TMEA: A Thermodynamically Motivated Framework for Functional Characterization of Biological Responses to System Acclimation




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Surprisal Analysis
2.3. Functional Annotation and Pathway Database
2.4. Gene Set Enrichment Analysis Based on Hypergeometric Function
2.5. Further Statistical Analysis and Visualization
3. Results
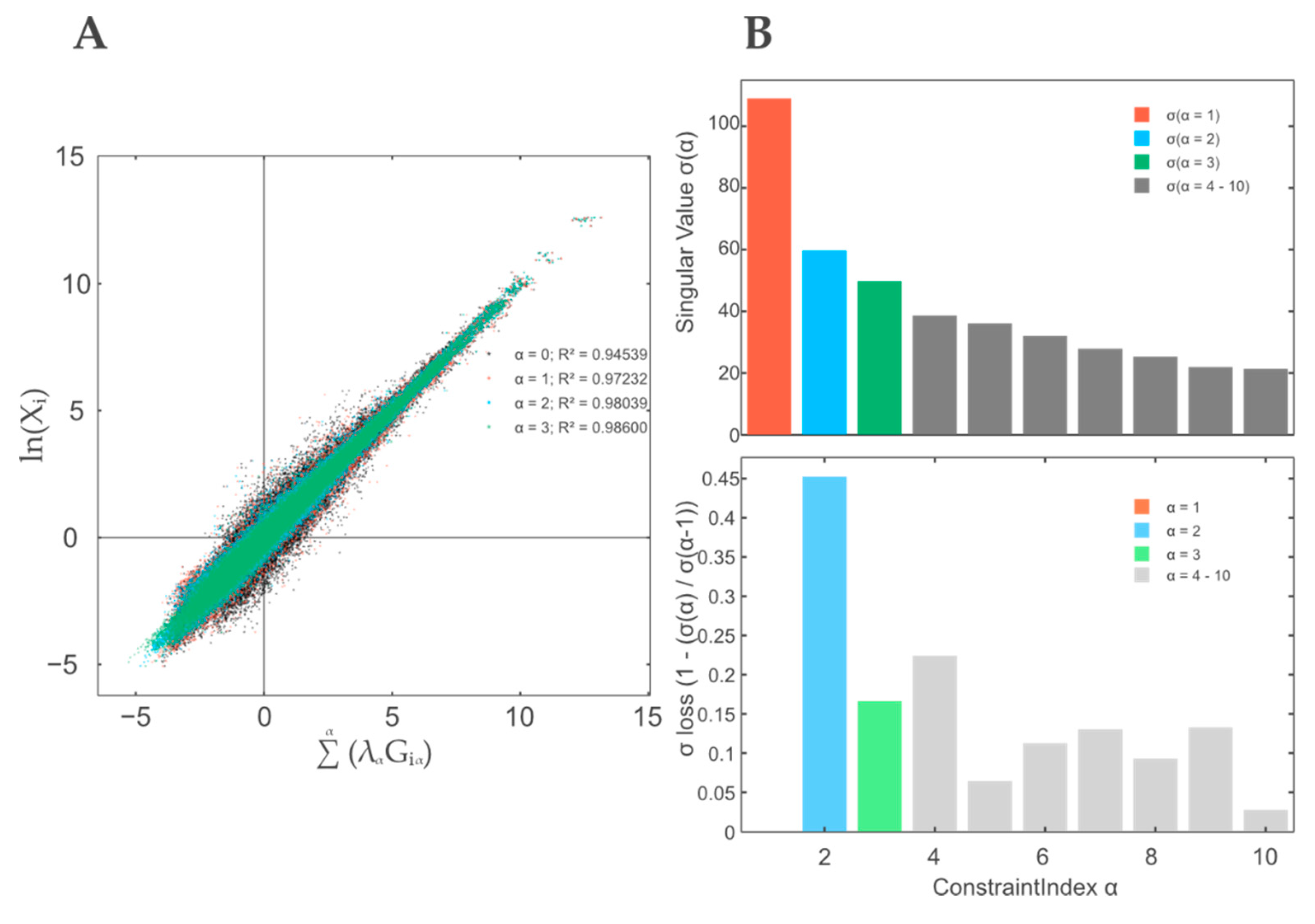
3.1. A Thermodynamic-Free Energy-Based Framework for the Functional Description of Biological Systems Not in Equilibrium Named TMEA
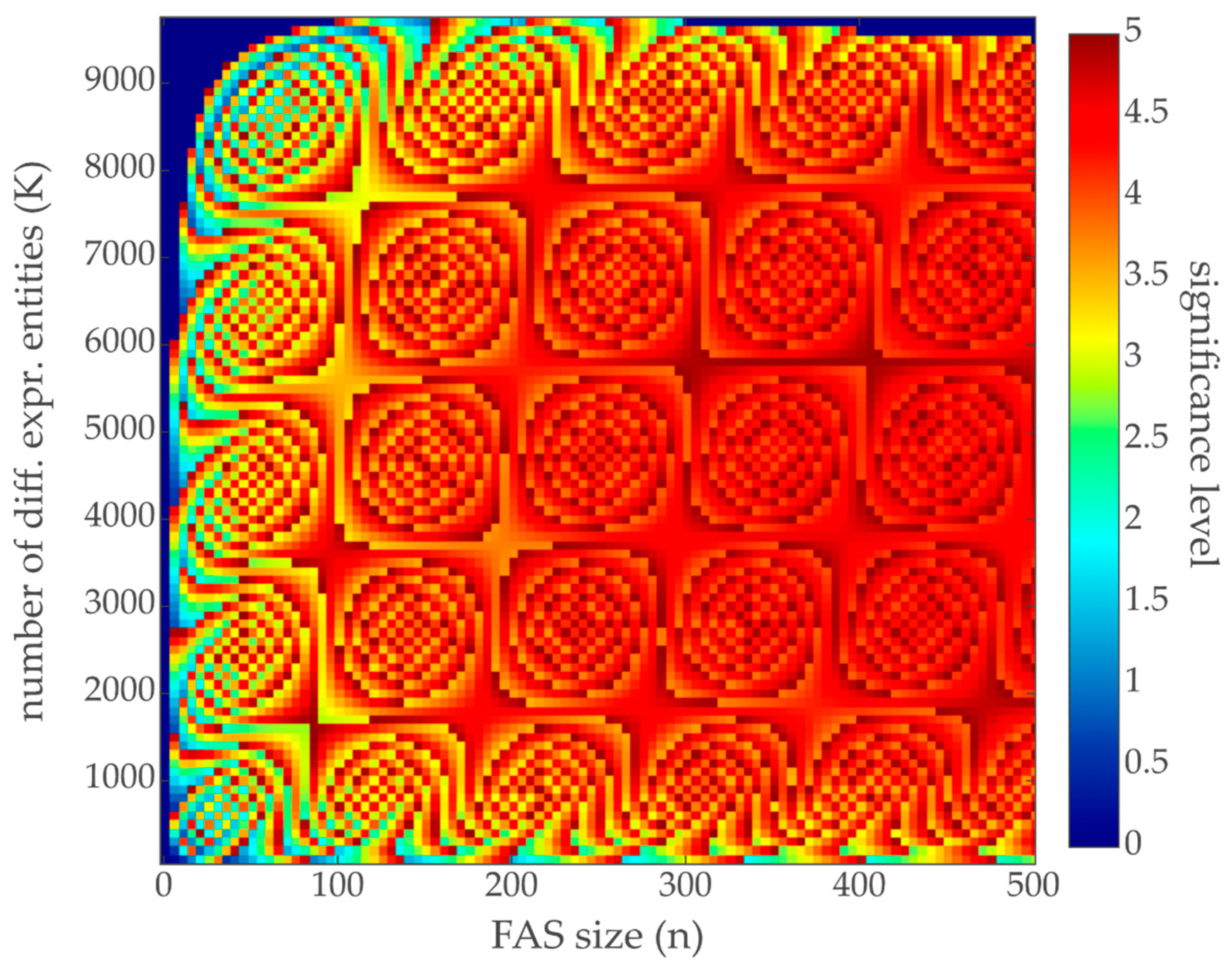
3.2. Contribution Weight Sums as Test Statistic
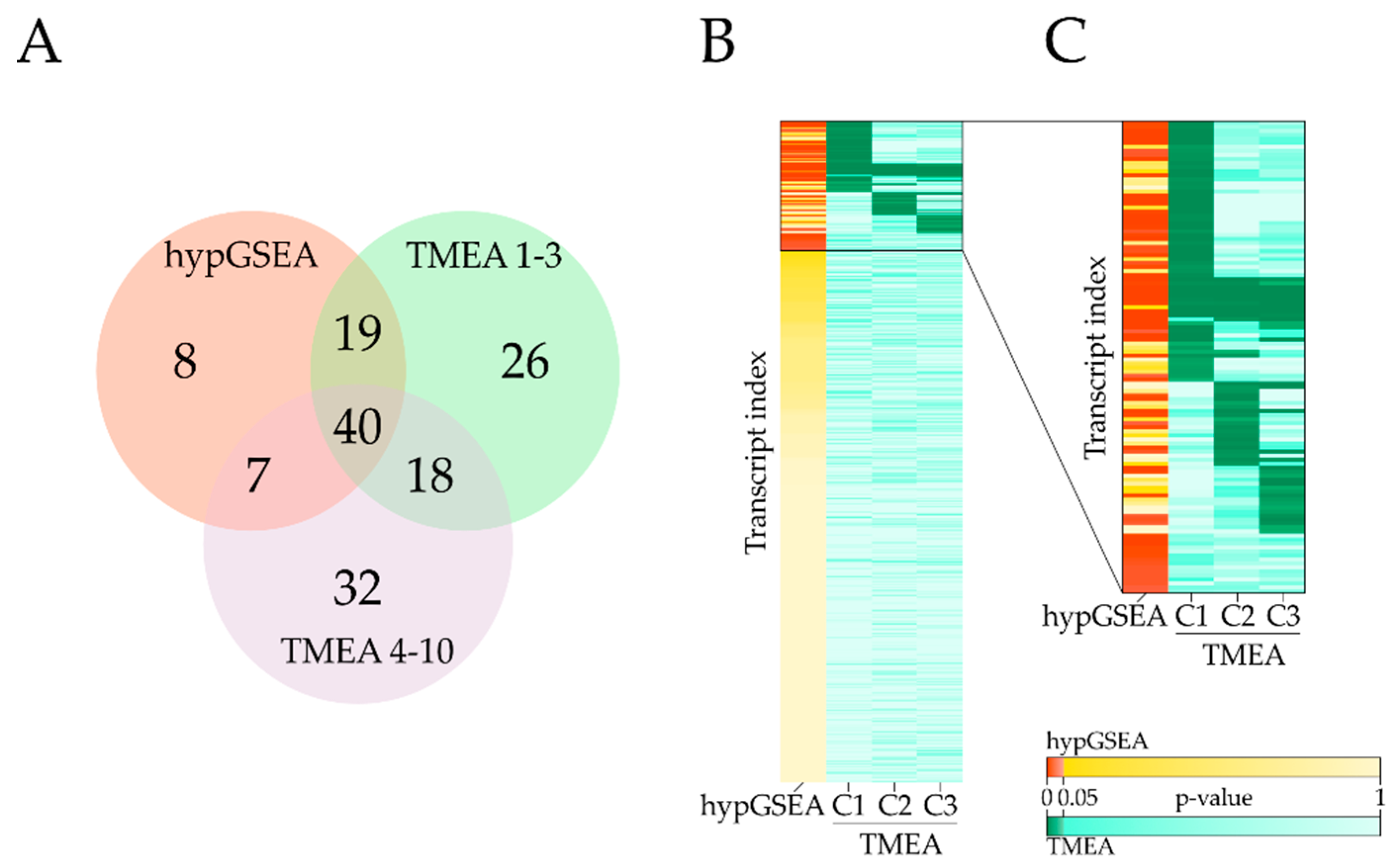
3.3. Comparison with Hypergeometric Test Based GSEA
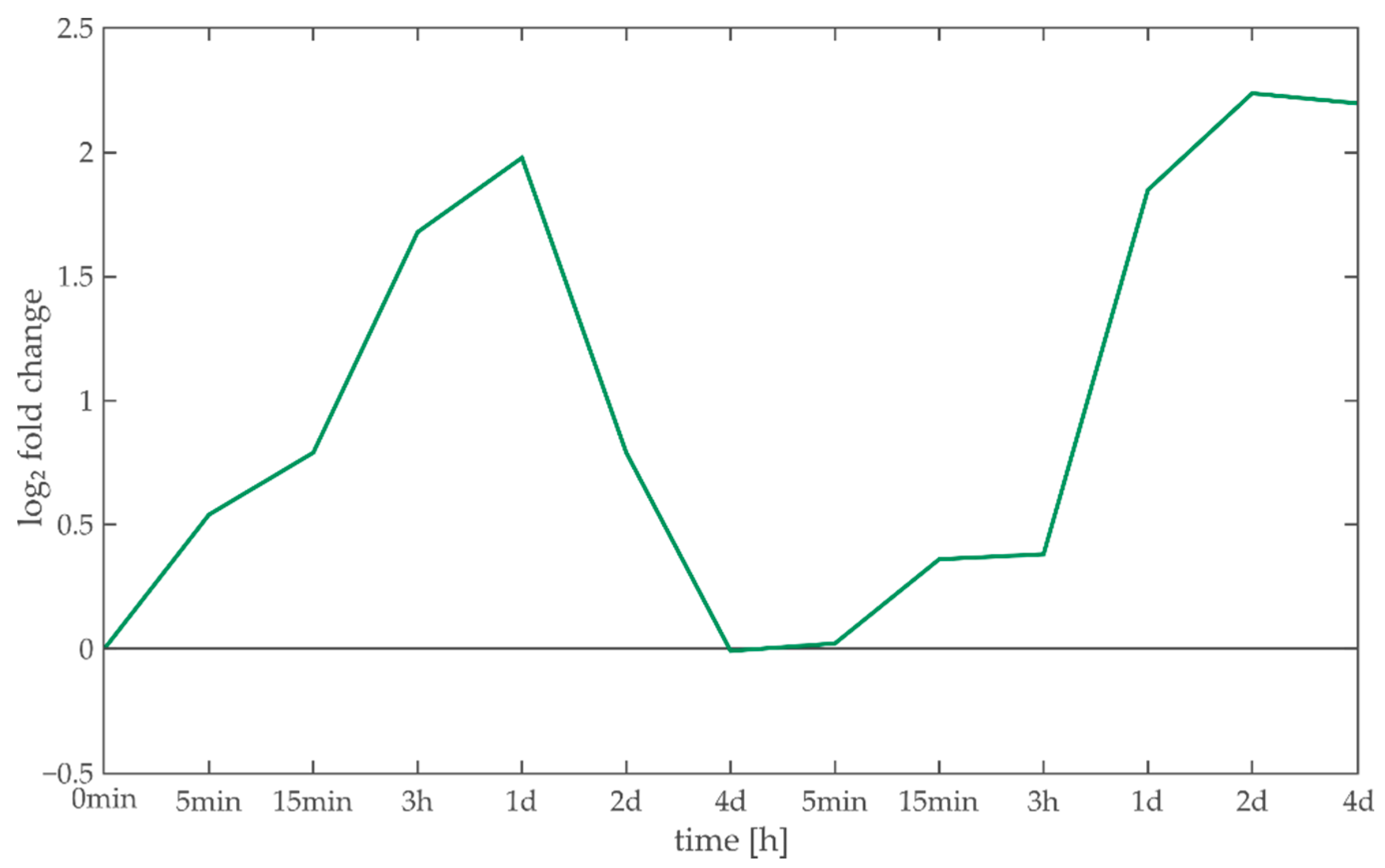
3.4. Case Study: Characterization of Light Acclimation in Arabidopsis thaliana
3.4.1. Anthocyanins
3.4.2. Myb-Related Transcription Factor Family
3.4.3. Ribosomes
3.4.4. Light/Calcium Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEG | Differentially expressed gene |
| FAS | Functionally annotated set |
| FCS | Functional Class Scoring |
| FDR | False discovery rate |
| GSEA | Gene set enrichment analysis |
| hypGSEA | Gene set enrichment analysis based on hypergeometric tests |
| TMEA | Thermodynamically motivated enrichment analysis |
| SA | Surprisal analysis |
| SS | Single-Sample |
| SVD | singular value decomposition |
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cID | MapMan Annotation (FAS) | cID | MapMan Annotation (FAS) |
|---|---|---|---|
| 1 | cell wall.cell wall proteins | 4 | major CHO metabolism |
| 1 | cell wall.cell wall proteins.AGPs | 4 | major CHO metabolism.degradation |
| 1 | cell wall.cell wall proteins.AGPs.AGP | 4 | major CHO metabolism.degradation.starch |
| 1 | cell wall.pectin*esterases.misc | 4 | misc.invertase/pectin methylesterase inhibitor family protein |
| 1 | lipid metabolism.FA desaturation | 4 | not assigned.no ontology.DC1 domain containing protein |
| 1 | lipid metabolism.FA desaturation.desaturase | 4 | not assigned.unknown |
| 1 | misc.beta 1,3 glucan hydrolases | 4 | RNA.regulation of transcription.AP2/EREBP, APETALA2/Ethylene-responsive element binding protein family |
| 1 | misc.beta 1,3 glucan hydrolases.glucan endo-1,3-beta-glucosidase | 4 | RNA.regulation of transcription.C2C2(Zn) CO-like, Constans-like zinc finger family |
| 1 | misc.glutathione S transferases | 4 | RNA.regulation of transcription.C2C2(Zn) DOF zinc finger family |
| 1 | misc.nitrilases, *nitrile lyases, berberine bridge enzymes, reticuline oxidases, troponine reductases | 4 | RNA.regulation of transcription.MYB-related transcription factor family |
