
Benzimidazoles Downregulate Mdm2 and MdmX and Activate p53 in MdmX Overexpressing Tumor Cells

 ,
,
Abstract
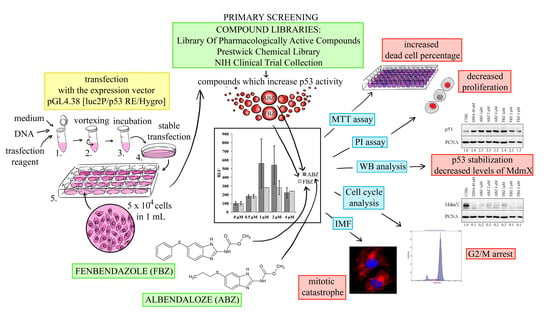
:
{kind=link}
{kind=link}
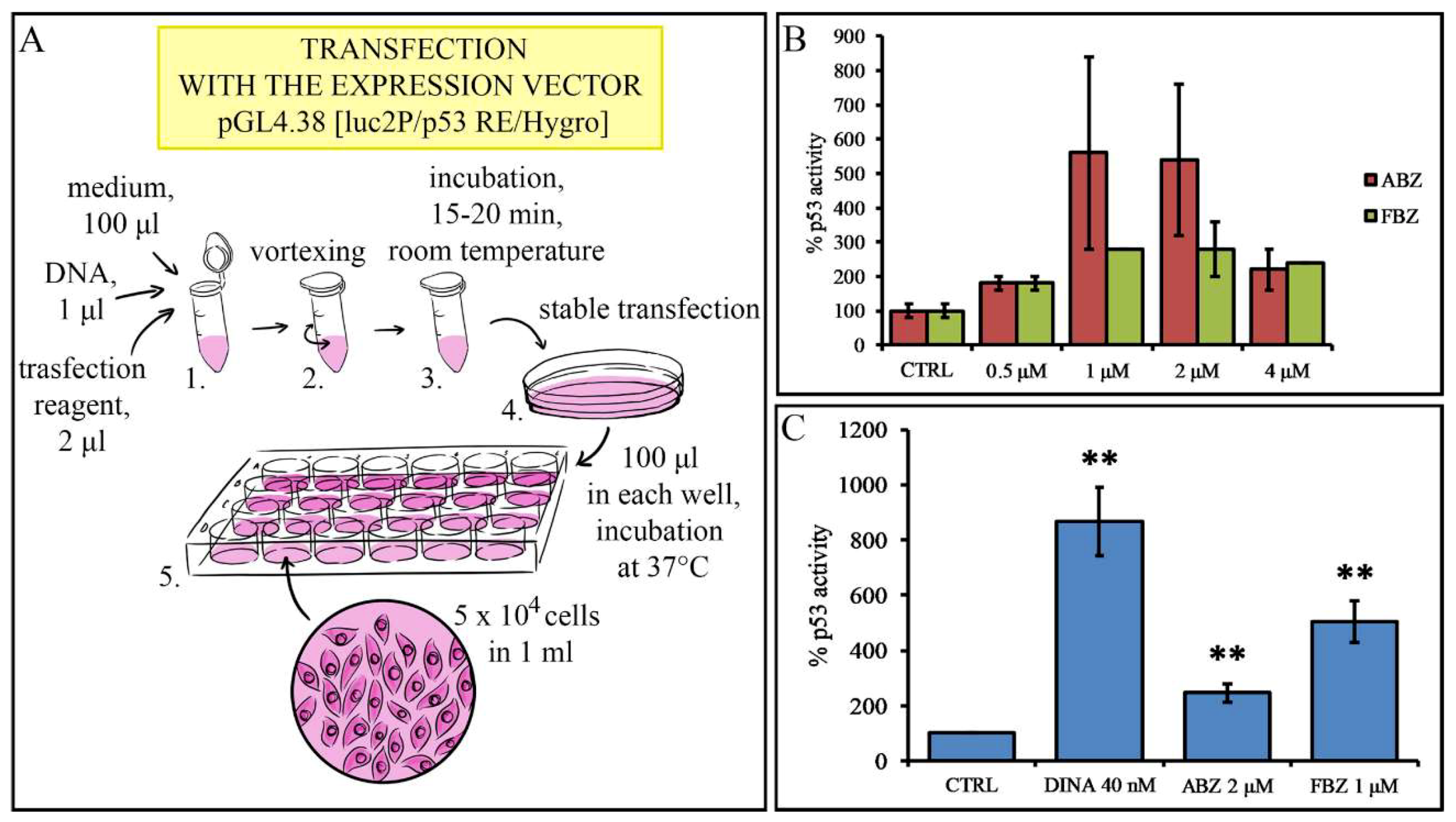{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
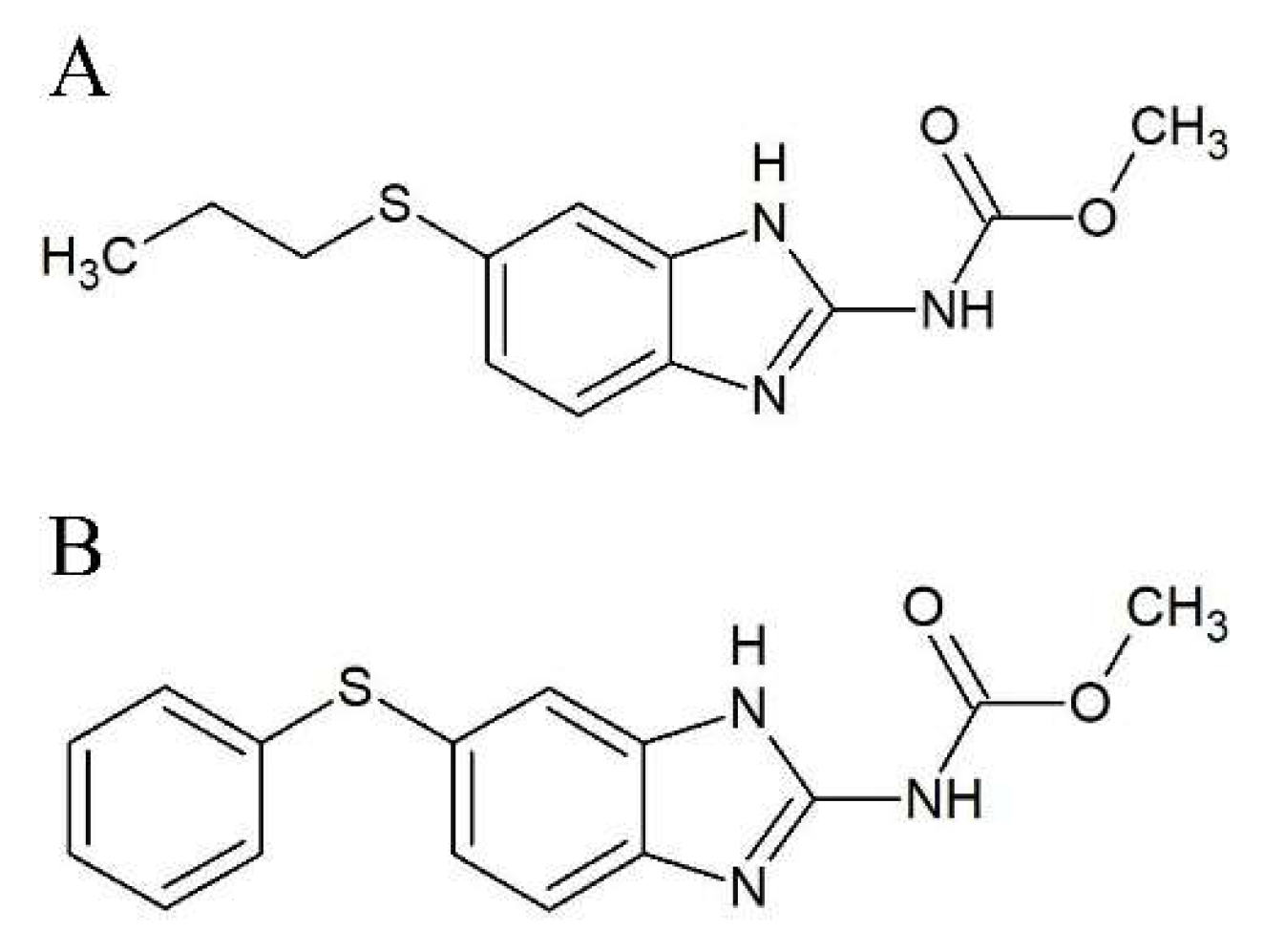
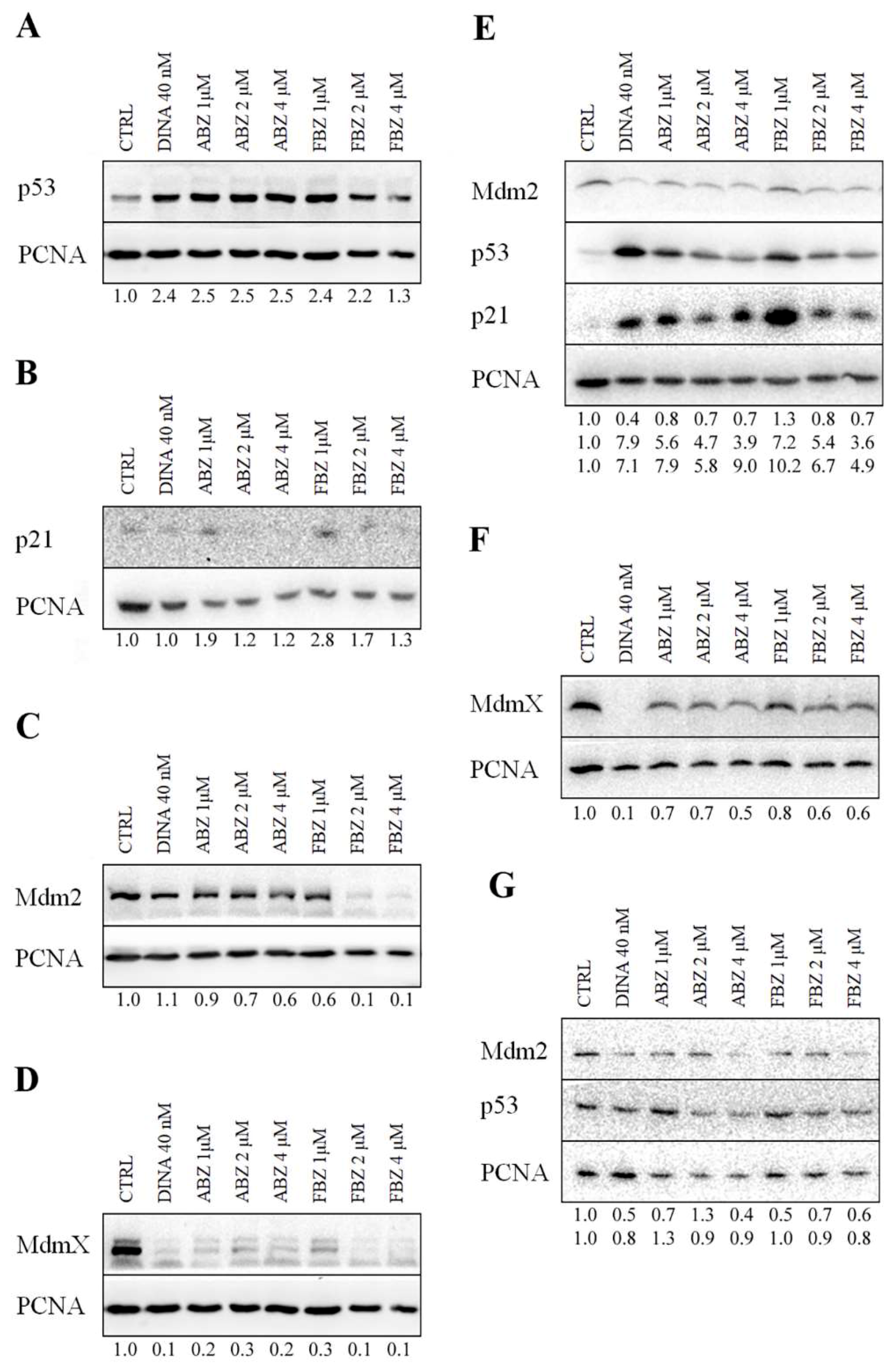
2.1. Effect on p53 Activity
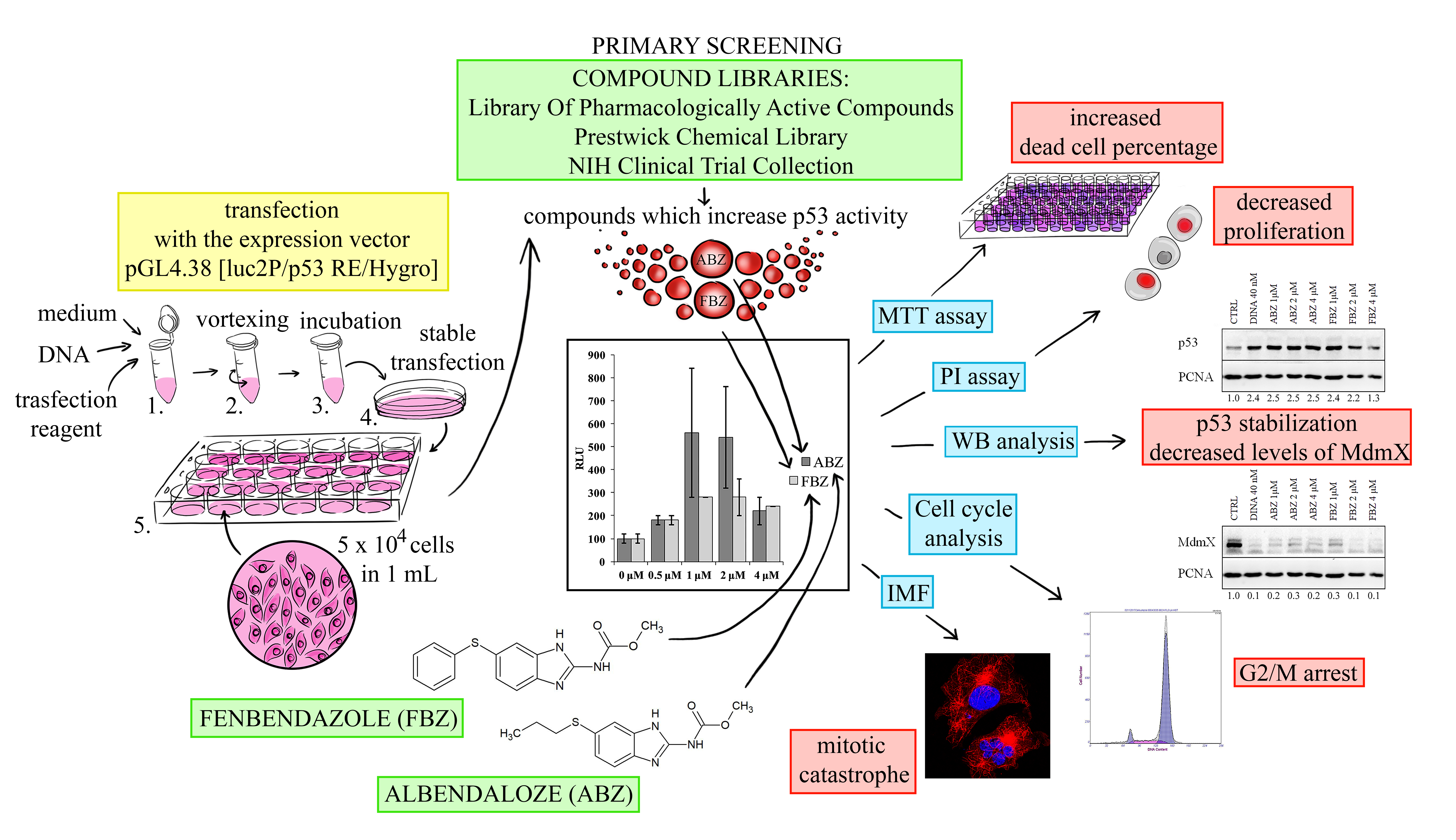
2.2. Mechanism of ABZ and FBZ Action
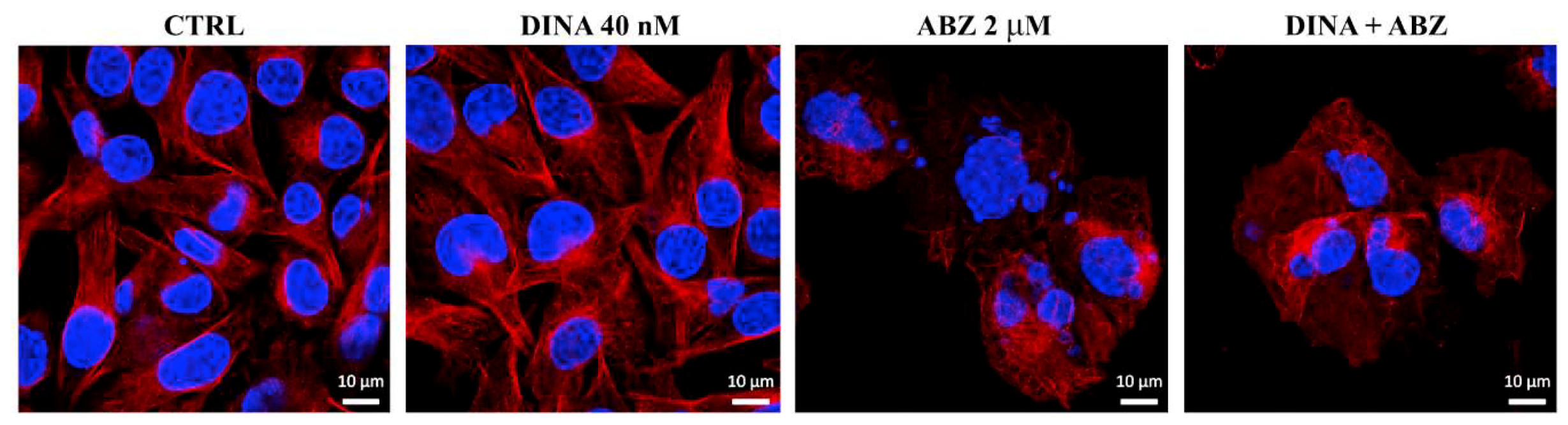
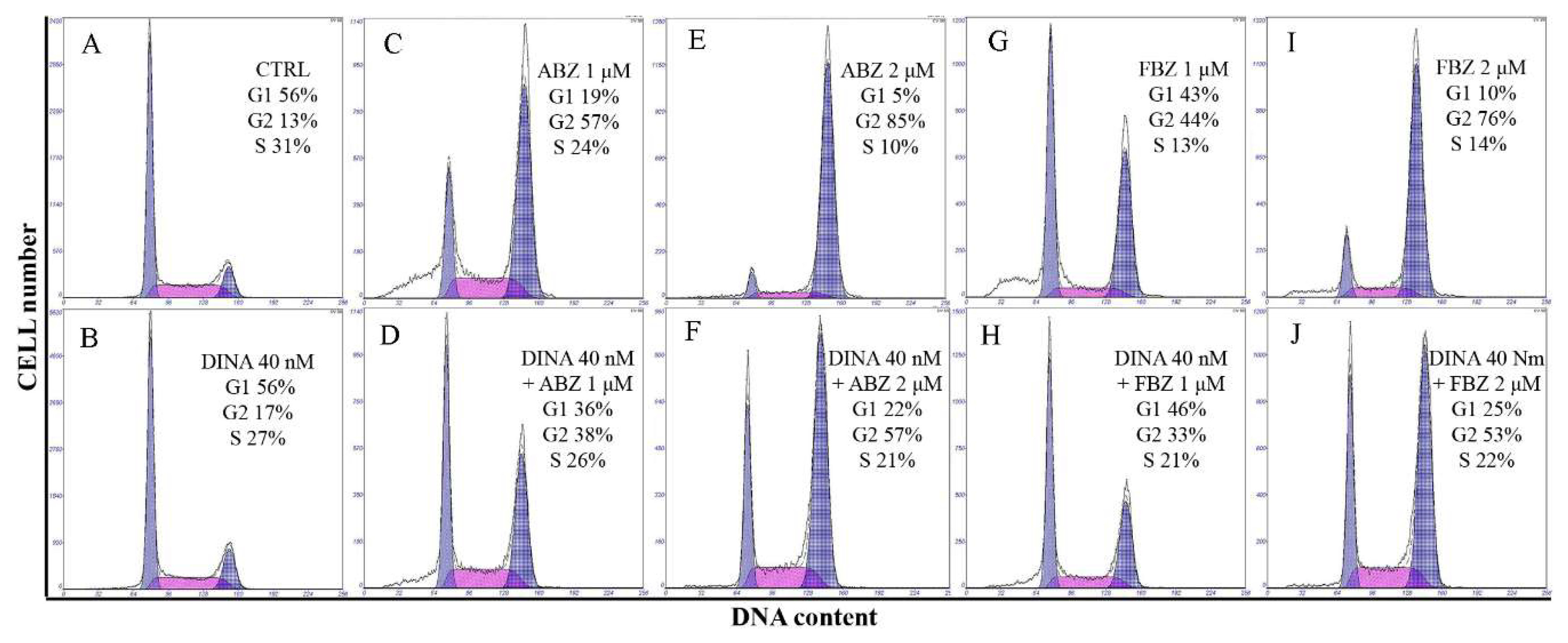
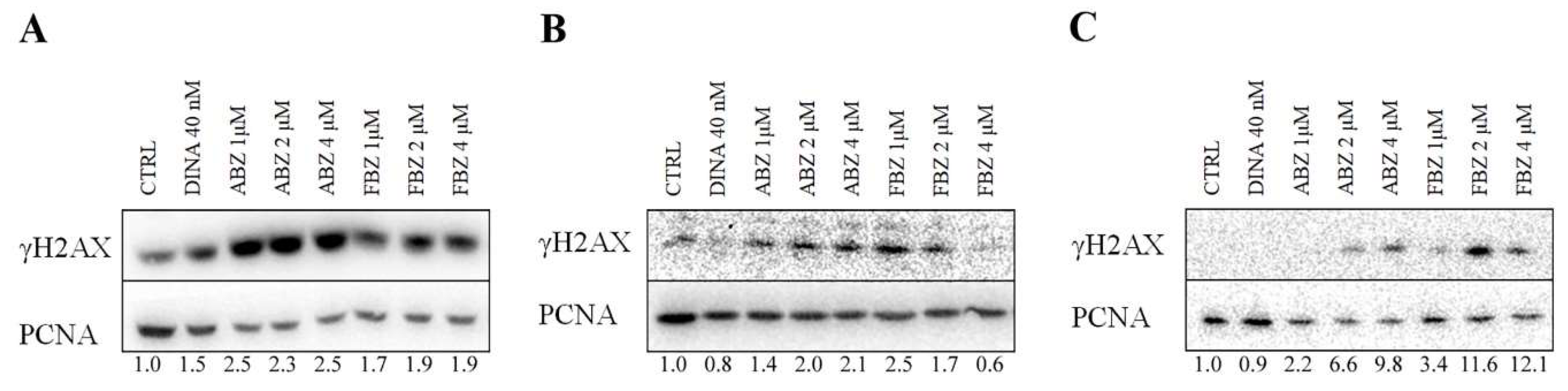
2.3. Effect on Microtubules, Cell Cycle, and DNA
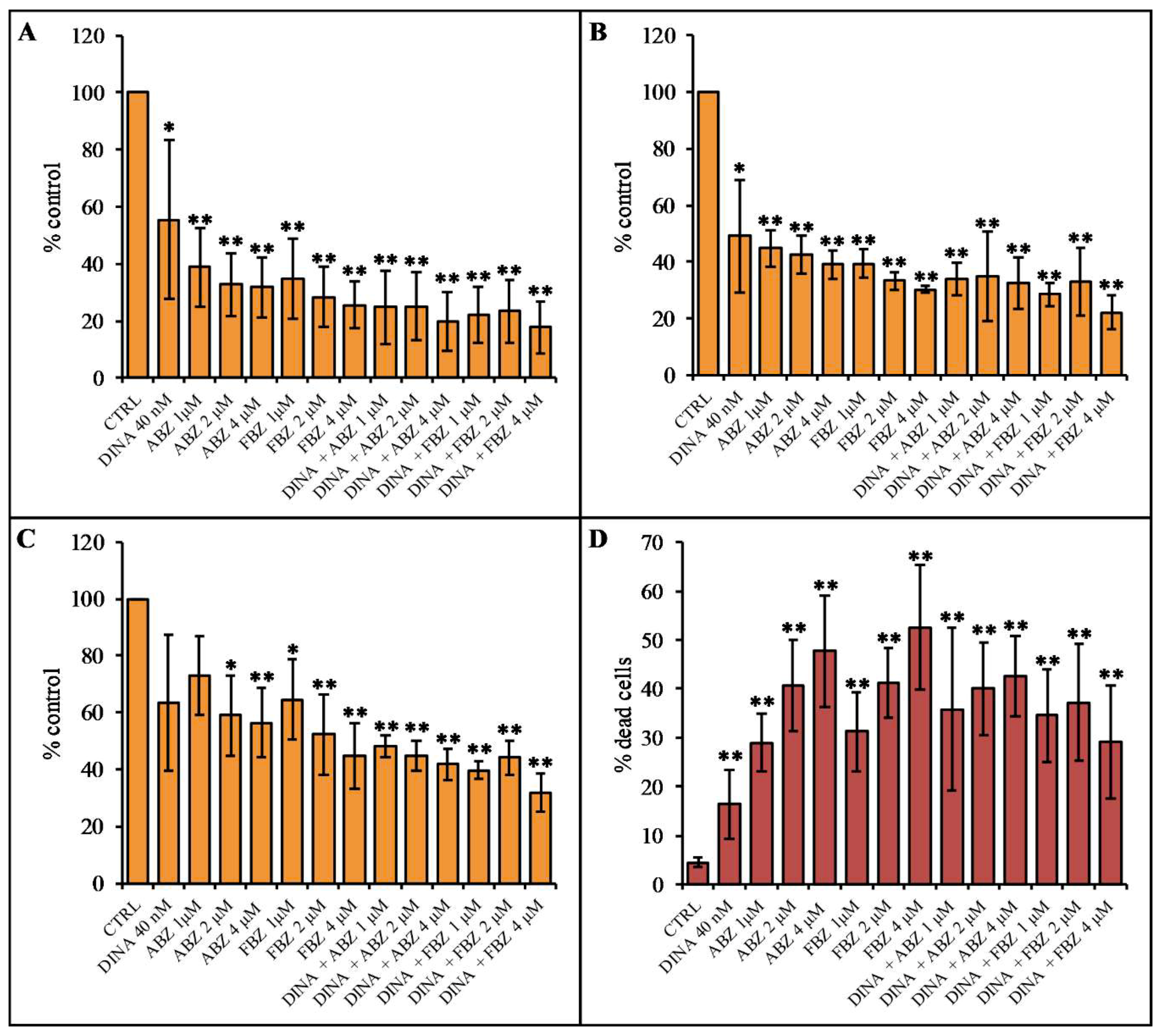
2.4. Effect on Cell Proliferation and Viability
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Cells
4.3. Primary Screening
4.4. Luciferase Assay and Bradford Assay
4.5. SDS-PAGE and Western Blot Analysis
4.6. MTT Assay
4.7. Propidium Iodide Exclusion Assay
4.8. Immunofluorescence and DAPI Staining
4.9. Cell Cycle Analysis
