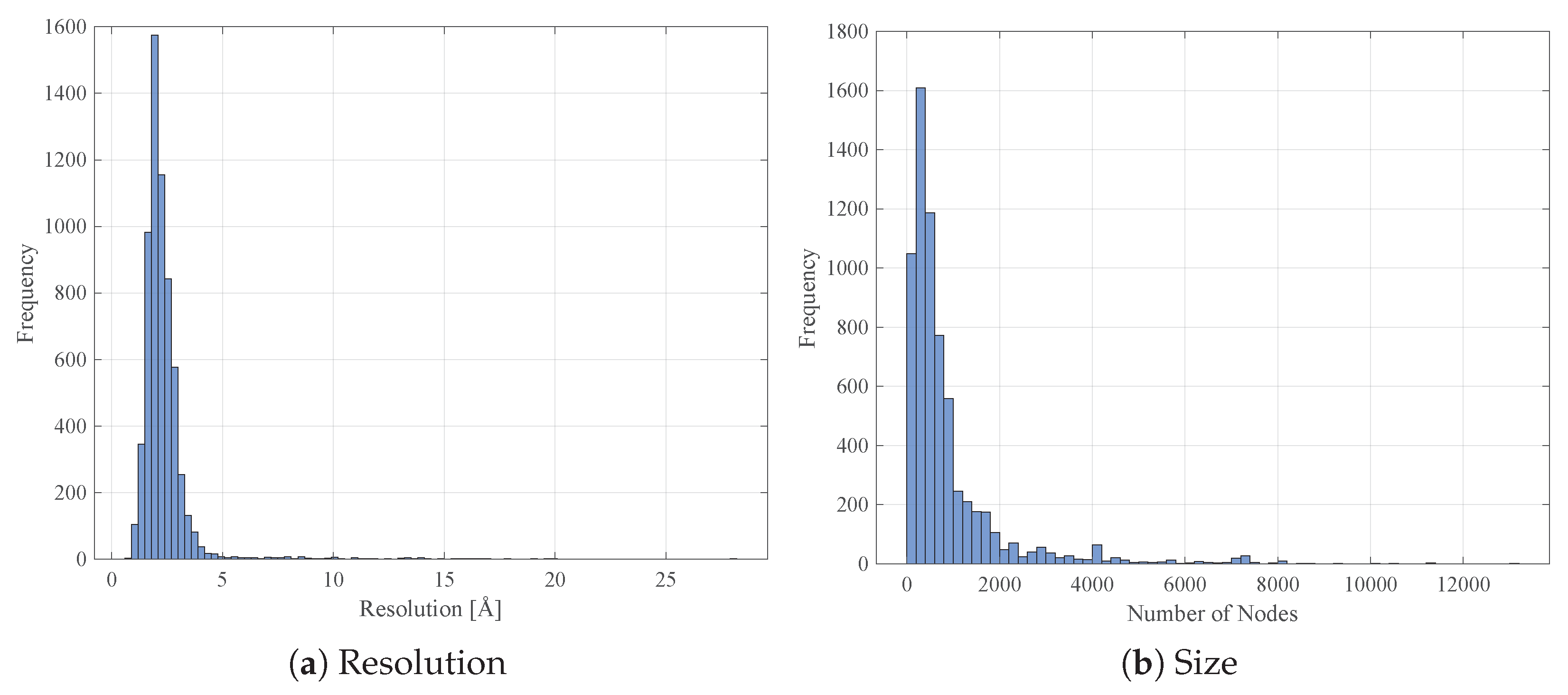
4.1. Data Collection and Pre-Processing
- .pdb files have been downloaded for all resolved proteins;
- information such as the EC number and the measurement resolution (if present) have been parsed from the .pdb file header;
- proteins having multiple EC numbers have been discarded.
- -carbon atoms 3D coordinates have been parsed from each .pdb file;
- In case of multiple equivalent models within the same .pdb file, only the first model is retained;
- Similarly, for atoms having alternate coordinate locations, only the first location is retained.
- The NetworkX library [76] (Python) for evaluating centrality measures () and the Vietoris–Rips complex ();