| 1 | misc.O-methyl transferases | 4 | RNA.regulation of transcription.Psudo ARR transcription factor family |
| 1 | misc.protease inhibitor/seed storage/lipid transfer protein (LTP) family protein | 4 | secondary metabolism.isoprenoids.terpenoids |
| 1 | nucleotide metabolism.synthesis.purine | 4 | secondary metabolism.phenylpropanoids.lignin biosynthesis |
| 1 | protein | 4 | stress.abiotic |
| 1 | protein.degradation.AAA type | 4 | stress.abiotic.cold |
| 1 | protein.synthesis | 4 | stress.biotic.respiratory burst |
| 1 | protein.synthesis.ribosomal protein | 4 | transport.sulfate |
| 1 | protein.synthesis.ribosomal protein.eukaryotic | 5 | cell wall |
| 1 | protein.synthesis.ribosomal protein.eukaryotic.40S subunit | 5 | cell wall.modification |
| 1 | protein.synthesis.ribosomal protein.eukaryotic.60S subunit | 5 | misc |
| 1 | protein.synthesis.ribosomal protein.prokaryotic.chloroplast | 5 | secondary metabolism |
| 1 | protein.synthesis.ribosomal protein.prokaryotic.chloroplast.50S subunit | 5 | secondary metabolism.flavonoids |
| 1 | protein.synthesis.ribosome biogenesis | 5 | secondary metabolism.flavonoids.anthocyanins |
| 1 | protein.synthesis.ribosome biogenesis.Pre-rRNA processing and modifications | 5 | secondary metabolism.flavonoids.anthocyanins.anthocyanin 5-aromatic acyltransferase |
| 1 | protein.synthesis.ribosome biogenesis.Pre-rRNA processing and modifications.snoRNPs | 5 | secondary metabolism.flavonoids.dihydroflavonols |
| 1 | protein.synthesis.ribosome biogenesis.Pre-rRNA processing and modifications.WD-repeat proteins | 5 | stress |
| 1 | redox.glutaredoxins | 5 | stress.biotic |
| 1 | RNA.regulation of transcription.ARR | 5 | transport |
| 1 | RNA.regulation of transcription.NAC domain transcription factor family | 6 | cell wall.degradation |
| 1 | RNA.regulation of transcription.WRKY domain transcription factor family | 6 | cell wall.degradation.mannan-xylose-arabinose-fucose |
| 1 | secondary metabolism.simple phenols | 6 | DNA.synthesis/chromatin structure.retrotransposon/transposase |
| 1 | signaling | 6 | DNA.synthesis/chromatin structure.retrotransposon/transposase.gypsy-like retrotransposon |
| 1 | signaling.in sugar and nutrient physiology | 6 | hormone metabolism |
| 1 | signaling.receptor kinases.DUF 26 | 6 | hormone metabolism.auxin |
| 1 | signaling.receptor kinases.misc | 6 | minor CHO metabolism |
| 1 | signaling.receptor kinases.wall associated kinase | 6 | minor CHO metabolism.trehalose |
| 1 | signaling.receptor kinases.wheat LRK10 like | 6 | minor CHO metabolism.trehalose.potential TPS/TPP |
| 1 | stress.biotic.PR-proteins.plant defensins | 6 | misc.gluco-, galacto- and mannosidases |
