The Flavonoid Kaempferol Ameliorates Streptozotocin-Induced Diabetes by Suppressing Hepatic Glucose Production

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
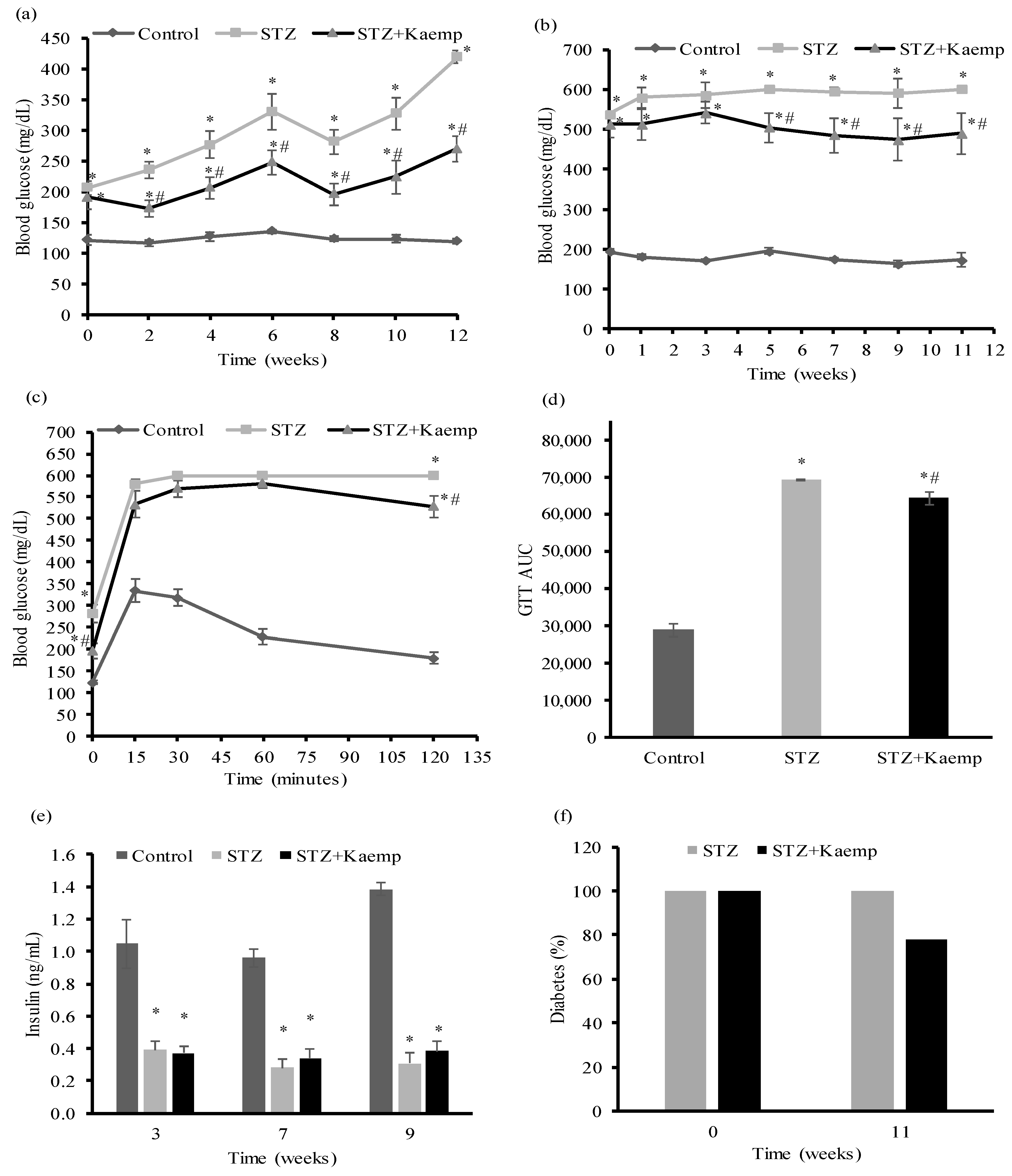
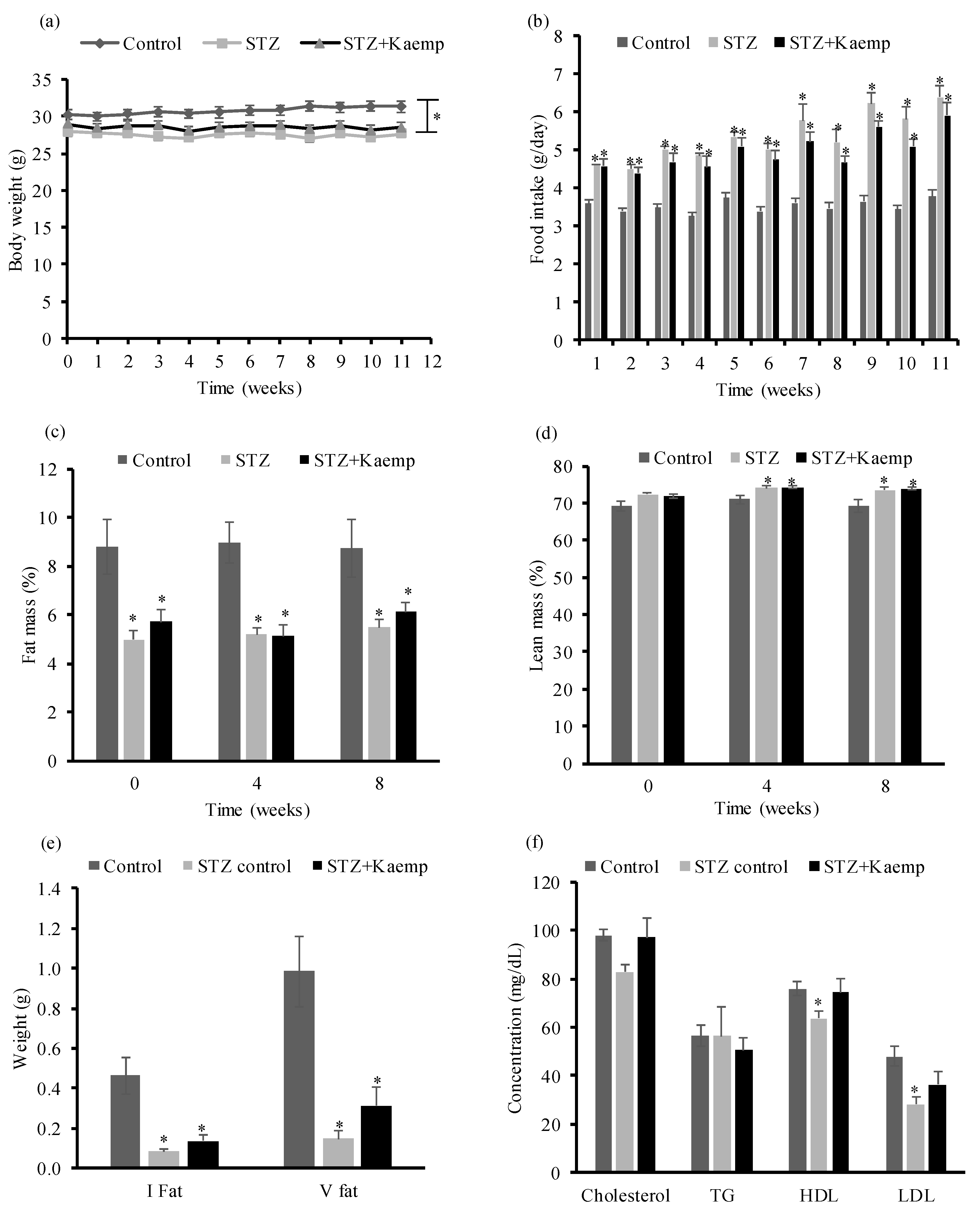
2.1. Kaempferol Treatment Ameliorated HyperGlycemia in Diabetic Mice
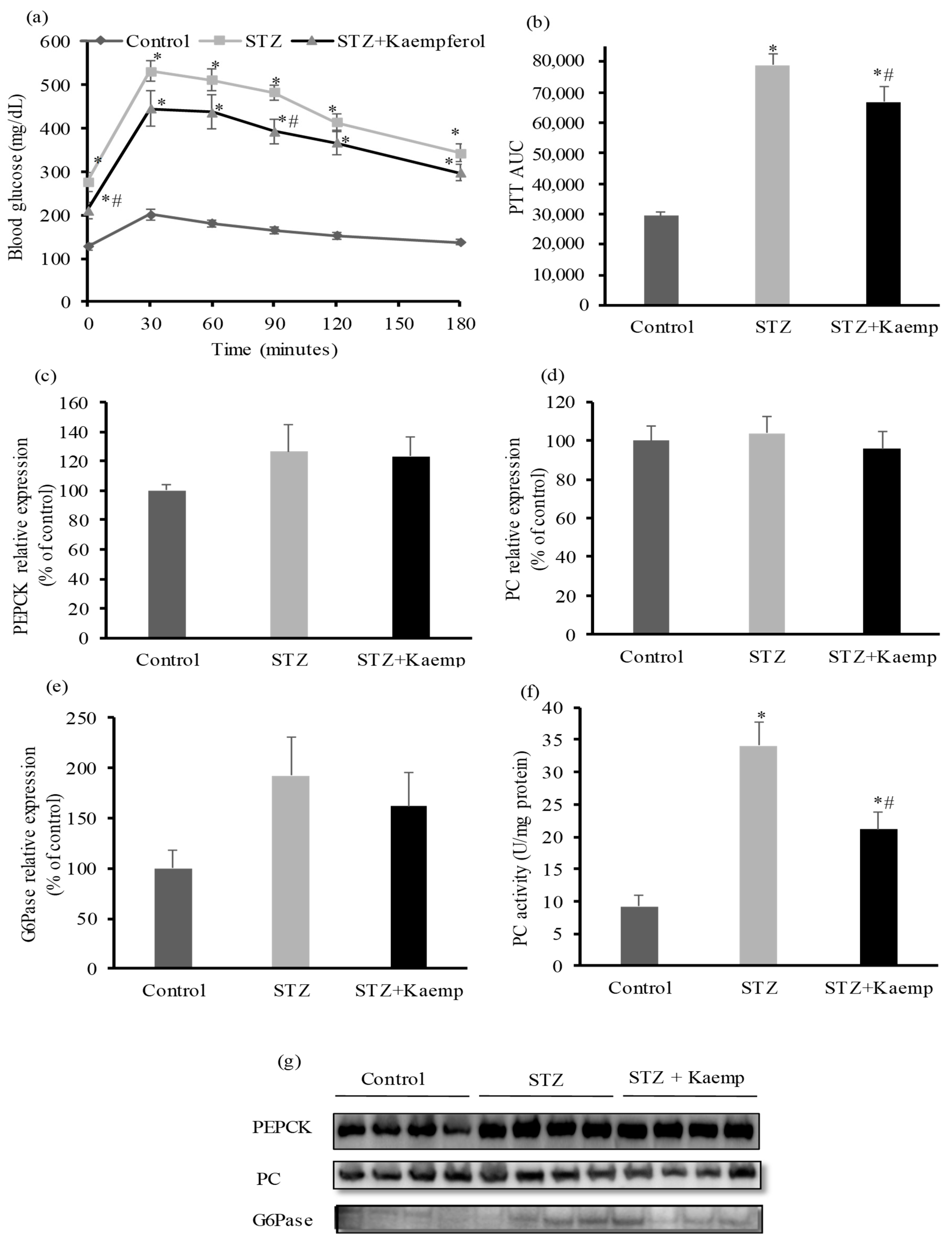
2.2. Kaempferol Suppressed Hepatic Gluconeogenesis
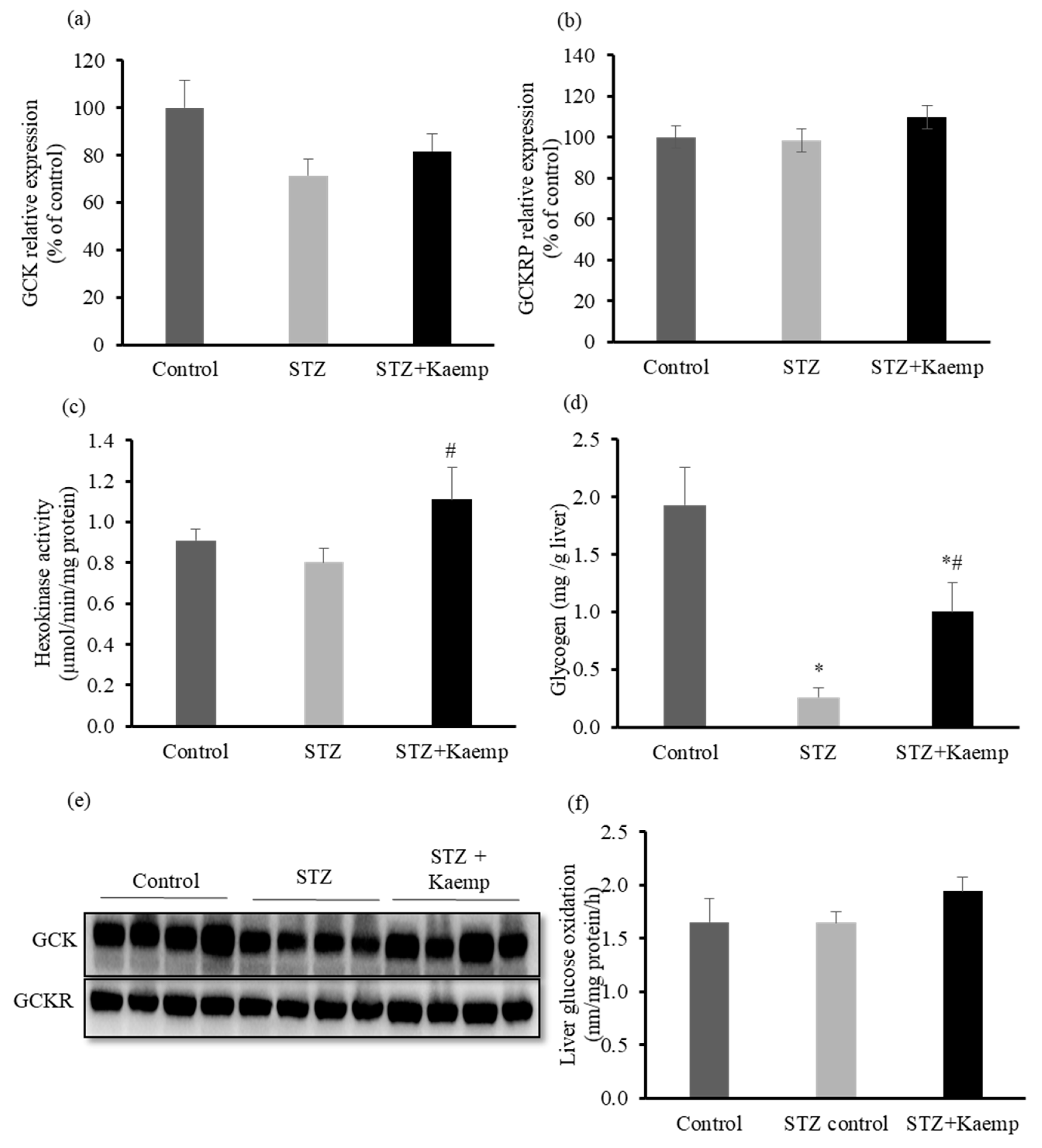
2.3. Kaempferol Increased GCK Activity and Glycogen Content in the l-Liver without Affecting Glucose Oxidation
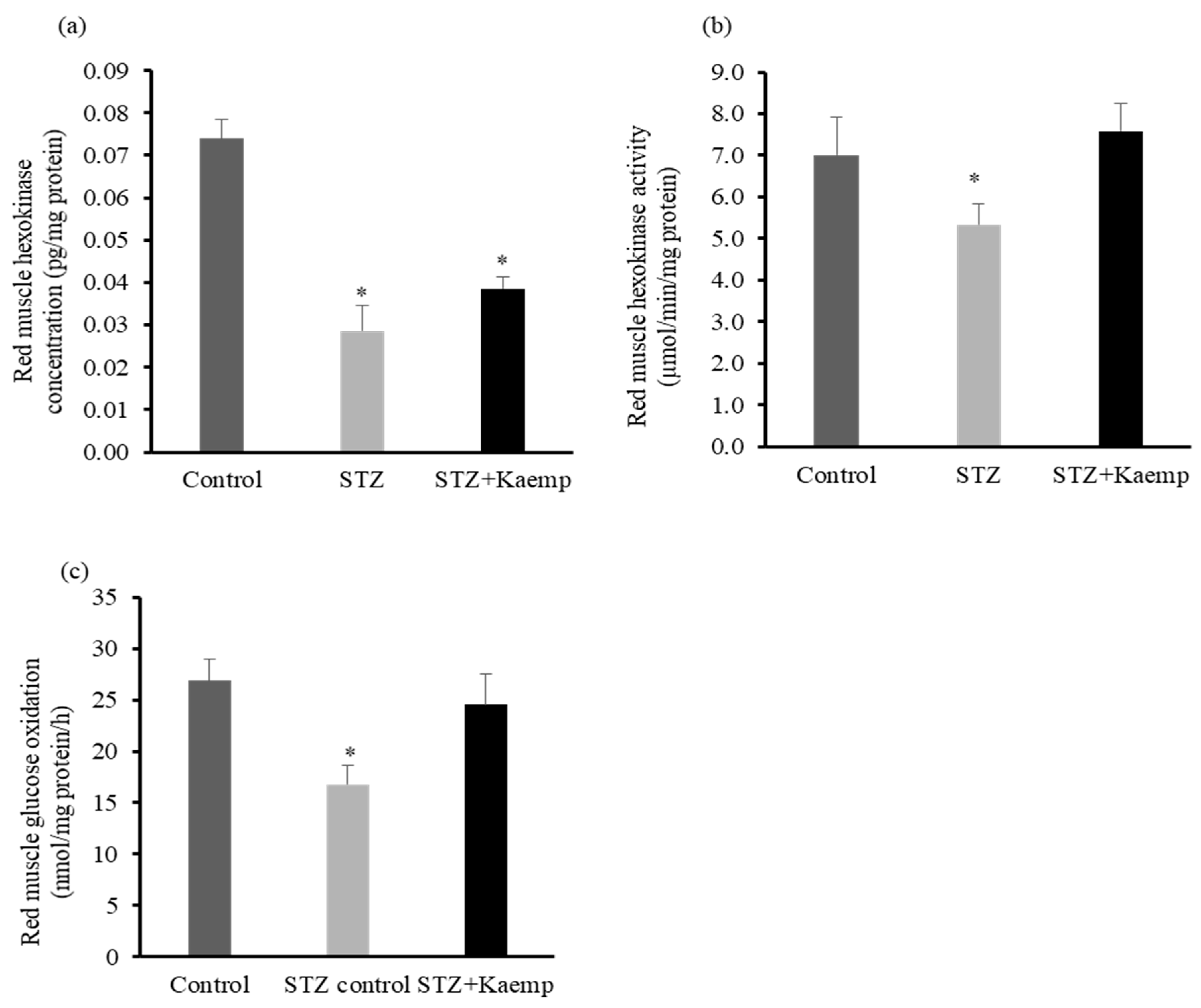
2.4. Kaempferol Increased Hexokinase Activity and Glucose Oxidation in Red Muscle
3. Discussion
4. Methods
4.1. Mice, STZ Administration, and Kaempferol Treatment
4.2. Metabolic Studies
4.3. Glucose Oxidation Assay
4.4. Enzyme Activity Assays
4.5. Glycogen Content Measurement
4.6. Western Blot Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Agius, L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 2008, 414, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pagliassotti, M.J.; Cherrington, A.D. Regulation of net hepatic glucose uptake in vivo. Annu. Rev. Physiol. 1992, 54, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, D.S.; Cardin, S.; Emshwiller, M.; Neal, D.; Chandramouli, V.; Schumann, W.C.; Landau, B.R.; Rossetti, L.; Cherrington, A.D. Small increases in insulin inhibit hepatic glucose production solely caused by an effect on glycogen metabolism. Diabetes 2001, 50, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Sindelar, D.K.; Chu, C.A.; Venson, P.; Donahue, E.P.; Neal, D.W.; Cherrington, A.D. Basal hepatic glucose production is regulated by the portal vein insulin concentration. Diabetes 1998, 47, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, B.; Gastaldelli, A.; Antoniello, S.; Auletta, M.; Pardo, F.; Ciociaro, D.; Guida, R.; Ferrannini, E.; Sacca, L. Splanchnic and leg substrate exchange after ingestion of a natural mixed meal in humans. Diabetes 1999, 48, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Scheen, A.J.; Albert, A.; Lefebvre, P.J. Effects of pulsatile delivery of insulin and glucagon in humans. Am. J. Physiol. 1989, 257, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Jitrapakdee, S.; Vidal-Puig, A.; Wallace, J.C. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell. Mol. Life Sci. 2006, 63, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Klover, P.J.; Mooney, R.A. Hepatocytes: Critical for glucose homeostasis. Int. J. Biochem. Cell Biol. 2004, 36, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, A.D. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes 1999, 48, 1198–1214. [Google Scholar] [CrossRef] [PubMed]
- Maggs, D.G.; Jacob, R.; Rife, F.; Lange, R.; Leone, P.; During, M.J.; Tamborlane, W.V.; Sherwin, R.S. Interstitial fluid concentrations of glycerol, glucose, and amino acids in human quadricep muscle and adipose tissue. Evidence for significant lipolysis in skeletal muscle. J. Clin. Investig. 1995, 96, 370–377. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010, 33 (Suppl. 1), S62–S69. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, V.A.; Yki-Jarvinen, H. Changes in muscle glucose metabolism in type 1 diabetes. Ann. Med. 1990, 22, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Morral, N.; McEvoy, R.; Dong, H.J.; Meseck, M.; Altomonte, J.; Thung, S.; Woo, S.L.C. Adenovirus-mediated expression of glucokinase in the liver as an adjuvant treatment for type 1 diabetes. Hum. Gene Ther. 2002, 13, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.K.; Fromm, H.J. Effect of Streptozotocin-Induced Diabetes and Insulin-Treatment on the Synthesis of Hexokinase-Ii in the Skeletal-Muscle of the Rat. Arch. Biochem. Biophys. 1986, 249, 61–69. [Google Scholar] [CrossRef]
- Hwang, J.H.; Perseghin, G.; Rothman, D.L.; Cline, G.W.; Magnusson, I.; Petersen, K.F.; Shulman, G.I. Impaired net hepatic glycogen synthesis in insulin-dependent diabetic subjects during mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy study. J. Clin. Investig. 1995, 95, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Control of glycaemia. Bailliere’s Clin. Endocrinol. Metab. 1993, 7, 551–586. [Google Scholar] [CrossRef]
- Gerich, J.E.; Lorenzi, M.; Bier, D.M.; Schneider, V.; Tsalikian, E.; Karam, J.H.; Forsham, P.H. Prevention of human diabetic ketoacidosis by somatostatin. Evidence for an essential role of glucagon. N. Eng. J. Med. 1975, 292, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, M.Y.; Du, X.Q.; Charron, M.J.; Unger, R.H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 2011, 60, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Tamborlane, W.V.; Beck, R.W.; Bode, B.W.; Buckingham, B.; Chase, H.P.; Clemons, R.; Fiallo-Scharer, R.; Fox, L.A.; Gilliam, L.K.; Hirsch, I.B.; et al. Continuous glucose monitoring and intensive treatment of type 1 diabetes. N. Engl. J. Med. 2008, 359, 1464–1476. [Google Scholar] [PubMed]