- The Rnetcarto (https://cran.r-project.org/package=rnetcarto) library (R) for network cartography ().
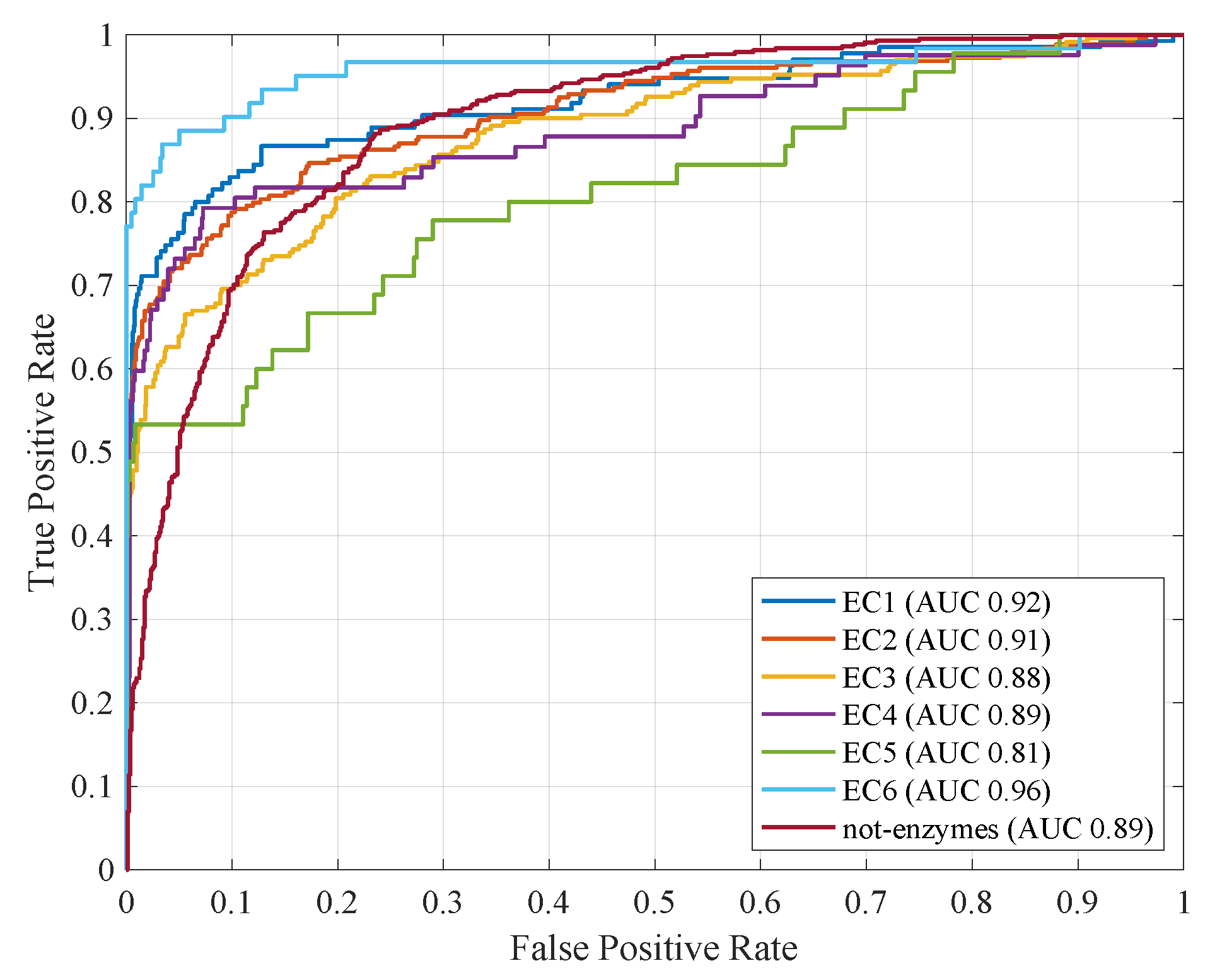
4.2. Computational Results with Fitness Function
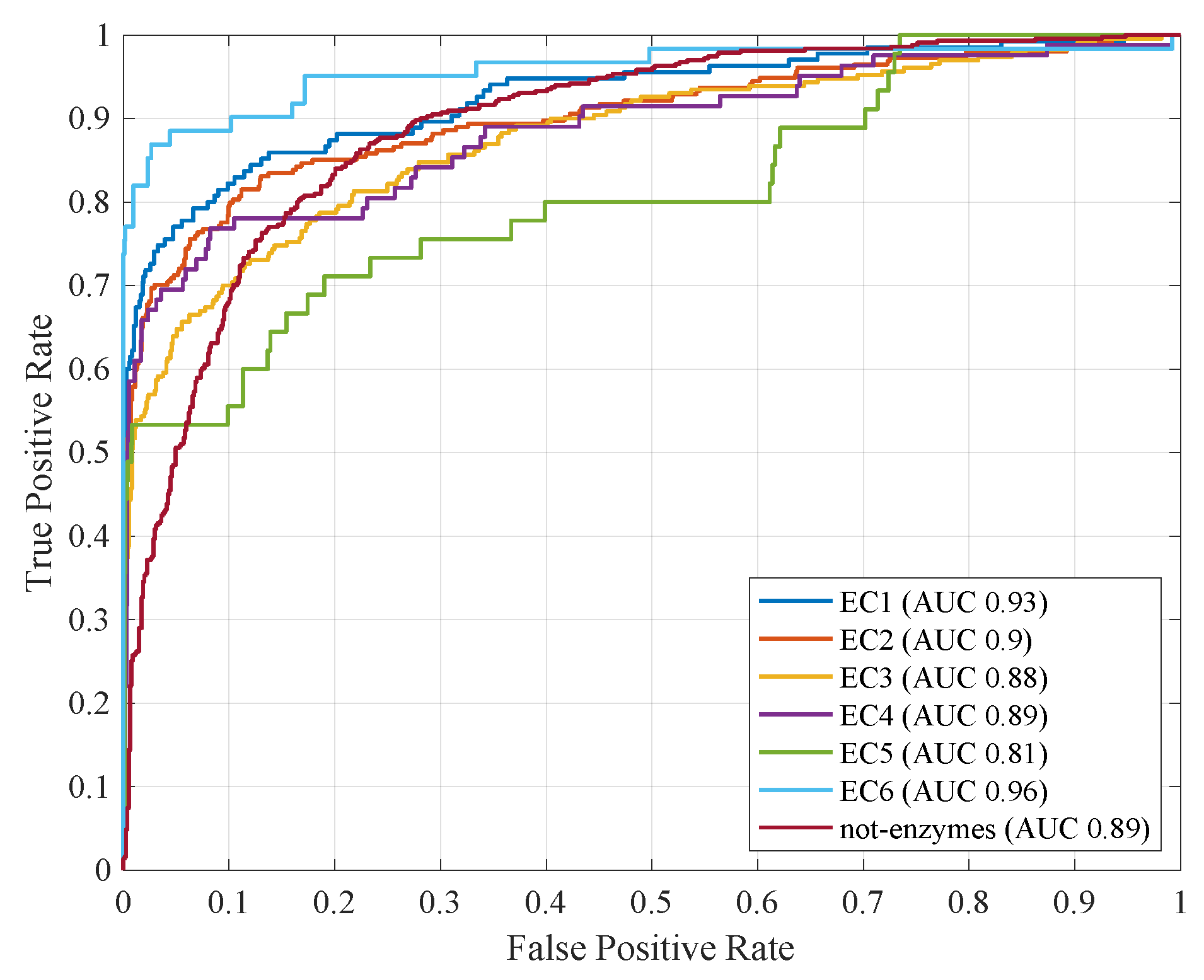
4.3. Computational Results with Fitness Function
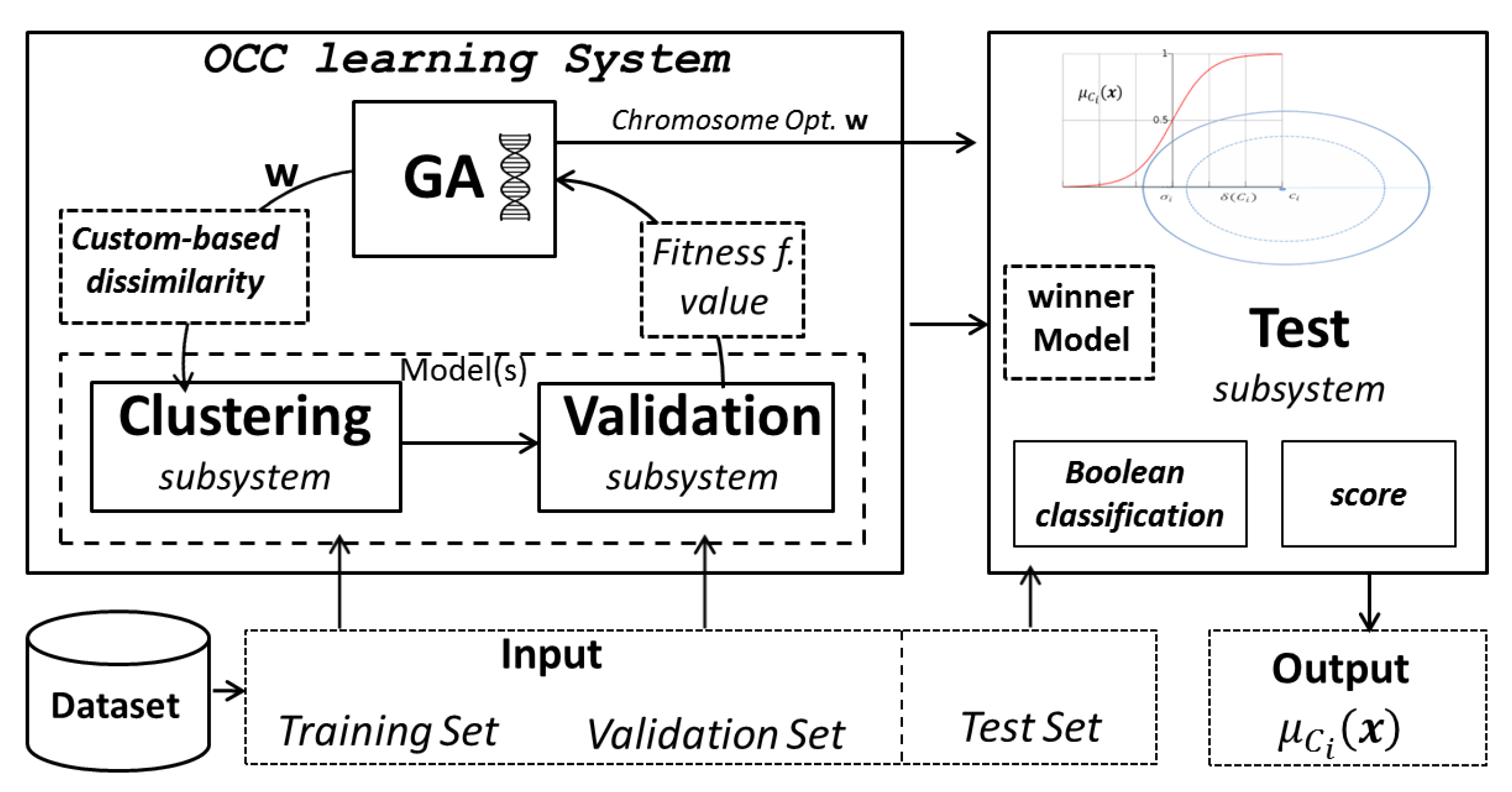
4.4. Benchmarking against a Clustering-Based One-Class Classifier
- Learning a cluster model of proteins through a suitable dataset divided into two disjoint sets, namely training and validation set;
- Using the learned model in order to recognise or classify unseen proteins drawn from the test set, assigning to each pattern a probability value.