| 1 | transport.Major Intrinsic Proteins | 6 | not assigned.no ontology.glycine rich proteins |
| 2 | amino acid metabolism.synthesis | 6 | not assigned.no ontology.pentatricopeptide (PPR) repeat-containing protein |
| 2 | amino acid metabolism.synthesis.aspartate family | 6 | PS.lightreaction |
| 2 | development.storage proteins | 6 | PS.lightreaction.photosystem II |
| 2 | hormone metabolism.auxin.induced-regulated-responsive-activated | 6 | PS.lightreaction.photosystem II.LHC-II |
| 2 | nucleotide metabolism.synthesis | 6 | secondary metabolism.flavonoids.chalcones |
| 2 | protein.synthesis.ribosomal protein.eukaryotic.60S subunit.L7A | 6 | secondary metabolism.flavonoids.flavonols |
| 2 | protein.synthesis.ribosomal protein.prokaryotic | 6 | secondary metabolism.phenylpropanoids |
| 2 | signaling.calcium | 6 | signaling.light |
| 2 | stress.biotic.receptors | 6 | transport.ABC transporters and multidrug resistance systems |
| 2 | transport.Major Intrinsic Proteins.PIP | 6 | transport.sugars |
| 3 | misc.cytochrome P450 | ||
| 3 | misc.GDSL-motif lipase | ||
| 3 | misc.peroxidases | ||
| 3 | signaling.receptor kinases | ||
| 3 | stress.biotic.PR-proteins |
References
- Ruffel, S.; Krouk, G.; Coruzzi, G.M. A systems view of responses to nutritional cues in Arabidopsis: Toward a paradigm shift for predictive network modeling. Plant Physiol. 2010, 152, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Anjum, N.A. Plant acclimation to environmental stress: A critical appraisal. Front. Plant Sci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Razzaq, A.; Mehmood, S.S.; Zou, X.; Zhang, X.; Lv, Y.; Xu, J. Impact of Climate Change on Crops Adaptation and Strategies to Tackle Its Outcome: A Review. Plants 2019, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minorsky, P.V. Achieving the in Silico Plant. Systems Biology and the Future of Plant Biological Research. Plant Physiol. 2003, 132, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Beine-Golovchuk, O.; Firmino, A.A.P.; Dąbrowska, A.; Schmidt, S.; Erban, A.; Walther, D.; Zuther, E.; Hincha, D.K.; Kopka, J. Plant Temperature Acclimation and Growth Rely on Cytosolic Ribosome Biogenesis Factor Homologs. Plant Physiol. 2018, 176, 2251–2276. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, P.; Bräutigam, A.; Buijs, V.A.; Tazelaar, A.O.E.; van der Werf, A.; Schlüter, U.; Reichart, G.-J.; Bolger, A.; Usadel, B.; Weber, A.P.M.; et al. Metabolic Adaptation, a Specialized Leaf Organ Structure and Vascular Responses to Diurnal N2 Fixation by Nostoc azollae Sustain the Astonishing Productivity of Azolla Ferns without Nitrogen Fertilizer. Front. Plant Sci. 2017, 8, 442. [Google Scholar] [CrossRef] [Green Version]