- Miean, K.H.; Mohamed, S. Flavonoid (myricetin, quercetin, kaempferol, luteolin, and apigenin) content of edible tropical plants. J. Agric. Food Chem. 2001, 49, 3106–3112. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Perez-Guerrero, C.; Lopez-Lazaro, M. A review on the dietary flavonoid kaempferol. Mini-Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Suh, K.S.; Choi, M.C.; Chon, S.; Oh, S.; Woo, J.T.; Kim, S.W.; Kim, J.W.; Kim, Y.S. Kaempferol protects HIT-T15 pancreatic beta cells from 2-deoxy-D-ribose-induced oxidative damage. Phytother. Res. 2010, 24, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.S.; Choi, E.M.; Kwon, M.; Chon, S.; Oh, S.; Woo, J.T.; Kim, S.W.; Kim, J.W.; Kim, Y.S. Kaempferol attenuates 2-deoxy-d-ribose-induced oxidative cell damage in MC3T3-E1 osteoblastic cells. Biol. Pharm. Bull. 2009, 32, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Hamalainen, M.; Nieminen, R.; Vuorela, P.; Heinonen, M.; Moilanen, E. Anti-inflammatory effects of flavonoids: genistein, kaempferol, quercetin, and daidzein inhibit STAT-1 and NF-kappaB activations, whereas flavone, isorhamnetin, naringenin, and pelargonidin inhibit only NF-kappaB activation along with their inhibitory effect on iNOS expression and NO production in activated macrophages. Mediators Inflamm. 2007, 2007, 45673. [Google Scholar] [PubMed]
- Crespo, I.; Garcia-Mediavilla, M.V.; Gutierrez, B.; Sanchez-Campos, S.; Tunon, M.J.; Gonzalez-Gallego, J. A comparison of the effects of kaempferol and quercetin on cytokine-induced pro-inflammatory status of cultured human endothelial cells. Br. J. Nutr. 2008, 100, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Olszanecki, R.; Bujak-Gizycka, B.; Madej, J.; Suski, M.; Wolkow, P.P.; Jawien, J.; Korbut, R. Kaempferol, but not resveratrol inhibits angiotensin converting enzyme. J. Physiol. Pharmacol. 2008, 59, 387–392. [Google Scholar] [PubMed]