- The pairwise distances between the test data and the k clusters centres (for OCC);
- The dot product between the test data and the support vectors (for MKMD).
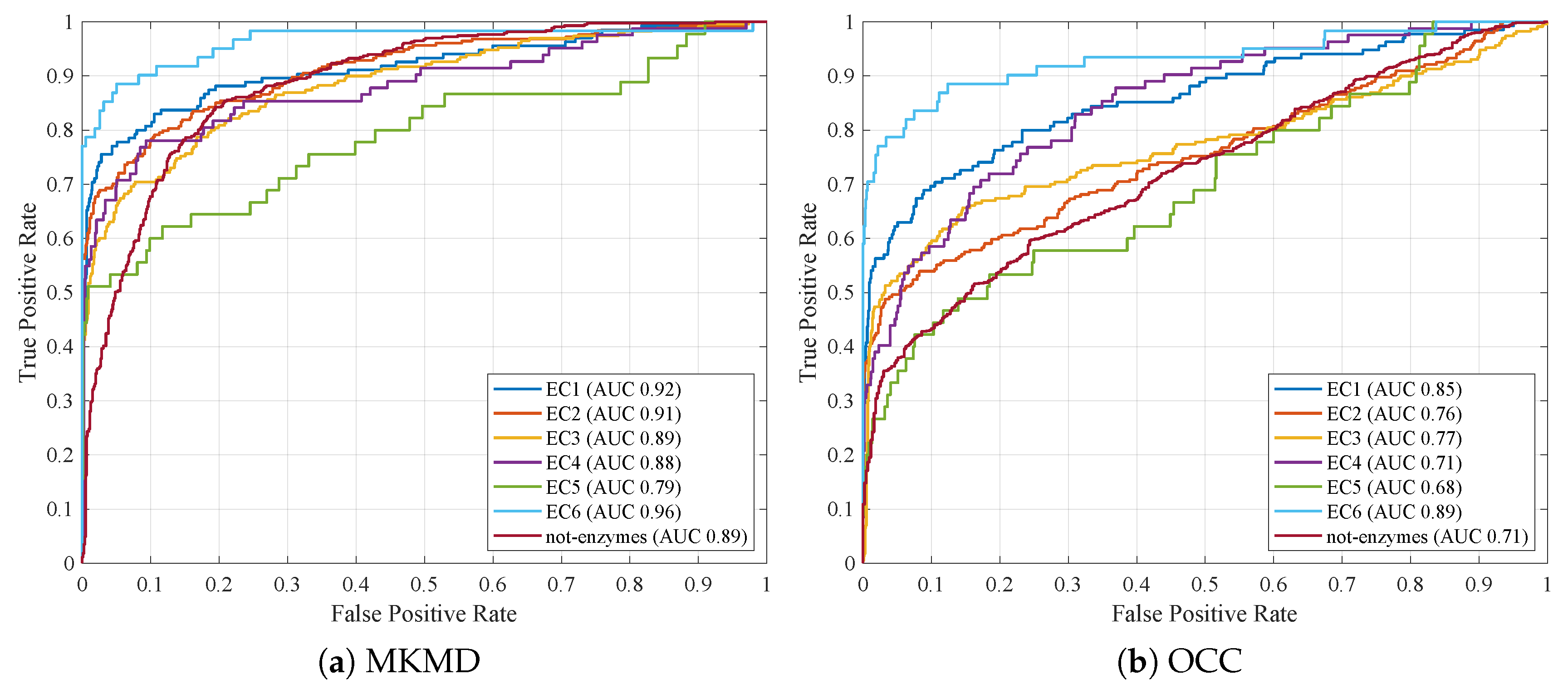
4.5. Comparing against Previous Works
- Avoiding to filter out PDB structures by considering only the best resolution for a given UniProt ID (as carried out also in this work) helps in improving classification models: indeed, performances from [44] are amongst the lowest ones;
- The proposed MKMD approach, regardless of the fitness function and/or representative selection, outperforms all competitors for all EC classes (including not-enzymes).
4.6. On the Knowledge Discovery Phase
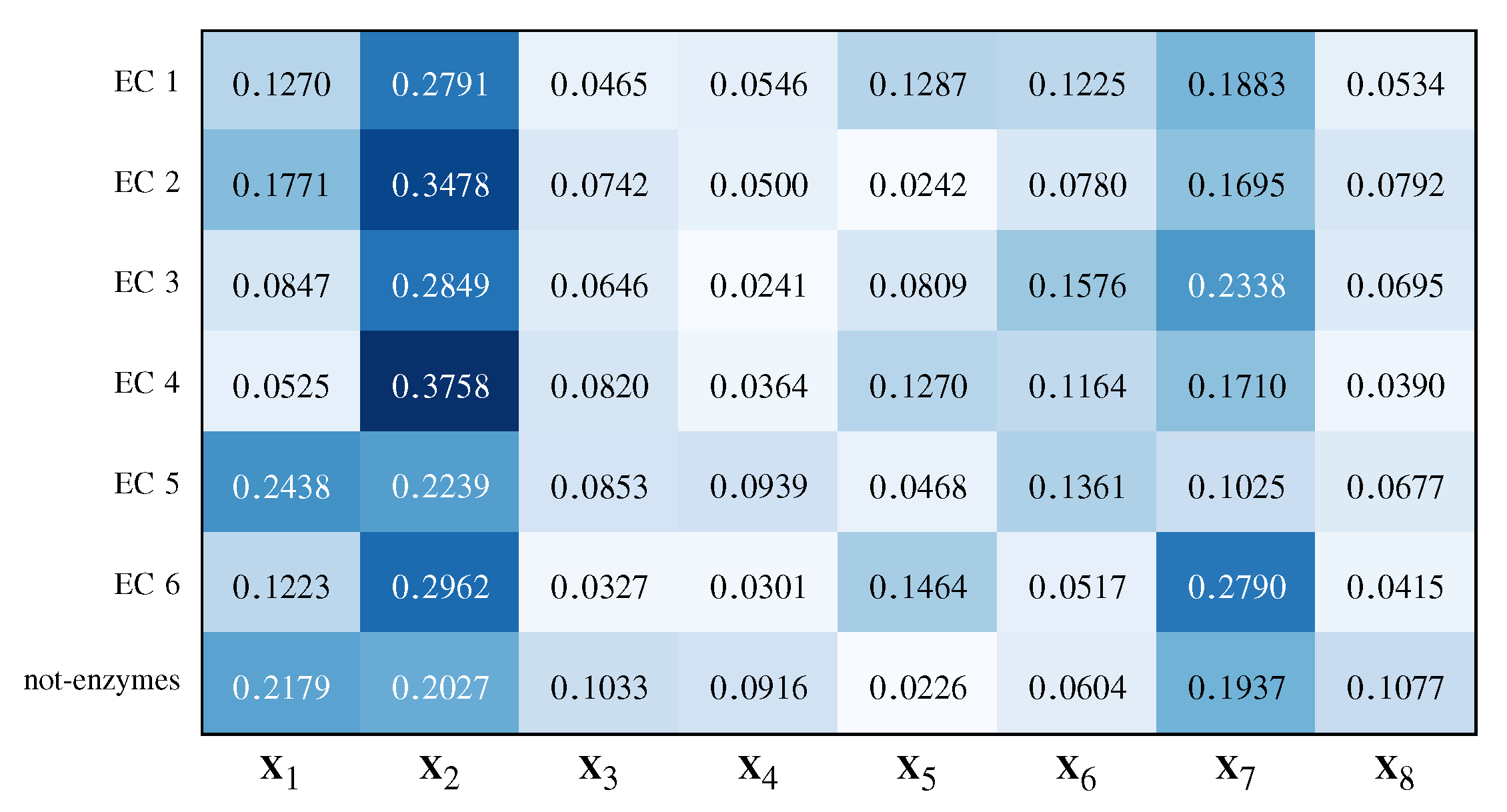
- By analysing the kernel weights , it is possible to determine the most important representations for the problem at hand;
- By analysing , namely the binary vector in charge of selecting prototypes from the dissimilarity space, it is possible to determine and analyse the patterns (proteins, in this case) elected as prototypes.