- Hemme, D.; Veyel, D.; Mühlhaus, T.; Sommer, F.; Jüppner, J.; Unger, A.-K.; Sandmann, M.; Fehrle, I.; Schnfelder, S.; Steup, M.; et al. Systems-Wide Analysis of Acclimation Responses to Long-Term Heat Stress and Recovery in the Photosynthetic Model Organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 4270–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettler, T.; Mühlhaus, T.; Hemme, D.; Schöttler, M.-A.; Rupprecht, J.; Idoine, A.; Veyel, D.; Pal, S.K.; Yaneva-Roder, L.; Winck, F.V.; et al. Systems Analysis of the Response of Photosynthesis, Metabolism, and Growth to an Increase in Irradiance in the Photosynthetic Model Organism Chlamydomonas reinhardtii. Plant Cell 2014, 26, 2310–2350. [Google Scholar] [CrossRef] [Green Version]
- Rademacher, N.; Wrobel, T.J.; Rossoni, A.W.; Kurz, S.; Bräutigam, A.; Weber, A.P.M.; Eisenhut, M. Transcriptional response of the extremophile red alga Cyanidioschyzon merolae to changes in CO2 concentrations. J. Plant Physiol. 2017, 217, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Schmollinger, S.; Mühlhaus, T.; Boyle, N.R.; Blaby, I.K.; Casero, D.; Mettler, T.; Moseley Jeffrey, L.; Kropat, J.; Sommer, F.; Strenkert, D.; et al. Nitrogen-Sparing Mechanisms in Chlamydomonas Affect the Transcriptome, the Proteome, and Photosynthetic Metabolism. Plant Cell 2014, 26, 1410–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valledor, L.; Furuhashi, T.; Hanak, A.-M.; Weckwerth, W. Systemic Cold Stress Adaptation of Chlamydomonas reinhardtii*. Mol. Cell Proteom. 2013, 12, 2032–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandalinas, S.I.; Sengupta, S.; Burks, D.; Azad, R.K.; Mittler, R. Identification and characterization of a core set of ROS wave-associated transcripts involved in the systemic acquired acclimation response of Arabidopsis to excess light. Plant J. 2019, 98, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Zuther, E.; Schaarschmidt, S.; Fischer, A.; Erban, A.; Pagter, M.; Mubeen, U.; Giavalisco, P.; Kopka, J.; Sprenger, H.; Hincha, D.K. Molecular signatures associated with increased freezing tolerance due to low temperature memory in Arabidopsis. Plant Cell Environ. 2019, 42, 854–873. [Google Scholar] [CrossRef]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Mller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. Cell Mol. Biol. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301-7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, P.D.; Ouzounis, C.A.; Moore-Kochlacs, C.; Goldovsky, L.; Kaipa, P.; Ahrén, D.; Tsoka, S.; Darzentas, N.; Kunin, V.; Lpez-Bigas, N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005, 33, 6083–6089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shahrour, F.; Díaz-Uriarte, R.; Dopazo, J. FatiGO: A web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004, 20, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Zeeberg, B.R.; Feng, W.; Wang, G.; Wang, M.D.; Fojo, A.T.; Sunshine, M.; Narasimhan, S.; Kane, D.W.; Reinhold, W.C.; Lababidi, S.; et al. GoMiner: A resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003, 4, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Storch, K.-F.; Lipan, O.; Kao, M.-C.J.; Weitz, C.J.; Wong, W.H. GoSurfer: A graphical interactive tool for comparative analysis of large gene sets in Gene Ontology space. Appl. Bioinform. 2004, 3, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Su, Z. EasyGO: Gene Ontology-based annotation and functional enrichment analysis tool for agronomical species. BMC Genom. 2007, 8, 246. [Google Scholar] [CrossRef]
- Zhang, B.; Schmoyer, D.; Kirov, S.; Snoddy, J. GOTree Machine (GOTM): A web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinform. 2004, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.-C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2007, 23, 401–407. [Google Scholar] [CrossRef]
- Pan, K.-H.; Lih, C.-J.; Cohen, S.N. Effects of threshold choice on biological conclusions reached during analysis of gene expression by DNA microarrays. Proc. Natl. Acad. Sci. USA 2005, 102, 8961–8965. [Google Scholar] [CrossRef] [Green Version]
- Tarca, A.L.; Bhatti, G.; Romero, R. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS ONE 2013, 8, e79217. [Google Scholar] [CrossRef]
- Shen, H.; West, M. Bayesian Modeling for Biological Pathway Annotation of Genomic Signatures; Department of Statistical Science, Duke University: Durham, NC, USA, 2008. [Google Scholar]
- Frost, H.R.; Li, Z.; Moore, J.H. Spectral gene set enrichment (SGSE). BMC Bioinform. 2015, 16, 70. [Google Scholar] [CrossRef] [Green Version]
- Dinu, I.; Potter, J.D.; Mueller, T.; Liu, Q.; Adewale, A.J.; Jhangri, G.S.; Einecke, G.; Famulski, K.S.; Halloran, P.; Yasui, Y. Improving gene set analysis of microarray data by SAM-GS. BMC Bioinform. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Simillion, C.; Liechti, R.; Lischer, H.E.L.; Ioannidis, V.; Bruggmann, R. Avoiding the pitfalls of gene set enrichment analysis with SetRank. BMC Bioinform. 2017, 18, 151. [Google Scholar] [CrossRef] [Green Version]
