Structural and Photophysical Properties of 2,1,3-Benzothiadiazole-Based Phosph(III)azane and Its Complexes


Abstract
:
1. Introduction
2. Results and Discussion
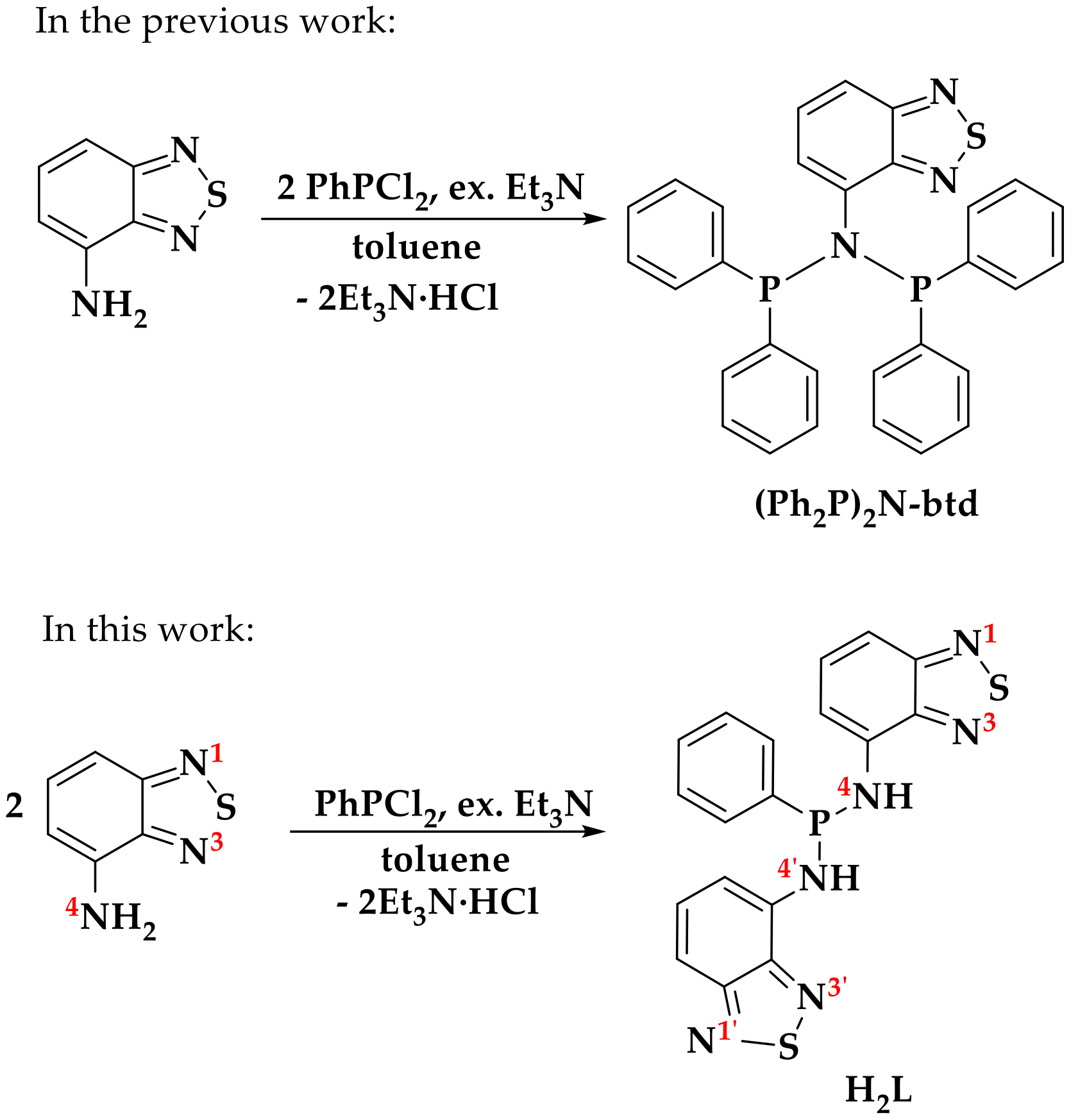
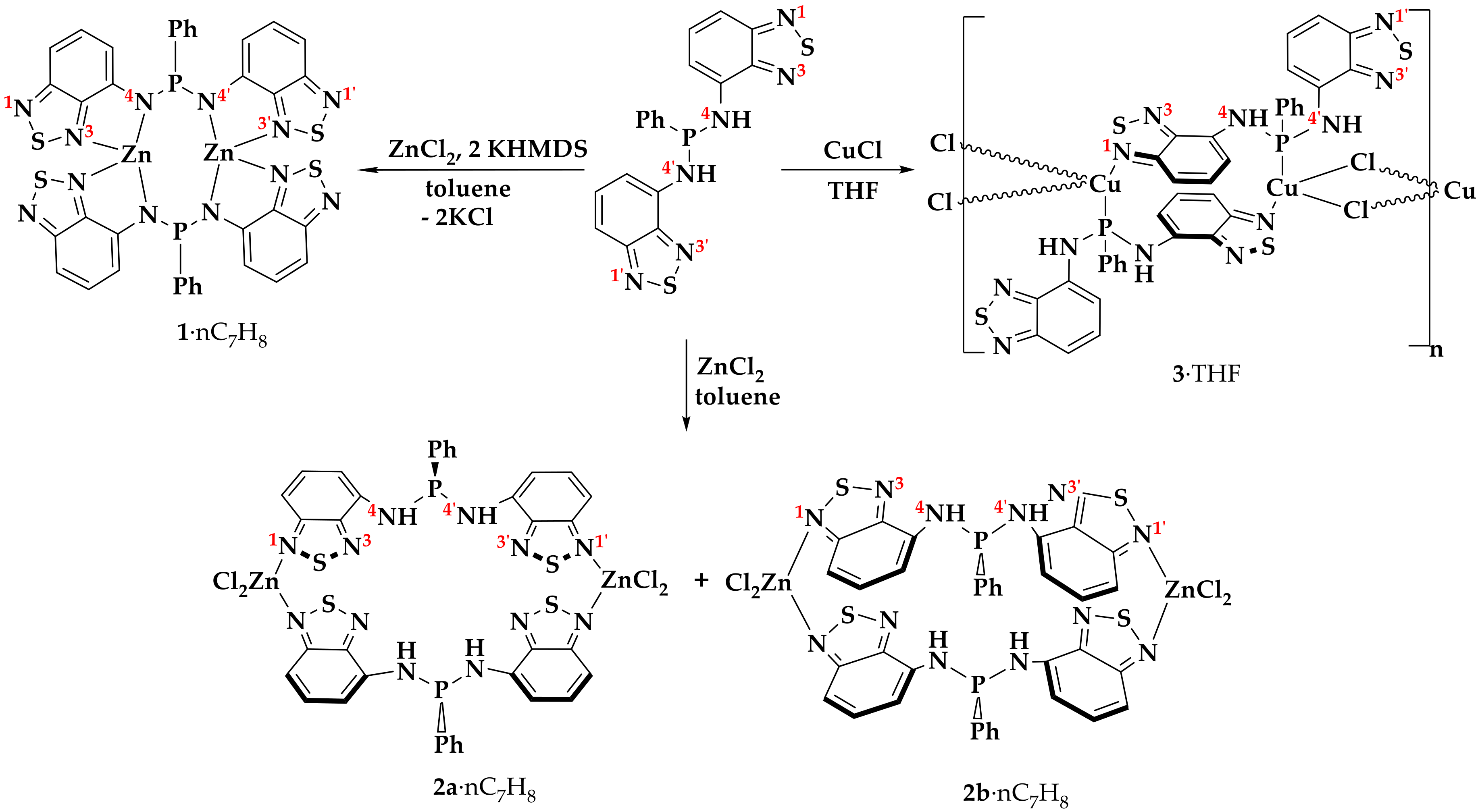