- To efficiently accomplish the task of being water soluble while maintaining a stable structure (or dynamics);
- To allow for an efficient spreading of the signal across amino-acid residues contact network so to sense relevant microenvironment changes and to reshape accordingly—allosteric effect, see [94].
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | Area Under the Curve |
| DME | Dissimilarity Matrix Embedding |
| MKMD | Multiple Kernels over Multiple Dissimilarities |
| OCC | One-Class Classification (also OCC_System) |
| PCN | Protein Contact Networks |
| PDB | Protein Data Bank |
| ROC | Receiver Operating Characteristic |
| SVM | Support Vector Machine |
Appendix A. Selected Representations
Appendix A.1. Betti Numbers
- Build the Vietoris–Rips neighbourhood graph : an undirected graph where edges between two nodes, say , are scored if ;
- The set of maximal cliques in form the Vietoris–Rips complex.
Appendix A.2. Centrality Measures
- The degree centrality [110] for node , defined as the percentage of nodes connected to it:where is the adjacency matrix, defined as in Equation (A22). The normalisation coefficient takes into account the maximum attainable degree in a simple graph, thus making the degree centrality in Equation (A4) independent from the number of nodes in the graph;
- The eigenvector centrality [110] highly rank nodes whether they are connected to other high-rank nodes. Formally, the eigenvector centrality for node is given by:where is a scalar constant. Equation (A5) can be re-written in matrix form as:Hence, the eigenvector centrality vector is the left-hand eigenvector of the adjacency matrix associated with the eigenvalue . According to the Perron–Frobenius theorem, by choosing as the largest (in absolute value) eigenvalue of , the solution is unique and all its entries are positive;
- The PageRank centrality [110] for node is given by:where is a scalar constant (usually ) and is the degree of node . It is worth remarking the difference between degree and degree centrality: the degree is the number of nodes connected to a given node (namely Equation (A4) without the normalisation term), whereas the degree centrality includes the normalisation term. As in the eigenvector centrality case, Equation (A7) can be re-written in matrix form as:where is a diagonal matrix whose ith element equals and is a vector whose elements are all equal to ;
- The Katz centrality [110,111] for node is given by:where controls the initial centrality (first neighbourhood weights) and attenuates the importance with respect to higher-order neighbours (in turn, is the largest eigenvalue of ). It is worth noting that if and , the Katz centrality equals the eigenvector centrality;
- The closeness centrality [110] for node is the inverse sum of shortest path distances between node and all other reachable nodes. Formally:where indicates the shortest path distance. The normalisation factor takes into account the graph size in order to allow comparison between nodes of graphs having different sizes, also in case of multiple connected components [112]. Indeed, n can be seen as the number of nodes in the connected component in which lies. In case of one connected component, the scale factor can be neglected since ;
- The betweenness centrality [110] quantifies how many times a given node acts as a bridge along the shortest paths between any two nodes:where is the number of shortest paths from to and is the number of shortest paths from to passing through . As in the closeness centrality case, it is often customary to normalise the betweenness centrality in order to avoid dependency from the number of nodes, thus:
- The edge betweenness centrality [113] is the edge counterpart of the “standard” (node) betweenness centrality as it quantifies how many times a given edge acts as a bridge along the shortest paths between two nodes:where is the number of shortest paths between nodes and passing through edge and is the total number of shortest paths between nodes and . As in the “standard” betweenness centrality, the edge betweenness centrality can be normalised as follows:
- The edge load centrality for edge is the edge-related counterpart of the load centrality (like betweenness vs. edge betweenness): it is defined as the percentage of the total number of shortest paths crossing edge ;
- The subgraph centrality [115] for node is the sum of (weighted) closed walks (i.e., connected subgraphs) starting and ending at (the longer the walk, the lower the weight). It can be evaluated thanks to the spectral decomposition of the adjacency matrix, which reads as where is a diagonal matrix containing the eigenvalues in increasing order and contains the corresponding unitary-length eigenvectors, thus:where and are the eigenvalue and eigenvector associated to node and indicates the value related to in the jth eigenvector;
- The Estrada Index [116] of a graph quantifies the compactness (or ‘folding’, since the Estrada Index was indeed originally proposed in order to study molecular 3D compactness) of a graph starting from the spectral decomposition of the adjacency matrix (as in the subgraph centrality):
- The harmonic centrality [117] is the sum of inverse shortest paths distances from a given node to all other nodes:
- The global reaching centrality [118] of a graph is the average (over all nodes) of the difference between the maximum local reaching centrality and each node’s local reaching centrality. Formally:where is the local reaching centrality of node and is the maximum local reaching centrality amongst all nodes. In turn, the local reaching centrality for a given node is defined as the percentage of nodes reachable from ;
- The average clustering coefficient [119] of a graph is given by:where is the clustering coefficient for node , defined as:where, in turn, is the number of triangles passing through node and is its degree;
- The average neighbour degree [120] of node is given by:where is the set of neighbours of node .
Appendix A.3. Energy and Laplacian Energy
Appendix A.4. Nodes Functional Cartography
Appendix A.5. Heat Content Invariant
Appendix A.6. Heat Kernel Trace
Appendix A.7. Size
Appendix A.8. Normalised Laplacian Spectral Density
Appendix B. Selected Prototypes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Notes/Description |
|---|---|
| 1KOF | Transferase |
| 1XFG | Transferase |
| 3E2R | Oxydoreductase |
| 4TS9 | Transferase |
| 1ZDM | Signalling Protein |
| 1MPG | Hydrolase |
| 1QQQ | Transferase |
| PDB ID | Notes/Description |
|---|---|
| 3EDC | LAC repressor (signalling protein) |
| 1DKL | Hydrolase |
| 1JKJ | Ligase |
| 2DBI | Unknown function |
| 3UCS | Chaperone |
| 1LX7 | Transferase |
| 2GAR | Transferase |
| 3ILI | Transferase |
| 1S08 | Transferase |
| 4IXM | Hydrolase |
| 4XTJ | Isomerase |
| 1KW1 | Lyase |
| 1BDH | Transcription factor (DNA-binding) |
| 4PC3 | Elongation factor (RNA-binding) |
| 5G1L | Isomerase |
| PDB ID | Notes/Description |
|---|---|
| 4RZS | Transcription factor (signalling protein) |
| 1ZDM | Signalling protein |
| 3I7R | Lyase |