- Prifti, E.; Zucker, J.-D.; Clement, K.; Henegar, C. FunNet: An integrative tool for exploring transcriptional interactions. Bioinformatics 2008, 24, 2636–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-H.; Kim, M.-S.; Han, Y.; Yi, G.-S. COFECO: Composite function annotation enriched by protein complex data. Nucleic Acids Res. 2009, 37, W350-5. [Google Scholar] [CrossRef] [Green Version]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Håkansson, P.; Johansson, M.; Nelander, S.; Fioretos, T. Threshold-free high-power methods for the ontological analysis of genome-wide gene-expression studies. Genome Biol. 2007, 8, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glansdorff, P.; Prigogine, I.V. Thermodynamic: Theory of Structure, Stability; Wiley: London, UK, 1971. [Google Scholar]
- Zadran, S.; Arumugam, R.; Herschman, H.; Phelps, M.E.; Levine, R.D. Surprisal analysis characterizes the free energy time course of cancer cells undergoing epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 13235–13240. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.D. Information Theory Approach to Molecular Reaction Dynamics. Annu. Rev. Phys. Chem. 1978, 29, 59–92. [Google Scholar] [CrossRef]
- Agmon, N.; Alhassid, Y.; Levine, R.D. An algorithm for finding the distribution of maximal entropy. J. Comput. Phys. 1979, 30, 250–258. [Google Scholar] [CrossRef]
- Kravchenko-Balasha, N.; Remacle, F.; Gross, A.; Rotter, V.; Levitzki, A.; Levine, R.D. Convergence of logic of cellular regulation in different premalignant cells by an information theoretic approach. BMC Syst. Biol. 2011, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.; Levine, R.D. Surprisal analysis of transcripts expression levels in the presence of noise: A reliable determination of the onset of a tumor phenotype. PLoS ONE 2013, 8, e61554. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Molina, A.; Kleine, T.; Schneider, K.; Mühlhaus, T.; Lehmann, M.; Leister, D. Translational Components Contribute to Acclimation Responses to High Light, Heat, and Cold in Arabidopsis. iScience 2020, 23, 101331. [Google Scholar] [CrossRef] [PubMed]
- Remacle, F.; Kravchenko-Balasha, N.; Levitzki, A.; Levine, R.D. Information-theoretic analysis of phenotype changes in early stages of carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 10324–10329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.; Li, C.M.; Remacle, F.; Levine, R.D. Free energy rhythms in Saccharomyces cerevisiae: A dynamic perspective with implications for ribosomal biogenesis. Biochemistry 2013, 52, 1641–1648. [Google Scholar] [CrossRef] [Green Version]
- Procaccia, I.; Levine, R.D. Potential work: A statistical-mechanical approach for systems in disequilibrium. J. Chem. Phys. 1976, 65, 3357–3364. [Google Scholar] [CrossRef]
- CSBiology. TMEA Package. 8/16/2020. Available online: https://github.com/CSBiology/TMEA (accessed on 16 August 2020).