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Review: MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. Febs Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–99. [Google Scholar] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Word Health Organization. World Cancer Report 2014; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2014; ISBN 978-92-832-0429-9. [Google Scholar]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef]
- Telleria, C.M. Drug repurposing for cancer therapy. J. Cancer Sci. Ther. 2012, 4, ix–xi. [Google Scholar] [CrossRef]
- Russell, G.J.; Lacey, E. Inhibition of [3H] mebendazole binding to tubulin by structurally diverse microtubule inhibitors which interact at the colchicine binding site. Biochem. Mol. Biol. Int. 1995, 35, 1153–1159. [Google Scholar]
- Nogales, E. Structural insights into microtubule function. Annu. Rev. Biochem. 2000, 69, 277–302. [Google Scholar] [CrossRef]
- Khalilzadeh, A.; Wangoo, K.T.; Morris, D.L.; Pourgholami, M.H. Epothilone-paclitaxel resistant leukemic cells CEM/dEpoB300 are sensitive to albendazole: Involvement of apoptotic pathways. Biochem. Pharmacol. 2007, 74, 407–414. [Google Scholar] [CrossRef]
- Chu, S.W.L.; Badar, S.; Morris, D.L.; Pourgholami, M.H. Potent inhibition of tubulin polymerisation and proliferation of paclitaxel-resistant 1A9PTX22 human ovarian cancer cells by albendazole. Anticancer Res. 2009, 29, 3791–3796. [Google Scholar]
- Upcroft, P.; Upcroft, J. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001, 14, 150–164. [Google Scholar] [CrossRef]
- Martin, R.J. Modes of action of anthelmintic drugs. Vet. J. 1997, 154, 11–34. [Google Scholar] [CrossRef]
- Castro, L.S.E.P.W.; Kviecinski, M.R.; Ourique, F.; Parisotto, E.B.; Grinevicius, V.M.A.S.; Correia, J.F.G.; Wilhelm Filho, D.; Pedrosa, R.C. Albendazole as a promising molecule for tumor control. Redox Biol. 2016, 10, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Seaton, A.; Higgins, C.; Mann, J.; Baron, A.; Bailly, C.; Neidle, S.; van den Berg, H. Mechanistic and anti-proliferative studies of two novel, biologically active bis-benzimidazoles. Eur. J. Cancer 2003, 39, 2548–2555. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Woon, L.; Almajd, R.; Akhter, J.; Bowery, P.; Morris, D.L. In vitro and in vivo suppression of growth of hepatocellular carcinoma cells by albendazole. Cancer Lett. 2001, 165, 43–49. [Google Scholar] [CrossRef]