2.1. Synthesis
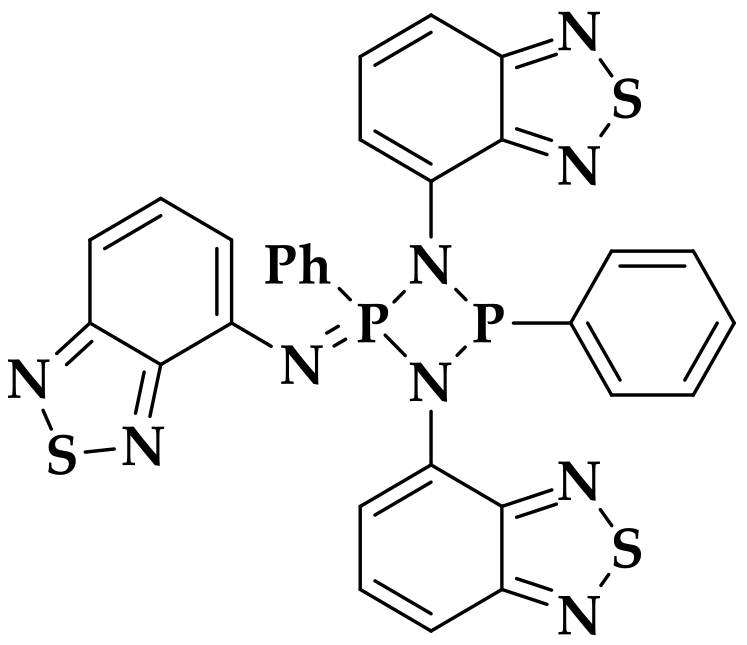