- Anderson, E.; Bai, Z.; Bischof, C.; Blackford, S.; Demmel, J.; Dongarra, J.; Du Croz, J.; Greenbaum, A.; Hammarling, S.; McKenney, A.; et al. LAPACK Users’ Guide, 3rd ed.; SIAM: Philadelphia, PA, USA, 1999. [Google Scholar]
- NCBO BioPortal. GoMapMan—Summary. 2016. Available online: https://bioportal.bioontology.org/ontologies/GMM (accessed on 14 August 2020).
- MapMan. MapManStore—Ath_AFFY_ATH1_TAIR10_Aug2012. 14/08/2020. Available online: https://mapman.gabipd.org/mapmanstore (accessed on 14 August 2020).
- KEGG. KEGG COMPOUND Database. 14/08/2020. Available online: https://www.genome.jp/kegg/compound (accessed on 14 August 2020).
- KEGG. KEGG BRITE: KEGG Orthology (KO)—Arabidopsis thaliana (thale cress). 14/08/2020. Available online: https://www.genome.jp/kegg-bin/get_htext?ath00001 (accessed on 14 August 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Whitehouse, N.; Cho, J.; Shaw, C. OntologyTraverser: An R package for GO analysis. Bioinformatics 2005, 21, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- CSBiology. FSharp.Stats. 13/08/2020. Available online: https://github.com/CSBiology/FSharp.Stats (accessed on 13 August 2020).
- CSBiology. BioFSharp. 13/08/2020. Available online: https://github.com/CSBiology/BioFSharp (accessed on 13 August 2020).
- Mühlhaus, T. FSharp.Plotly. 13/08/2020. Available online: https://github.com/muehlhaus/FSharp.Plotly (accessed on 13 August 2020).
- Knijnenburg, T.A.; Wessels, L.F.A.; Reinders, M.J.T.; Shmulevich, I. Fewer permutations, more accurate P-values. Bioinformatics 2009, 25, i161–i168. [Google Scholar] [CrossRef] [Green Version]
- Emmott, E.; Jovanovic, M.; Slavov, N. Ribosome Stoichiometry: From Form to Function. Trends Biochem. Sci. 2019, 44, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Agresti, A.; Min, Y. On small-sample confidence intervals for parameters in discrete distributions. Biometrics 2001, 57, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Harvaux, M.; Kloppstech, K. The protective functions of carotenoid and flavonoid pigments against excess visible radiation at chilling temperature investigated in Arabidopsis npq and tt mutants. Planta 2001, 213, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Trojak, M.; Skowron, E. Role of anthocyanins in highlight stress response. World Sci. News 2017, 81, 150–168. [Google Scholar]
- Gould, K.S.; Dudle, D.A.; Neufeld, H.S. Why some stems are red: Cauline anthocyanins shield photosystem II against high light stress. J. Exp. Bot. 2010, 61, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.-Q.; Chow, W.S.; Su, L.-J.; Peng, X.-X.; Peng, C.-L. Protective effect of supplemental anthocyanins on Arabidopsis leaves under high light. Physiol. Plant 2010, 138, 215–225. [Google Scholar] [CrossRef]
- Page, M.; Sultana, N.; Paszkiewicz, K.; Florance, H.; Smirnoff, N. The influence of ascorbate on anthocyanin accumulation during high light acclimation in Arabidopsis thaliana: Further evidence for redox control of anthocyanin synthesis. Plant Cell Environ. 2012, 35, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Grayer, R.J. Anthocyanins and other flavonoids. Nat. Prod. Rep. 2004, 21, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, K.; Sun, Z.; Yan, M.; Chen, C.; Zhang, X.; Tang, Y.; Wu, Y. LNK1 and LNK2 Corepressors Interact with the MYB3 Transcription Factor in Phenylpropanoid Biosynthesis. Plant Physiol. 2017, 174, 1348–1358. [Google Scholar] [CrossRef]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, e0152. [Google Scholar] [CrossRef] [Green Version]
- Lamers, J.; van der Meer, T.; Testerink, C. How Plants Sense and Respond to Stressful Environments. Plant Physiol. 2020, 182, 1624–1635. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Jones, J.D.G. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Rossini, S.; Casazza, A.P.; Engelmann, E.C.M.; Havaux, M.; Jennings, R.C.; Soave, C. Suppression of both ELIP1 and ELIP2 in Arabidopsis does not affect tolerance to photoinhibition and photooxidative stress. Plant Physiol. 2006, 141, 1264–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine, T.; Kindgren, P.; Benedict, C.; Hendrickson, L.; Strand, A. Genome-wide gene expression analysis reveals a critical role for CRYPTOCHROME1 in the response of Arabidopsis to high irradiance. Plant Physiol. 2007, 144, 1391–1406. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.A.; Cloix, C.; Jiang, G.H.; Kaiserli, E.; Herzyk, P.; Kliebenstein, D.J.; Jenkins, G.I. A UV-B-specific signaling component orchestrates plant UV protection. Proc. Natl. Acad. Sci. USA 2005, 102, 18225–18230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayami, N.; Sakai, Y.; Kimura, M.; Saito, T.; Tokizawa, M.; Iuchi, S.; Kurihara, Y.; Matsui, M.; Nomoto, M.; Tada, Y.; et al. The Responses of Arabidopsis Early Light-Induced Protein2 to Ultraviolet B, High Light, and Cold Stress Are Regulated by a Transcriptional Regulatory Unit Composed of Two Elements. Plant Physiol. 2015, 169, 840–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutin, C.; Nussaume, L.; Moise, N.; Moya, I.; Kloppstech, K.; Havaux, M. Early light-induced proteins protect Arabidopsis from photooxidative stress. Proc. Natl. Acad. Sci. USA 2003, 100, 4921–4926. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkova-Chevolleau, T.; Franck, F.; Alawady, A.E.; Dall’Osto, L.; Carrière, F.; Bassi, R.; Grimm, B.; Nussaume, L.; Havaux, M. The light stress-induced protein ELIP2 is a regulator of chlorophyll synthesis in Arabidopsis thaliana. Plant J. 2007, 50, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Mahajan, S. Calcium signaling network in plants: An overview. Plant Signal Behav. 2007, 2, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.; Brownlee, C.; Harper, J.F. Communicating with calcium. Plant Cell 1999, 11, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Gagneur, J.; Robinson, P.N. GOing Bayesian: Model-based gene set analysis of genome-scale data. Nucleic Acids Res. 2010, 38, 3523–3532. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Rosenfeld, R.; Simon, I.; Nau, G.J.; Bar-Joseph, Z. A probabilistic generative model for GO enrichment analysis. Nucleic Acids Res. 2008, 36, e109. [Google Scholar] [CrossRef]
- Raghavan, N.; Amaratunga, D.; Cabrera, J.; Nie, A.; Qin, J.; McMillian, M. On methods for gene function scoring as a means of facilitating the interpretation of microarray results. J. Comput. Biol. 2006, 13, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, A.; Komatsu, S. Impact of Post-Translational Modifications of Crop Proteins under Abiotic Stress. Proteomes 2016, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bhattacharya, S.; Pi, J.; Clewell, R.A.; Carmichael, P.L.; Andersen, M.E. Adaptive Posttranslational Control in Cellular Stress Response Pathways and Its Relationship to Toxicity Testing and Safety Assessment. Toxicol. Sci. 2015, 147, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaert, K.A.; Perez, E.; Rumin, J.; Giltay, A.; Carone, M.; Coosemans, N.; Radoux, M.; Eppe, G.; Levine, R.D.; Remacle, F.; et al. Metabolic, Physiological, and Transcriptomics Analysis of Batch Cultures of the Green Microalga Chlamydomonas Grown on Different Acetate Concentrations. Cells 2019, 8, 1367. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, K.A.; Manoharan-Basil, S.S.; Perez, E.; Levine, R.D.; Remacle, F.; Remacle, C. Surprisal analysis of genome-wide transcript profiling identifies differentially expressed genes and pathways associated with four growth conditions in the microalga Chlamydomonas. PLoS ONE 2018, 13, e0195142. [Google Scholar] [CrossRef]
- Willamme, R.; Alsafra, Z.; Arumugam, R.; Eppe, G.; Remacle, F.; Levine, R.D.; Remacle, C. Metabolomic analysis of the green microalga Chlamydomonas reinhardtii cultivated under day/night conditions. J. Biotechnol. 2015, 215, 20–26. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Stone, B.A.B.; Bowerman, A.F.; Eichten, S.R.; Pogson, B.J. Excess Light Priming in Arabidopsis thaliana Genotypes with Altered DNA Methylomes. G3 2019, 9, 3611–3621. [Google Scholar] [CrossRef] [Green Version]
- Remacle, F.; Goldstein, A.; Levine, R. Multivariate Surprisal Analysis of Gene Expression Levels. Entropy 2016, 18, 445. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, K.; Venn, B.; Mühlhaus, T. TMEA: A Thermodynamically Motivated Framework for Functional Characterization of Biological Responses to System Acclimation. Entropy 2020, 22, 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/e22091030
Schneider K, Venn B, Mühlhaus T. TMEA: A Thermodynamically Motivated Framework for Functional Characterization of Biological Responses to System Acclimation. Entropy. 2020; 22(9):1030. https://0-doi-org.brum.beds.ac.uk/10.3390/e22091030
Chicago/Turabian StyleSchneider, Kevin, Benedikt Venn, and Timo Mühlhaus. 2020. "TMEA: A Thermodynamically Motivated Framework for Functional Characterization of Biological Responses to System Acclimation" Entropy 22, no. 9: 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/e22091030