| 1HW5 | Signalling protein |
| 1SO5 | Lyase |
| PDB ID | Notes/Description |
|---|---|
| 2BWX | Hydrolase |
| 3UWM | Oxydoreductase |
| 2H71 | Electron transport |
| 1D7A | Lyase |
| 4DAP | DNA-binding |
| 1SPV | Structural genomics, unknown function |
| 1EXD | Ligase + RNA-binding |
| 1X83 | Isomerase |
| 3ILJ | Transferase |
| 2D4U | Signalling protein |
| 1JNW | Oxydoreductase |
| 1TRE | Oxydoreductase |
| 1ZPT | Oxydoreductase |
| 3LGU | Hydrolase |
| 1IB6 | Oxydoreductase |
| 3C0U | Structural genomics, unknown function |
| 5GT2 | Oxydoreductase |
| 2RN2 | Hydrolase |
| 4L4Z | Transcription regulator |
| 3CMR | Hydrolase |
| 1NQF | Transport protein |
| 1GPQ | Hydrolase |
| 4ODM | Isomerase + chaperone |
| 2NPG | Transport protein |
| 2UAG | Ligase |
| 1OVG | Transferase |
| 3AVU | Transferase |
| 1RBV | Hydrolase |
| 5AB1 | Cell adhesion |
| 1TMM | Transferase |
| 4NIY | Hydrolase |
| 4WR3 | Isomerase |
| PDB ID | Notes/Description |
|---|---|
| 4ITX | Lyase |
| 2BWW | Hydrolase |
| 5IU6 | Transferase |
| 1ODD | Gene regulatory |
| 5G5G | Oxydoreductase |
| 1G7X | Transferase |
| 2E0Y | Transferase |
| 2SCU | Ligase |
| 1HO4 | Hydrolase |
| 3RGM | Transport Protein |
| 1OAC | Oxydoreductase |
| 5MUC | Oxydoreductase |
| 3OGD | Hydrolase + DNA binding |
| 4K34 | Membrane protein |
| 1Q0L | Oxydoreductase |
| 1G58 | Isomerase |
| 5M3B | Transport protein |
| 2WOH | Oxydoreductase |
| 2PJP | Translation regulation (RNA-binding) |
| PDB ID | Notes/Description |
|---|---|
| 2OLQ | Lyase |
| 1JDI | Isomerase |
| 4NIG | Oxydoreductase + DNA-binding |
| 5T03 | Transferase |
| 5FNN | Oxydoreductase |
| 2Z9D | Oxydoreductase |
| 2V3Z | Hydrolase |
| 4ARI | Ligase + RNA-binding |
| 3LBS | Transport protein |
| 4QGS | Oxydoreductase |
| 5B7F | Oxydoreductase |
| 2ABH | Transferase |
| PDB ID | Notes/Description |
|---|---|
| 1SPA | Transferase |
| 2YH9 | Membrane protein |
| 1NQF | Transport protein |
| 1LDI | Transport protein |
| 1TIK | Hydrolase |
| 1MWI | Hydrolase + DNA-binding |
| 1GEW | Transferase |
| 5CKH | Hydrolase |
| 3ABQ | Lyase |
| 3B6M | Oxydoreductase |
References
- Bianchi, F.M.; Scardapane, S.; Livi, L.; Uncini, A.; Rizzi, A. An interpretable graph-based image classifier. In Proceedings of the 2014 International Joint Conference on Neural Networks (IJCNN), Beijing, China, 6–11 July 2014; pp. 2339–2346. [Google Scholar] [CrossRef]
- Bianchi, F.M.; Scardapane, S.; Rizzi, A.; Uncini, A.; Sadeghian, A. Granular Computing Techniques for Classification and Semantic Characterization of Structured Data. Cogn. Comput. 2016, 8, 442–461. [Google Scholar] [CrossRef]
- Del Vescovo, G.; Rizzi, A. Online Handwriting Recognition by the Symbolic Histograms Approach. In Proceedings of the 2007 IEEE International Conference on Granular Computing (GRC 2007), San Jose, CA, USA, 2–4 November 2007; p. 686. [Google Scholar] [CrossRef]
- Giuliani, A.; Filippi, S.; Bertolaso, M. Why network approach can promote a new way of thinking in biology. Front. Genet. 2014, 5, 83. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, L.; De Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein contact networks: An emerging paradigm in chemistry. Chem. Rev. 2012, 113, 1598–1613. [Google Scholar] [CrossRef]
- Krishnan, A.; Zbilut, J.P.; Tomita, M.; Giuliani, A. Proteins as networks: Usefulness of graph theory in protein science. Curr. Protein Pept. Sci. 2008, 9, 28–38. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, L.; Giuliani, A. Protein–Protein Interactions: The Structural Foundation of Life Complexity; American Cancer Society: Atlanta, GA, USA, 2017; pp. 1–12. [Google Scholar] [CrossRef]
- Wuchty, S. Scale-Free Behavior in Protein Domain Networks. Mol. Biol. Evol. 2001, 18, 1694–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, A.; Giuliani, A.; Rizzi, A. Granular Computing Techniques for Bioinformatics Pattern Recognition Problems in Non-metric Spaces. In Computational Intelligence for Pattern Recognition; Pedrycz, W., Chen, S.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 53–81. [Google Scholar]
- Ieracitano, C.; Mammone, N.; Hussain, A.; Morabito, F.C. A novel multi-modal machine learning based approach for automatic classification of EEG recordings in dementia. Neural Netw. 2020, 123, 176–190. [Google Scholar] [CrossRef]
- Cinti, A.; Bianchi, F.M.; Martino, A.; Rizzi, A. A Novel Algorithm for Online Inexact String Matching and its FPGA Implementation. Cogn. Comput. 2019. [Google Scholar] [CrossRef] [Green Version]
- Bunke, H. On a relation between graph edit distance and maximum common subgraph. Pattern Recognit. Lett. 1997, 18, 689–694. [Google Scholar] [CrossRef]
- Bargiela, A.; Pedrycz, W. Granular Computing: An Introduction; Kluwer Academic Publishers: Boston, MA, USA, 2003. [Google Scholar]
- Pedrycz, W. Granular computing: An introduction. In Proceedings of the 9th IFSA World Congress and 20th NAFIPS International Conference, Vancouver, BC, Canada, 25–28 July 2001; Volume 3, pp. 1349–1354. [Google Scholar] [CrossRef]
- Bargiela, A.; Pedrycz, W. Granular Computing. In Handbook on Computational Intelligence; World Scientific Publishers: Singapore, 2016; Chapter 2; pp. 43–66. [Google Scholar] [CrossRef]
- Singh, P.K. Similar Vague Concepts Selection Using Their Euclidean Distance at Different Granulation. Cogn. Comput. 2018, 10, 228–241. [Google Scholar] [CrossRef]
- Lin, T.Y.; Yao, Y.Y.; Zadeh, L.A. Data Mining, Rough Sets and Granular Computing; Springer: Berlin/Heidelberg, Germany, 2013; Volume 95. [Google Scholar]
- Bianchi, F.M.; Livi, L.; Rizzi, A.; Sadeghian, A. A Granular Computing approach to the design of optimized graph classification systems. Soft Comput. 2014, 18, 393–412. [Google Scholar] [CrossRef]
- Rizzi, A.; Del Vescovo, G.; Livi, L.; Frattale Mascioli, F.M. A new Granular Computing approach for sequences representation and classification. In Proceedings of the 2012 International Joint Conference on Neural Networks (IJCNN), Brisbane, Australia, 10–15 June 2012; pp. 1–8. [Google Scholar] [CrossRef]
- Del Vescovo, G.; Rizzi, A. Automatic Classification of Graphs by Symbolic Histograms. In Proceedings of the 2007 IEEE International Conference on Granular Computing (GRC 2007), San Jose, CA, USA, 2–4 November 2007; p. 410. [Google Scholar] [CrossRef]
- Baldini, L.; Martino, A.; Rizzi, A. Stochastic Information Granules Extraction for Graph Embedding and Classification. In Proceedings of the 11th International Joint Conference on Computational Intelligence—Volume 1: NCTA, (IJCCI 2019), Vienna, Austria, 17–19 September 2019; pp. 391–402. [Google Scholar] [CrossRef]
- Martino, A.; Giuliani, A.; Todde, V.; Bizzarri, M.; Rizzi, A. Metabolic networks classification and knowledge discovery by information granulation. Comput. Biol. Chem. 2020, 84, 107187. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Frattale Mascioli, F.M.; Rizzi, A. On the Optimization of Embedding Spaces via Information Granulation for Pattern Recognition. In Proceedings of the 2020 International Joint Conference on Neural Networks (IJCNN), Glasgow, UK, 19–24 July 2020. [Google Scholar]