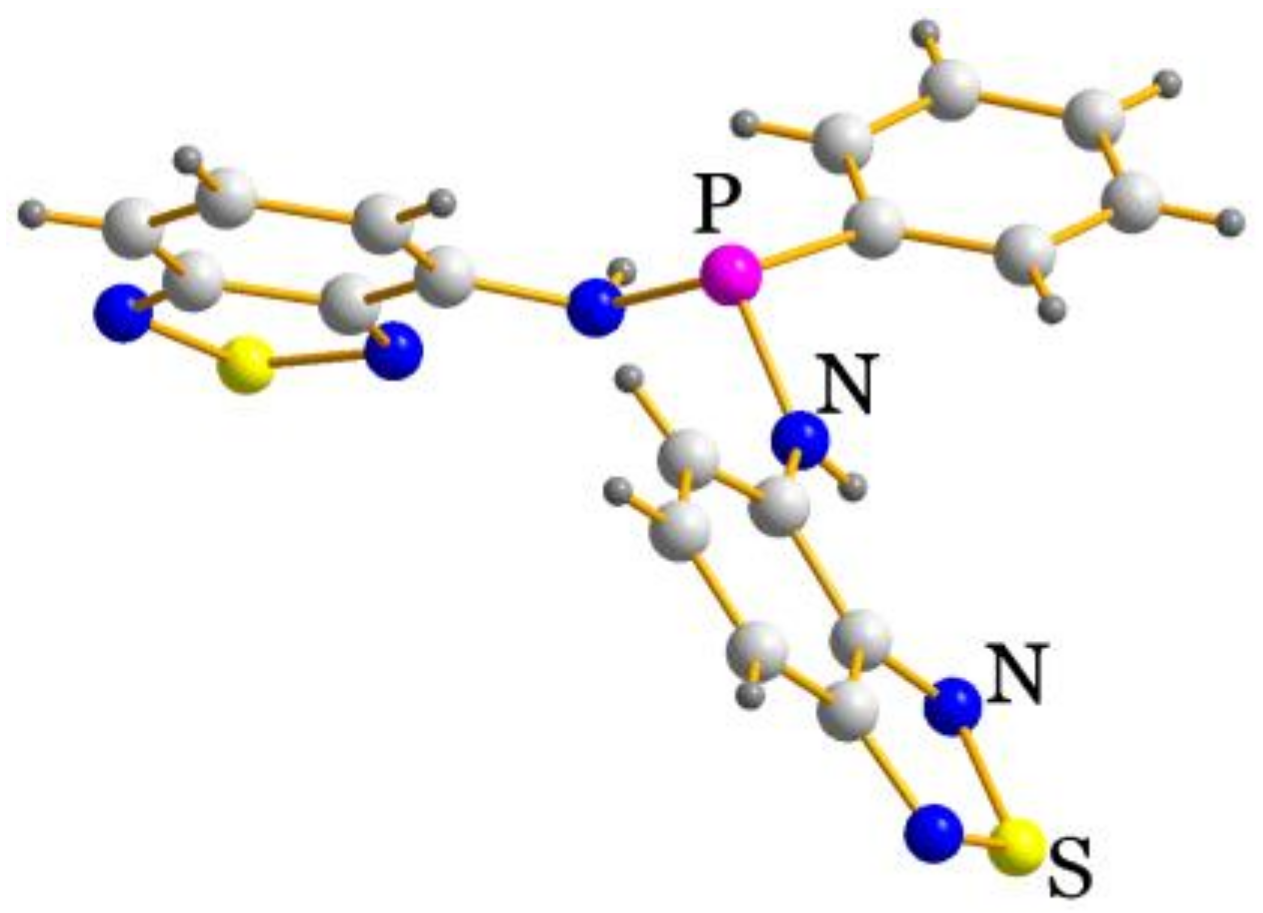
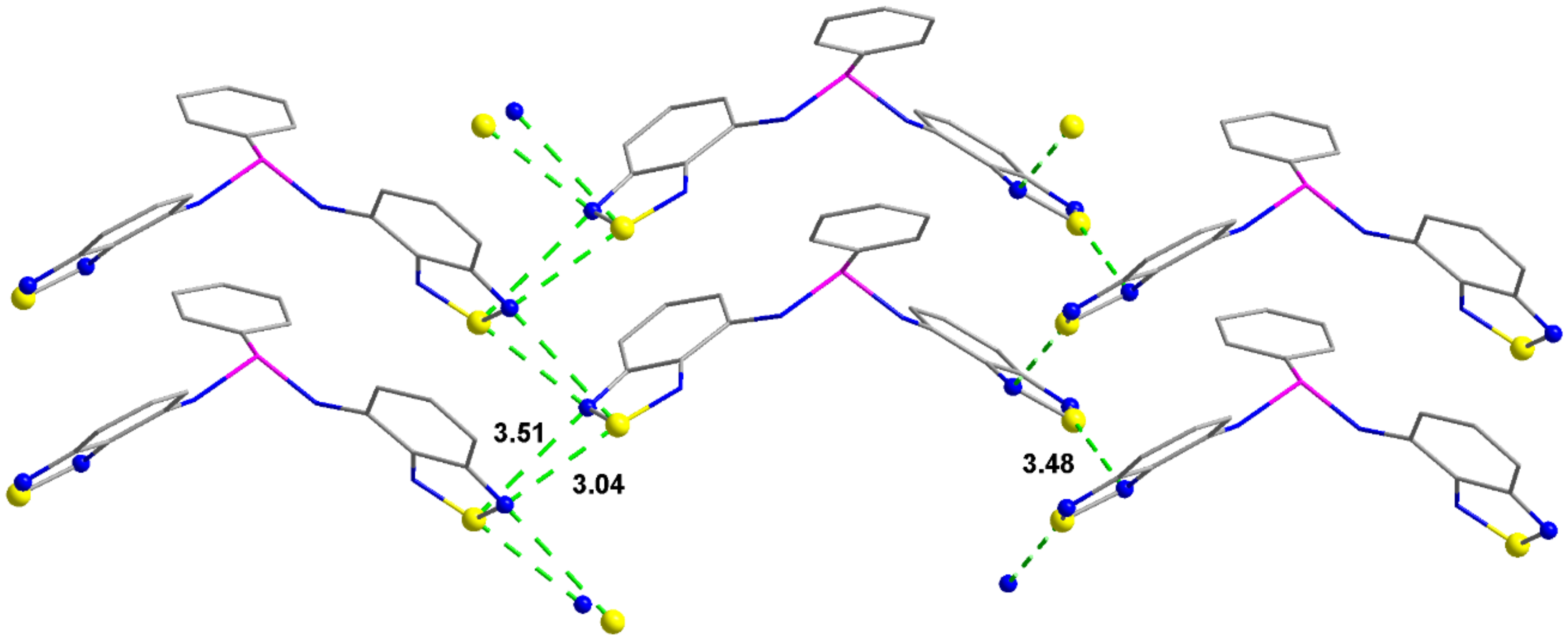