- Králová, V.; Hanušová, V.; Rudolf, E.; Čáňová, K.; Skálová, L. Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro. J. Pharm. Pharmacol. 2016, 68, 208–218. [Google Scholar] [CrossRef]
- Gao, P.; Dang, C.V.; Watson, J. Unexpected antitumorigenic effect of fenbendazole when combined with supplementary vitamins. J. Am. Assoc. Lab. Anim. Sci. 2008, 47, 37–40. [Google Scholar]
- Kotala, V.; Uldrijan, S.; Horky, M.; Trbusek, M.; Strnad, M.; Vojtesek, B. Potent induction of wild-type p53-dependent transcription in tumour cells by a synthetic inhibitor of cyclin-dependent kinases. Cell. Mol. Life Sci. 2001, 58, 1333–1339. [Google Scholar] [CrossRef]
- Desai, B.M.; Villanueva, J.; Nguyen, T.-T.K.; Lioni, M.; Xiao, M.; Kong, J.; Krepler, C.; Vultur, A.; Flaherty, K.T.; Nathanson, K.L.; et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS ONE 2013, 8, e59588. [Google Scholar] [CrossRef]
- Ghasemi, F.; Black, M.; Pinto, N.; Ruicci, K.M.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C.; et al. Repurposing albendazole: New potential as a chemotherapeutic agent with preferential activity against HPV-negative head and neck squamous cell cancer. Oncotarget 2017, 8, 71512–71519. [Google Scholar] [CrossRef]
- Doudican, N.; Rodriguez, A.; Osman, I.; Orlow, S.J. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol. Cancer Res. 2008, 6, 1308–1315. [Google Scholar] [CrossRef]
- Čáňová, K.; Rudolf, E.; Rozkydalová, L.; Vokurková, D. Flubendazole induces mitotic catastrophe and apoptosis in melanoma cells. Toxicol. in Vitro 2018, 46, 313–322. [Google Scholar] [CrossRef]
- Hanušová, V.; Králová, V.; Skálová, L.; Matoušková, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef]
- Patel, K.; Doudican, N.A.; Schiff, P.B.; Orlow, S.J. Albendazole sensitizes cancer cells to ionizing radiation. Radiat Oncol 2011, 6, 160. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Cai, Z.Y.; Badar, S.; Wangoo, K.; Poruchynsky, M.S.; Morris, D.L. Potent inhibition of tumoral hypoxia-inducible factor 1alpha by albendazole. BMC Cancer 2010, 10, 143. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Khachigian, L.M.; Fahmy, R.G.; Badar, S.; Wang, L.; Chu, S.W.L.; Morris, D.L. Albendazole inhibits endothelial cell migration, tube formation, vasopermeability, VEGF receptor-2 expression and suppresses retinal neovascularization in ROP model of angiogenesis. Biochem. Biophys. Res. Commun. 2010, 397, 729–734. [Google Scholar] [CrossRef]
- Dogra, N.; Kumar, A.; Mukhopadhyay, T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, 11926. [Google Scholar] [CrossRef]
- Soderlind, K.; Gorodetsky, B.; Singh, A.; Bachur, N.; Miller, G.; Lown, J. Bis-benzimidazole anticancer agents: Targeting human tumour helicases. Anti-Cancer Drug Des. 1999, 14, 19–36. [Google Scholar]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef]
- Giannakakou, P.; Nakano, M.; Nicolaou, K.C.; O’Brate, A.; Yu, J.; Blagosklonny, M.V.; Greber, U.F.; Fojo, T. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc. Natl. Acad. Sci. USA 2002, 99, 10855–10860. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.P.; Larroquette, C.A.; Agrawal, K.C. Potential radiosensitizing agents. 5. 2-substituted benzimidazole derivatives. J. Med. Chem. 1982, 25, 1342–1346. [Google Scholar] [CrossRef]