- Loizzo, M.R.; Said, A.; Tundis, R.; Rashed, K.; Statti, G.A.; Hufner, A.; Menichini, F. Inhibition of angiotensin converting enzyme (ACE) by flavonoids isolated from Ailanthus excelsa (Roxb) (Simaroubaceae). Phytother. Res. 2007, 21, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Shun, C.T.; Chen, T.M.; Chen, Y.H. 3-O-beta-d-glucosyl-(1→6)-beta-D-glucosyl-kaempferol isolated from Sauropus androgenus reduces body weight gain in Wistar rats. Biol. Pharm. Bull. 2006, 29, 2510–2513. [Google Scholar] [CrossRef] [PubMed]
- Da-Silva, W.S.; Harney, J.W.; Kim, B.W.; Li, J.; Bianco, S.D.; Crescenzi, A.; Christoffolete, M.A.; Huang, S.A.; Bianco, A.C. The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes 2007, 56, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, A.Y.; Li, M.; Chen, C.; Yao, Q. Ginkgo biloba extract kaempferol inhibits cell proliferation and induces apoptosis in pancreatic cancer cells. J. Surg. Res. 2008, 148, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Lakka, A.; Tsakalof, A.; Simos, G. The dietary flavonoid kaempferol effectively inhibits HIF-1 activity and hepatoma cancer cell viability under hypoxic conditions. Biochem. Biophys. Res. Commun. 2010, 398, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Kim, J.H.; Song, K.; Kim, S.H.; Yoon, J.H.; Kim, K.S. Kaempferol and quercetin, components of Ginkgo biloba extract (EGb 761), induce caspase-3-dependent apoptosis in oral cavity cancer cells. Phytother. Res. 2010, 24 (Suppl. 1), S77–S82. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.G.; Fleischer, N.; Shin, S.I. Insulin-dependent diabetes mellitus induced by subdiabetogenic doses of streptozotocin: obligatory role of cell-mediated autoimmune processes. Proc. Natl. Acad. Sci. USA 1980, 77, 6129–6133. [Google Scholar] [CrossRef] [PubMed]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Leiter, E.H. Selecting the “right” mouse model for metabolic syndrome and type 2 diabetes research. Methods Mol. Biol. 2009, 560, 1–17. [Google Scholar] [PubMed]
- Newsholme, E.A.; Dimitriadis, G. Integration of biochemical and physiologic effects of insulin on glucose metabolism. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. 2), S122–S134. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, M.J. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell 1989, 57, 305–315. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Printz, R.L.; Cusi, K.A.; Kinchington, P.; O’Doherty, R.M.; Osawa, H.; Sewell, C.; Consoli, A.; Granner, D.K.; DeFronzo, R.A. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am. J. Physiol. 1995, 269, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.D.; Leighton, B.; Parry-Billings, M.; West, D.; Newsholme, E.A. Effects of hypothyroidism on the sensitivity of glycolysis and glycogen synthesis to insulin in the soleus muscle of the rat. Biochem. J. 1989, 257, 369–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Like, A.A.; Rossini, A.A. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 1976, 193, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, R.; Sakurai, H.; Sasaki, H.; Faloona, G.; Valverde, I.; Baetens, D.; Orci, L.; Unger, R. Glucagon: Role in the hyperglycemia of diabetes mellitus. Science 1975, 187, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1975, 1, 14–16. [Google Scholar] [CrossRef]
- Iynedjian, P.B.; Jotterand, D.; Nouspikel, T.; Asfari, M.; Pilot, P.R. Transcriptional induction of glucokinase gene by insulin in cultured liver cells and its repression by the glucagon-cAMP system. J. Biol. Chem. 1989, 264, 21824–21829. [Google Scholar] [PubMed]
- Arden, C.; Petrie, J.L.; Tudhope, S.J.; Al-Oanzi, Z.; Claydon, A.J.; Beynon, R.J.; Towle, H.C.; Agius, L. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes 2011, 60, 3110–3120. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.H.; Yun, J.M. Fisetin inhibits hyperglycemia-induced proinflammatory cytokine production by epigenetic mechanisms. Evid. Based Complement. Alternat. Med. 2012, 2012, 639469. [Google Scholar] [CrossRef] [PubMed]
- Selvi, R.B.; Swaminathan, A.; Chatterjee, S.; Shanmugam, M.K.; Li, F.; Ramakrishnan, G.B.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Inhibition of p300 lysine acetyltransferase activity by luteolin reduces tumor growth in head and neck squamous cell carcinoma (HNSCC) xenograft mouse model. Oncotarget 2015, 6, 43806–43818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.M.; Kim, T.H.; Jo, S.H.; Kim, M.Y.; Ahn, Y.H. Acetylation of glucokinase regulatory protein decreases glucose metabolism by suppressing glucokinase activity. Sci. Rep. 2015, 5, 17395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Inaba, Y.; Kimura, K.; Matsumoto, M.; Kaneko, S.; Kasuga, M.; Inoue, H. Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat. Commun. 