- Martino, A.; Giuliani, A.; Rizzi, A. (Hyper)Graph Embedding and Classification via Simplicial Complexes. Algorithms 2019, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Pękalska, E.; Duin, R.P. The Dissimilarity Representation for Pattern Recognition: Foundations and Applications; World Scientific: London, UK, 2005. [Google Scholar]
- Pękalska, E.; Duin, R.P.; Paclík, P. Prototype selection for dissimilarity-based classifiers. Pattern Recognit. 2006, 39, 189–208. [Google Scholar] [CrossRef]
- Duin, R.P.; Pękalska, E. The dissimilarity space: Bridging structural and statistical pattern recognition. Pattern Recognit. Lett. 2012, 33, 826–832. [Google Scholar] [CrossRef]
- Shawe-Taylor, J.; Cristianini, N. Kernel Methods for Pattern Analysis; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Schölkopf, B.; Smola, A.J. Learning with Kernels: Support Vector Machines, Regularization, Optimization, and Beyond; MIT Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Mercer, J. Functions of positive and negative type, and their connection with the theory of integral equations. R. Soc. 1909, 209, 415–446. [Google Scholar]
- Cover, T.M. Geometrical and statistical properties of systems of linear inequalities with applications in pattern recognition. IEEE Trans. Electron. Comput. 1965, EC-14, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Boser, B.E.; Guyon, I.; Vapnik, V. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory, Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Haasdonk, B. Feature space interpretation of SVMs with indefinite kernels. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Laub, J.; Müller, K.R. Feature Discovery in Non-Metric Pairwise Data. J. Mach. Learn. Res. 2004, 5, 801–818. [Google Scholar]
- Ong, C.S.; Mary, X.; Canu, S.; Smola, A.J. Learning with Non-positive Kernels. In Proceedings of the Twenty-first International Conference on Machine Learning, Banff, AL, Canada, 4–8 July 2004. [Google Scholar] [CrossRef]
- Chen, Y.; Gupta, M.R.; Recht, B. Learning kernels from indefinite similarities. In Proceedings of the 26th Annual International Conference on Machine Learning, Montreal, QC, Canada, 14–18 June 2009; pp. 145–152. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Garcia, E.K.; Gupta, M.R.; Rahimi, A.; Cazzanti, L. Similarity-based classification: Concepts and algorithms. J. Mach. Learn. Res. 2009, 10, 747–776. [Google Scholar]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livi, L.; Giuliani, A.; Sadeghian, A. Characterization of graphs for protein structure modeling and recognition of solubility. Curr. Bioinform. 2016, 11, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Livi, L.; Giuliani, A.; Rizzi, A. Toward a multilevel representation of protein molecules: Comparative approaches to the aggregation/folding propensity problem. Inf. Sci. 2016, 326, 134–145. [Google Scholar] [CrossRef] [Green Version]
- De Santis, E.; Martino, A.; Rizzi, A.; Frattale Mascioli, F.M. Dissimilarity Space Representations and Automatic Feature Selection for Protein Function Prediction. In Proceedings of the 2018 International Joint Conference on Neural Networks (IJCNN), Rio de Janeiro, Brazil, 8–13 July 2018; pp. 1–8. [Google Scholar] [CrossRef]
- Martino, A.; Maiorino, E.; Giuliani, A.; Giampieri, M.; Rizzi, A. Supervised Approaches for Function Prediction of Proteins Contact Networks from Topological Structure Information. In Image Analysis, Proceedings of the 20th Scandinavian Conference, SCIA 2017, Tromsø, Norway, 12–14 June 2017; Sharma, P., Bianchi, F.M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 285–296. [Google Scholar]
- Martino, A.; Rizzi, A.; Frattale Mascioli, F.M. Supervised Approaches for Protein Function Prediction by Topological Data Analysis. In Proceedings of the 2018 International Joint Conference on Neural Networks (IJCNN), Rio de Janeiro, Brazil, 8–13 July 2018; pp. 1–8. [Google Scholar] [CrossRef]
- Livi, L.; Maiorino, E.; Giuliani, A.; Rizzi, A.; Sadeghian, A. A generative model for protein contact networks. J. Biomol. Struct. Dyn. 2016, 34, 1441–1454. [Google Scholar] [CrossRef] [Green Version]
- Livi, L.; Maiorino, E.; Pinna, A.; Sadeghian, A.; Rizzi, A.; Giuliani, A. Analysis of heat kernel highlights the strongly modular and heat-preserving structure of proteins. Phys. A Stat. Mech. Its Appl. 2016, 441, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, E.; Livi, L.; Giuliani, A.; Sadeghian, A.; Rizzi, A. Multifractal characterization of protein contact networks. Phys. A Stat. Mech. Its Appl. 2015, 428, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, E.; Rizzi, A.; Sadeghian, A.; Giuliani, A. Spectral reconstruction of protein contact networks. Phys. A Stat. Mech. Its Appl. 2017, 471, 804–817. [Google Scholar] [CrossRef]
- Webb, E.C. Enzyme Nomenclature 1992. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes, 6th ed.; Academic Press: Cambridge, MA, USA, 1992. [Google Scholar]
- Livi, L.; Rizzi, A.; Sadeghian, A. Optimized dissimilarity space embedding for labeled graphs. Inf. Sci. 2014, 266, 47–64. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2006. [Google Scholar]
- Vapnik, V. Statistical Learning Theory; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Sonnenburg, S.; Rätsch, G.; Schäfer, C.; Schölkopf, B. Large scale multiple kernel learning. J. Mach. Learn. Res. 2006, 7, 1531–1565. [Google Scholar]
- Lewis, D.P.; Jebara, T.; Noble, W.S. Nonstationary kernel combination. In Proceedings of the 23rd International Conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 553–560. [Google Scholar] [CrossRef] [Green Version]
- Lanckriet, G.R.; Cristianini, N.; Bartlett, P.; Ghaoui, L.E.; Jordan, M.I. Learning the kernel matrix with semidefinite programming. J. Mach. Learn. Res. 2004, 5, 27–72. [Google Scholar]
- Gönen, M.; Alpaydin, E. Localized multiple kernel learning. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; pp. 352–359. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Mohri, M.; Rostamizadeh, A. Learning non-linear combinations of kernels. In Advances in Neural Information Processing Systems 22, Proceedings of the 23rd Annual Conference on Neural Information Processing Systems 2009, Vancouver, BC, Canada, 7–10 December 2009; Curran Associates Inc.: Nice, France, 2009; pp. 396–404. [Google Scholar]
- Bach, F.R.; Lanckriet, G.R.; Jordan, M.I. Multiple kernel learning, conic duality, and the SMO algorithm. In Proceedings of the Twenty-first International Conference on Machine Learning, Banff, AL, Canada, 4–8 July 2004; p. 6. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Chen, Y.; Kwok, J.T.Y. Building sparse multiple-kernel SVM classifiers. IEEE Trans. Neural Netw. 2009, 20, 827–839. [Google Scholar] [CrossRef]
- Gönen, M.; Alpaydın, E. Multiple kernel learning algorithms. J. Mach. Learn. Res. 2011, 12, 2211–2268. [Google Scholar]