- Duan, Y.-T.; Wang, Z.-C.; Sang, Y.-L.; Tao, X.-X.; Zhu, H.-L. Exploration of structure-based on imidazole core as antibacterial agents. Curr. Top Med. Chem. 2013, 13, 3118–3130. [Google Scholar] [CrossRef]
- Dogra, N.; Mukhopadhyay, T. Impairment of the ubiquitin-proteasome pathway by methyl N-(6-phenylsulfanyl-1H-benzimidazol-2-yl)carbamate leads to a potent cytotoxic effect in tumor cells: A novel antiproliferative agent with a potential therapeutic implication. J. Biol. Chem. 2012, 287, 30625–30640. [Google Scholar] [CrossRef]
- Valianatos, G.; Valcikova, B.; Growkova, K.; Verlande, A.; Mlcochova, J.; Radova, L.; Stetkova, M.; Vyhnakova, M.; Slaby, O.; Uldrijan, S. A small molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein. PLoS ONE 2017, 12, 1. [Google Scholar] [CrossRef]
- Hammerová, J.; Uldrijan, S.; Táborská, E.; Slaninová, I. Benzo[c]phenanthridine alkaloids exhibit strong anti-proliferative activity in malignant melanoma cells regardless of their p53 status. J. Dermatol. Sci. 2011, 62, 22–35. [Google Scholar] [CrossRef]
- Slaninová, I.; Březinová, L.; Koubíková, L.; Slanina, J. Dibenzocyclooctadiene lignans overcome drug resistance in lung cancer cells—Study of structure–activity relationship. Toxicology in Vitro 2009, 23, 1047–1054. [Google Scholar] [CrossRef]
Sample Availability: Samples albendazole and fenbendazole are available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrkvová, Z.; Uldrijan, S.; Pombinho, A.; Bartůněk, P.; Slaninová, I. Benzimidazoles Downregulate Mdm2 and MdmX and Activate p53 in MdmX Overexpressing Tumor Cells. Molecules 2019, 24, 2152. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112152
Mrkvová Z, Uldrijan S, Pombinho A, Bartůněk P, Slaninová I. Benzimidazoles Downregulate Mdm2 and MdmX and Activate p53 in MdmX Overexpressing Tumor Cells. Molecules. 2019; 24(11):2152. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112152
Chicago/Turabian StyleMrkvová, Zuzana, Stjepan Uldrijan, Antonio Pombinho, Petr Bartůněk, and Iva Slaninová. 2019. "Benzimidazoles Downregulate Mdm2 and MdmX and Activate p53 in MdmX Overexpressing Tumor Cells" Molecules 24, no. 11: 2152. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24112152