2018, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Peng, J.; An, H.; He, Q.; Boronina, T.; Guo, S.; White, M.F.; Cole, P.A.; He, L. Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dihingia, A.; Ozah, D.; Ghosh, S.; Sarkar, A.; Baruah, P.K.; Kalita, J.; Sil, P.C.; Manna, P. Vitamin K1 inversely correlates with glycemia and insulin resistance in patients with type 2 diabetes (T2D) and positively regulates SIRT1/AMPK pathway of glucose metabolism in liver of T2D mice and hepatocytes cultured in high glucose. J. Nutr. Biochem. 2018, 52, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.C.; Cherrington, A.D.; Wasserman, D.H. Regulation of hepatic and peripheral glucose disposal. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 343–364. [Google Scholar] [CrossRef]
- Saccomani, M.P.; Bonadonna, R.C.; Bier, D.M.; DeFronzo, R.A.; Cobelli, C. A model to measure insulin effects on glucose transport and phosphorylation in muscle: A three-tracer study. Am. J. Physiol. 1996, 270 Pt 1, E170–E185. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Mitrakou, A.; Marsh, H.; Schwenk, F.; Benn, J.; Sonnenberg, G.; Arcangeli, M.; Aoki, T.; Sorensen, J.; Berger, M.; et al. Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Investig. 1988, 81, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Price, T.B.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Direct Measurement of Change in Muscle Glycogen Concentration after a Mixed Meal in Normal Subjects. Am. J. Physiol. 1993, 265, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Manchester, J.; Salmons, S.; Lawrence, J.C., Jr. Glucose transporters in single skeletal muscle fibers. Relationship to hexokinase and regulation by contractile activity. J. Biol. Chem. 1994, 269, 12963–12967. [Google Scholar] [PubMed]
- Halseth, A.E.; Bracy, D.P.; Wasserman, D.H. Overexpression of hexokinase II increases insulinand exercise-stimulated muscle glucose uptake in vivo. Am. J. Physiol. 1999, 276 Pt 1, E70–E77. [Google Scholar] [CrossRef]
- Dullaart, R.P.F. Plasma-Lipoprotein Abnormalities in Type-1 (Insulin-Dependent) Diabetes-Mellitus. Neth. J. Med. 1995, 46, 44–54. [Google Scholar] [CrossRef]