- Horn, R.A.; Johnson, C.R. Matrix Analysis; Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Schölkopf, B.; Smola, A.J.; Williamson, R.C.; Bartlett, P.L. New support vector algorithms. Neural Comput. 2000, 12, 1207–1245. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E. Genetic Algorithms in Search, Optimization and Machine Learning, 1st ed.; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1989. [Google Scholar]
- Rojas, S.A.; Fernandez-Reyes, D. Adapting multiple kernel parameters for support vector machines using genetic algorithms. In Proceedings of the 2005 IEEE Congress on Evolutionary Computation, Scotland, UK, 2–5 September 2005; Volume 1, pp. 626–631. [Google Scholar] [CrossRef] [Green Version]
- Phienthrakul, T.; Kijsirikul, B. Evolving Hyperparameters of Support Vector Machines Based on Multi-Scale RBF Kernels. In Proceedings of the International Conference on Intelligent Information Processing, Adelaide, Australia, 20–23 September 2006; pp. 269–278. [Google Scholar] [CrossRef] [Green Version]
- Beyer, H.G.; Schwefel, H.P. Evolution strategies—A comprehensive introduction. Nat. Comput. 2002, 1, 3–52. [Google Scholar] [CrossRef]
- Youden, W.J. Index for rating diagnostic tests. Cancer 1950, 3, 32–35. [Google Scholar] [CrossRef]
- Powers, D.M.W. Evaluation: From precision, recall and f-measure to roc., informedness, markedness & correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Cokelaer, T.; Pultz, D.; Harder, L.M.; Serra-Musach, J.; Saez-Rodriguez, J. BioServices: A common Python package to access biological Web Services programmatically. Bioinformatics 2013, 29, 3241–3242. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Raschka, S. BioPandas: Working with molecular structures in pandas DataFrames. J. Open Source Softw. 2017, 2. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.; Schult, D. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference (SciPy), Pasadena, CA, USA, 19–24 August 2008; pp. 11–15. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliphant, T.E. Python for scientific computing. Comput. Sci. Eng. 2007, 9. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27:1–27:27. [Google Scholar] [CrossRef]
- Fawcett, T. An Introduction to ROC Analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- De Santis, E.; Livi, L.; Sadeghian, A.; Rizzi, A. Modeling and recognition of smart grid faults by a combined approach of dissimilarity learning and one-class classification. Neurocomputing 2015, 170, 368–383. [Google Scholar] [CrossRef] [Green Version]
- De Santis, E.; Rizzi, A.; Sadeghian, A. A cluster-based dissimilarity learning approach for localized fault classification in Smart Grids. Swarm Evol. Comput. 2018, 39, 267–278. [Google Scholar] [CrossRef]
- De Santis, E.; Paschero, M.; Rizzi, A.; Frattale Mascioli, F.M. Evolutionary Optimization of an Affine Model for Vulnerability Characterization in Smart Grids. In Proceedings of the 2018 International Joint Conference on Neural Networks (IJCNN), Rio de Janeiro, Brazil, 8–13 July 2018; pp. 1–8. [Google Scholar] [CrossRef]
- Khan, S.S.; Madden, M.G. A Survey of Recent Trends in One Class Classification. In Artificial Intelligence and Cognitive Science; Coyle, L., Freyne, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 6206, pp. 188–197. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, M.A.F.; Clifton, D.A.; Clifton, L.; Tarassenko, L. A review of novelty detection. Signal Process. 2014, 99, 215–249. [Google Scholar] [CrossRef]
- Martino, A.; Rizzi, A.; Frattale Mascioli, F.M. Efficient Approaches for Solving the Large-Scale k-medoids Problem. In Proceedings of the 9th International Joint Conference on Computational Intelligence—Volume 1, Madeira, Portugal, 1–3 November2017; pp. 338–347. [Google Scholar] [CrossRef]
- Martino, A.; Rizzi, A.; Frattale Mascioli, F.M. Distance Matrix Pre-Caching and Distributed Computation of Internal Validation Indices in k-medoids Clustering. In Proceedings of the 2018 International Joint Conference on Neural Networks (IJCNN), Rio de Janeiro, Brazil, 8–13 July 2018; pp. 1–8. [Google Scholar] [CrossRef]
- Martino, A.; Rizzi, A.; Frattale Mascioli, F.M. Efficient Approaches for Solving the Large-Scale k-Medoids Problem: Towards Structured Data. In Computational Intelligence, Proceedings of the 9th International Joint Conference, IJCCI 2017 Funchal-Madeira, Portugal, 1–3 November 2017; Sabourin, C., Merelo, J.J., Madani, K., Warwick, K., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 199–219. [Google Scholar] [CrossRef]
- Mendel, J.M. Fuzzy logic systems for engineering: A tutorial. Proc. IEEE 1995, 83, 345–377. [Google Scholar] [CrossRef] [Green Version]
- Martino, A.; De Santis, E.; Baldini, L.; Rizzi, A. Calibration Techniques for Binary Classification Problems: A Comparative Analysis. In Proceedings of the 11th International Joint Conference on Computational Intelligence—Volume 1, Vienna, Austria, 17–19 September 2019; pp. 487–495. [Google Scholar] [CrossRef]
- Martino, A. Pattern Recognition Techniques for Modelling Complex Systems in Non-Metric Domains. Ph.D. Thesis, University of Rome “La Sapienza”, Rome, Italy, 2020. [Google Scholar]
- Branden, C.I.; Tooze, J. Introduction to Protein Structure; Garland Publishing Inc.: New York, NY, USA, 1991. [Google Scholar]
- Giuliani, A.; Benigni, R.; Zbilut, J.P.; Webber, C.L.; Sirabella, P.; Colosimo, A. Nonlinear Signal Analysis Methods in the Elucidation of Protein Sequence-Structure Relationships. Chem. Rev. 2002, 102, 1471–1492. [Google Scholar] [CrossRef]
- Di Paola, L.; Giuliani, A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015, 31, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Devore, J.L.; Peck, R. Statistics: The Exploration and Analysis of Data, 4th ed.; Brooks/Cole: Pacific Grove, CA, USA, 2001. [Google Scholar]
- Bartz, A.E. Basic Statistical Concepts; Macmillan Pub Co.: New York, NY, USA, 1988. [Google Scholar]
- Guarnera, E.; Berezovsky, I.N. Allosteric sites: Remote control in regulation of protein activity. Curr. Opin. Struct. Biol. 2016, 37, 1–8. [Google Scholar] [CrossRef]
- Negre, C.F.A.; Morzan, U.N.; Hendrickson, H.P.; Pal, R.; Lisi, G.P.; Loria, J.P.; Rivalta, I.; Ho, J.; Batista, V.S. Eigenvector centrality for characterization of protein allosteric pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E12201–E12208. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, L. Topological Data Analysis. Annu. Rev. Stat. Its Appl. 2018, 5, 501–532. [Google Scholar] [CrossRef] [Green Version]
- Estrada, E.; Rodriguez-Velazquez, J.A. Complex networks as hypergraphs. arXiv 2005, arXiv:physics/0505137. [Google Scholar]
- Horak, D.; Maletić, S.; Rajković, M. Persistent homology of complex networks. J. Stat. Mech. Theory Exp. 2009, 2009, p03034. [Google Scholar] [CrossRef]
- Barbarossa, S.; Sardellitti, S. Topological Signal Processing over Simplicial Complexes. IEEE Trans. Signal Process. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ghrist, R.W. Elementary Applied Topology; Createspace: Seattle, WA, USA, 2014. [Google Scholar]
- Hausmann, J.C. On the Vietoris-Rips complexes and a cohomology theory for metric spaces. Ann. Math. Stud. 1995, 138, 175–188. [Google Scholar]
- Zomorodian, A.; Carlsson, G. Computing persistent homology. Discret. Comput. Geom. 2005, 33, 249–274. [Google Scholar] [CrossRef] [Green Version]