- Winocour, P.H.; Durrington, P.N.; Ishola, M.; Anderson, D.C. Lipoprotein abnormalities in insulin-dependent diabetes mellitus. Lancet 1986, 1, 1176–1178. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Wilson, D.L.; LeBoeuf, R.C. Increased atherosclerosis in streptozotocin-induced diabetic mice. J. Clin. Investig. 1996, 97, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, A.C.; Feitosa-Filho, G.S.; Freitas, F.R.; Wajchenberg, B.L.; Maranhao, R.C. Lipoprotein metabolism in patients with type 1 diabetes under intensive insulin treatment. Lipids Health Disease 2013, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, G.; Tilly-Kiesi, M.K.; Patti, L.; Taskinen, M.R.; Pacioni, D.; Cassader, M.; Riccardi, G.; Rivellese, A.A. Effects of dietary cholesterol on plasma lipoproteins and their subclasses in IDDM patients. Diabetologia 1998, 41, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burcelin, R.; Eddouks, M.; Maury, J.; Kande, J.; Assan, R.; Girard, J. Excessive glucose production, rather than insulin resistance, accounts for hyperglycaemia in recent-onset streptozotocin-diabetic rats. Diabetologia 1995, 38, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, J.J.; Matsuda, M.; DeFronzo, R.A.; Bressler, R. In vitro and in vivo suppression of gluconeogenesis by inhibition of pyruvate carboxylase. Biochem. Pharmacol. 1997, 53, 67–74. [Google Scholar] [CrossRef]
- Attwood, P.V. The structure and the mechanism of action of pyruvate carboxylase. Int. J. Biochem. Cell Biol. 1995, 27, 231–249. [Google Scholar] [CrossRef]
- Utter, M.F.; Keech, D.B. Formation of oxaloacetate from pyruvate and carbon dioxide. J. Biol. Chem. 1960, 235, 17–18. [Google Scholar]
- Ju, Y.; Untereiner, A.; Wu, L.; Yang, G. H2S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells. Biochim. Biophys. Acta 2015, 1850, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Beddow, S.A.; Vatner, D.F.; Majumdar, S.K.; Cantley, J.L.; Guebre-Egziabher, F.; Fat, I.; Guigni, B.; Jurczak, M.J.; Birkenfeld, A.L.; et al. Targeting Pyruvate Carboxylase Reduces Gluconeogenesis and Adiposity and Improves Insulin Resistance. Diabetes 2013, 62, 2183–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhalidy, H.; Moore, W.; Zhang, Y.; McMillan, R.; Wang, A.; Ali, M.; Suh, K.S.; Zhen, W.; Cheng, Z.; Jia, Z.; et al. Small Molecule Kaempferol Promotes Insulin Sensitivity and Preserved Pancreatic beta -Cell Mass in Middle-Aged Obese Diabetic Mice. J. Diabetes Res. 2015, 2015, 532984. [Google Scholar] [CrossRef] [PubMed]
- Cortright, R.N.; Sandhoff, K.M.; Basilio, J.L.; Berggren, J.R.; Hickner, R.C.; Hulver, M.W.; Dohm, G.L.; Houmard, J.A. Skeletal muscle fat oxidation is increased in African-American and white women after 10 days of endurance exercise training. Obesity 2006, 14, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Hulver, M.W.; Berggren, J.R.; Carper, M.J.; Miyazaki, M.; Ntambi, J.M.; Hoffman, E.P.; Thyfault, J.P.; Stevens, R.; Dohm, G.L.; Houmard, J.A.; et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005, 2, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhalidy, H.; Moore, W.; Wang, A.; Luo, J.; McMillan, R.P.; Wang, Y.; Zhen, W.; Hulver, M.W.; Liu, D. Kaempferol ameliorates hyperglycemia through suppressing hepatic gluconeogenesis and enhancing hepatic insulin sensitivity in diet-induced obese mice. J. Nutr. Biochem. 2018, 58, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.; Morris, J.G. Pyruvate carboxylase in Rhodopseudomonas spheroides. J. Gen. Microbiol. 1969, 59, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhang, W.; Zhen, W.; Lum, H.; Nadler, J.; Bassaganya-Riera, J.; Jia, Z.; Wang, Y.; Misra, H.; Liu, D. Genistein induces pancreatic beta-cell proliferation through activation of multiple signaling pathways and prevents insulin-deficient diabetes in mice. Endocrinology 2010, 151, 3026–3037. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhalidy, H.; Moore, W.; Wang, Y.; Luo, J.; McMillan, R.P.; Zhen, W.; Zhou, K.; Liu, D. The Flavonoid Kaempferol Ameliorates Streptozotocin-Induced Diabetes by Suppressing Hepatic Glucose Production. Molecules 2018, 23, 2338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092338
Alkhalidy H, Moore W, Wang Y, Luo J, McMillan RP, Zhen W, Zhou K, Liu D. The Flavonoid Kaempferol Ameliorates Streptozotocin-Induced Diabetes by Suppressing Hepatic Glucose Production. Molecules. 2018; 23(9):2338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092338
Chicago/Turabian StyleAlkhalidy, Hana, Will Moore, Yao Wang, Jing Luo, Ryan P. McMillan, Wei Zhen, Kequan Zhou, and Dongmin Liu. 2018. "The Flavonoid Kaempferol Ameliorates Streptozotocin-Induced Diabetes by Suppressing Hepatic Glucose Production" Molecules 23, no. 9: 2338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23092338