- Zomorodian, A. Fast construction of the Vietoris-Rips complex. Comput. Graph. 2010, 34, 263–271. [Google Scholar] [CrossRef]
- Munkres, J.R. Elements of Algebraic Topology; Addison-Wesley: Cambridge, MA, USA, 1984. [Google Scholar]
- Artin, M. Algebra; Prentice Hall: Englewood Cliffs, NJ, USA, 1991. [Google Scholar]
- Newman, M.E.J. Networks: An Introduction; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Katz, L. A new status index derived from sociometric analysis. Psychometrika 1953, 18, 39–43. [Google Scholar] [CrossRef]
- Wasserman, S.; Faust, K. Social Network Analysis: Methods and Applications; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Brandes, U. On variants of shortest-path betweenness centrality and their generic computation. Soc. Netw. 2008, 30, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.I.; Kahng, B.; Kim, D. Universal behavior of load distribution in scale-free networks. Phys. Rev. Lett. 2001, 87, 278701. [Google Scholar] [CrossRef] [Green Version]
- Estrada, E.; Rodriguez-Velazquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, E. Characterization of 3D molecular structure. Chem. Phys. Lett. 2000, 319, 713–718. [Google Scholar] [CrossRef]
- Boldi, P.; Vigna, S. Axioms for centrality. Internet Math. 2014, 10, 222–262. [Google Scholar] [CrossRef] [Green Version]
- Mones, E.; Vicsek, L.; Vicsek, T. Hierarchy measure for complex networks. PLoS ONE 2012, 7, e33799. [Google Scholar] [CrossRef] [PubMed]
- Saramäki, J.; Kivelä, M.; Onnela, J.P.; Kaski, K.; Kertesz, J. Generalizations of the clustering coefficient to weighted complex networks. Phys. Rev. E 2007, 75, 027105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrat, A.; Barthélemy, M.; Pastor-Satorras, R.; Vespignani, A. The architecture of complex weighted networks. Proc. Natl. Acad. Sci. USA 2004, 101, 3747–3752. [Google Scholar] [CrossRef] [Green Version]
- Gutman, I.; Zhou, B. Laplacian energy of a graph. Linear Algebra Appl. 2006, 414, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Guimera, R.; Amaral, L.A.N. Functional cartography of complex metabolic networks. Nature 2005, 433, 895. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Hancock, E.R.; Wilson, R.C. Graph characteristics from the heat kernel trace. Pattern Recognit. 2009, 42, 2589–2606. [Google Scholar] [CrossRef]
- Xiao, B.; Hancock, E.R. Graph clustering using heat content invariants. In Proceedings of the Iberian Conference on Pattern Recognition and Image Analysis, Estoril, Portugal, 7–9 June 2005; pp. 123–130. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Butler, S. Algebraic aspects of the normalized Laplacian. In Recent Trends in Combinatorics; Springer: Berlin/Heidelberg, Germany, 2016; pp. 295–315. [Google Scholar] [CrossRef]
- Parzen, E. On estimation of a probability density function and mode. Ann. Math. Stat. 1962, 33, 1065–1076. [Google Scholar] [CrossRef]
- Scott, D.W. On optimal and data-based histograms. Biometrika 1979, 66, 605–610. [Google Scholar] [CrossRef]







| Total | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | EC1 | EC2 | EC3 | EC4 | EC5 | EC6 | not-enzymes | |
| Count | 540 | 1017 | 919 | 329 | 182 | 244 | 1726 | 4957 |
| Percentage | 10.89 | 20.52 | 18.54 | 6.64 | 3.67 | 4.92 | 34.82 | 100% |
| Parameter | Bounds | Contraints |
|---|---|---|
| by definition | ||
| Class | Performances | Complexity | ||||
|---|---|---|---|---|---|---|
| Accuracy | Precision | Recall | Informedness | AUC | Sparsity | |
| 1 (EC1) | 0.95 | 0.87 | 0.68 | 0.83 | 0.92 | 49.43 |
| 2 (EC2) | 0.91 | 0.88 | 0.66 | 0.82 | 0.90 | 49.62 |
| 3 (EC3) | 0.90 | 0.84 | 0.58 | 0.78 | 0.88 | 49.48 |
| 4 (EC4) | 0.97 | 0.90 | 0.56 | 0.78 | 0.88 | 49.42 |
| 5 (EC5) | 0.98 | 0.83 | 0.44 | 0.72 | 0.78 | 50.78 |
| 6 (EC6) | 0.99 | 0.94 | 0.76 | 0.88 | 0.95 | 49.28 |
| 7 (not-enzymes) | 0.82 | 0.77 | 0.70 | 0.79 | 0.89 | 50.52 |
| Class | Performances | Complexity | ||||
|---|---|---|---|---|---|---|
| Accuracy | Precision | Recall | Informedness | AUC | Sparsity | |
| 1 (EC1) | 0.95 | 0.86 | 0.69 | 0.84 | 0.92 | 33.08 |
| 2 (EC2) | 0.91 | 0.88 | 0.67 | 0.82 | 0.90 | 32.48 |
| 3 (EC3) | 0.90 | 0.83 | 0.57 | 0.77 | 0.87 | 29.94 |
| 4 (EC4) | 0.97 | 0.88 | 0.54 | 0.77 | 0.88 | 33.89 |
| 5 (EC5) | 0.98 | 0.85 | 0.45 | 0.73 | 0.79 | 35.54 |
| 6 (EC6) | 0.98 | 0.91 | 0.76 | 0.88 | 0.95 | 35.38 |
| 7 (not-enzymes) | 0.82 | 0.77 | 0.69 | 0.79 | 0.88 | 33.37 |
| Class | Classifier | Performances | ||||
|---|---|---|---|---|---|---|
| Accuracy | Precision | Recall | Informedness | AUC | ||
| 1 (EC1) | OCC | 0.92 | 0.97 | 0.35 | 0.67 | 0.85 |
| MKMD | 0.95 | 0.88 | 0.67 | 0.83 | 0.91 | |
| 2 (EC2) | OCC | 0.83 | 0.87 | 0.45 | 0.69 | 0.76 |
| MKMD | 0.91 | 0.89 | 0.66 | 0.82 | 0.91 | |
| 3 (EC3) | OCC | 0.83 | 0.86 | 0.49 | 0.70 | 0.77 |
| MKMD | 0.90 | 0.84 | 0.57 | 0.77 | 0.88 | |
| 4 (EC4) | OCC | 0.68 | 0.60 | 0.78 | 0.61 | 0.72 |
| MKMD | 0.97 | 0.89 | 0.53 | 0.76 | 0.87 | |
| 5 (EC5) | OCC | 0.85 | 0.75 | 0.37 | 0.62 | 0.69 |
| MKMD | 0.98 | 0.82 | 0.44 | 0.72 | 0.78 | |
| 6 (EC6) | OCC | 0.97 | 0.96 | 0.57 | 0.78 | 0.88 |
| MKMD | 0.99 | 0.92 | 0.77 | 0.88 | 0.95 | |
| 7 (not-enzymes) | OCC | 0.68 | 0.60 | 0.78 | 0.61 | 0.72 |
| MKMD | 0.82 | 0.78 | 0.68 | 0.79 | 0.88 | |
| Approach | EC1 | EC2 | EC3 | EC4 | EC5 | EC6 | Not-Enzymes |
|---|---|---|---|---|---|---|---|
| DME + Logistic Regression [44] | – | – | – | – | – | – | 0.62 |
| DME + SVM [44] | – | – | – | – | – | – | 0.64 |
| DME + Naïve Bayes [44] | – | – | – | – | – | – | 0.62 |
| DME + Decision Tree [44] | – | – | – | – | – | – | 0.60 |
| DME + Neural Network [44] | – | – | – | – | – | – | 0.63 |
| OCC [44] | – | – | – | – | – | – | 0.63 |
| Feature Generation via Betti Numbers + SVM [46] | 0.79 | 0.75 | 0.73 | 0.73 | 0.46 | 0.77 | 0.77 |
| Feature Generation via Spectral Density + SVM [45] | 0.85 | 0.82 | 0.85 | 0.81 | 0.59 | 0.81 | 0.82 |
| MKMD with (Table 3) | 0.92 | 0.90 | 0.88 | 0.88 | 0.78 | 0.95 | 0.89 |
| MKMD with (Table 4) | 0.92 | 0.90 | 0.87 | 0.88 | 0.79 | 0.95 | 0.88 |
| MKMD with no representative selection (Table 5) | 0.91 | 0.91 | 0.88 | 0.87 | 0.78 | 0.95 | 0.88 |
| OCC (Table 5) | 0.85 | 0.76 | 0.77 | 0.72 | 0.69 | 0.88 | 0.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martino, A.; De Santis, E.; Giuliani, A.; Rizzi, A. Modelling and Recognition of Protein Contact Networks by Multiple Kernel Learning and Dissimilarity Representations. Entropy 2020, 22, 794. https://0-doi-org.brum.beds.ac.uk/10.3390/e22070794
Martino A, De Santis E, Giuliani A, Rizzi A. Modelling and Recognition of Protein Contact Networks by Multiple Kernel Learning and Dissimilarity Representations. Entropy. 2020; 22(7):794. https://0-doi-org.brum.beds.ac.uk/10.3390/e22070794
Chicago/Turabian StyleMartino, Alessio, Enrico De Santis, Alessandro Giuliani, and Antonello Rizzi. 2020. "Modelling and Recognition of Protein Contact Networks by Multiple Kernel Learning and Dissimilarity Representations" Entropy 22, no. 7: 794. https://0-doi-org.brum.beds.ac.uk/10.3390/e22070794