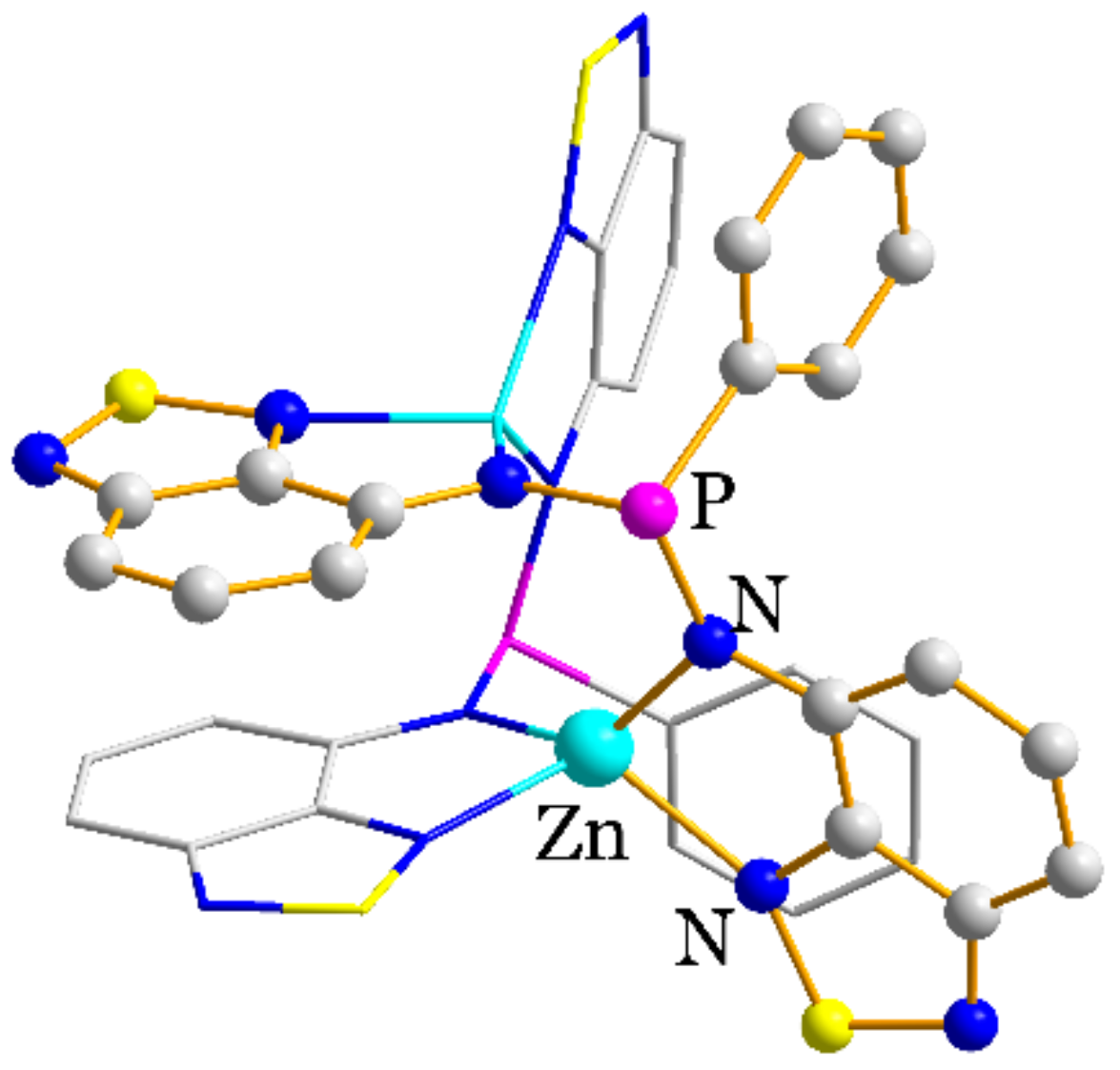
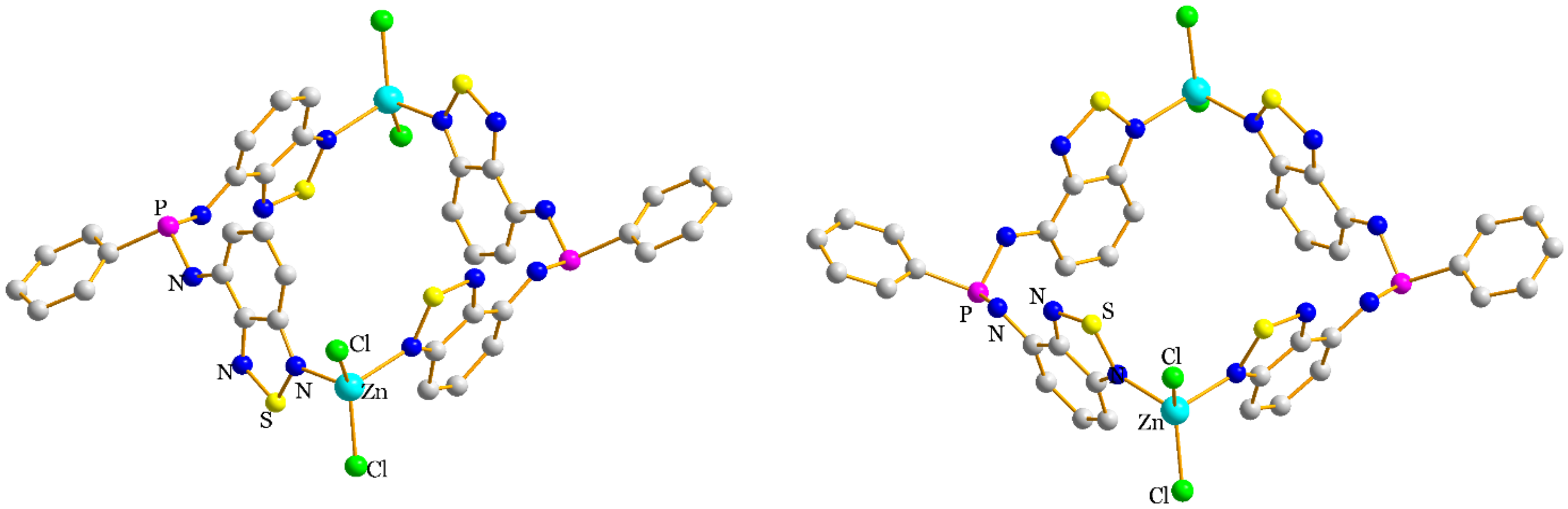
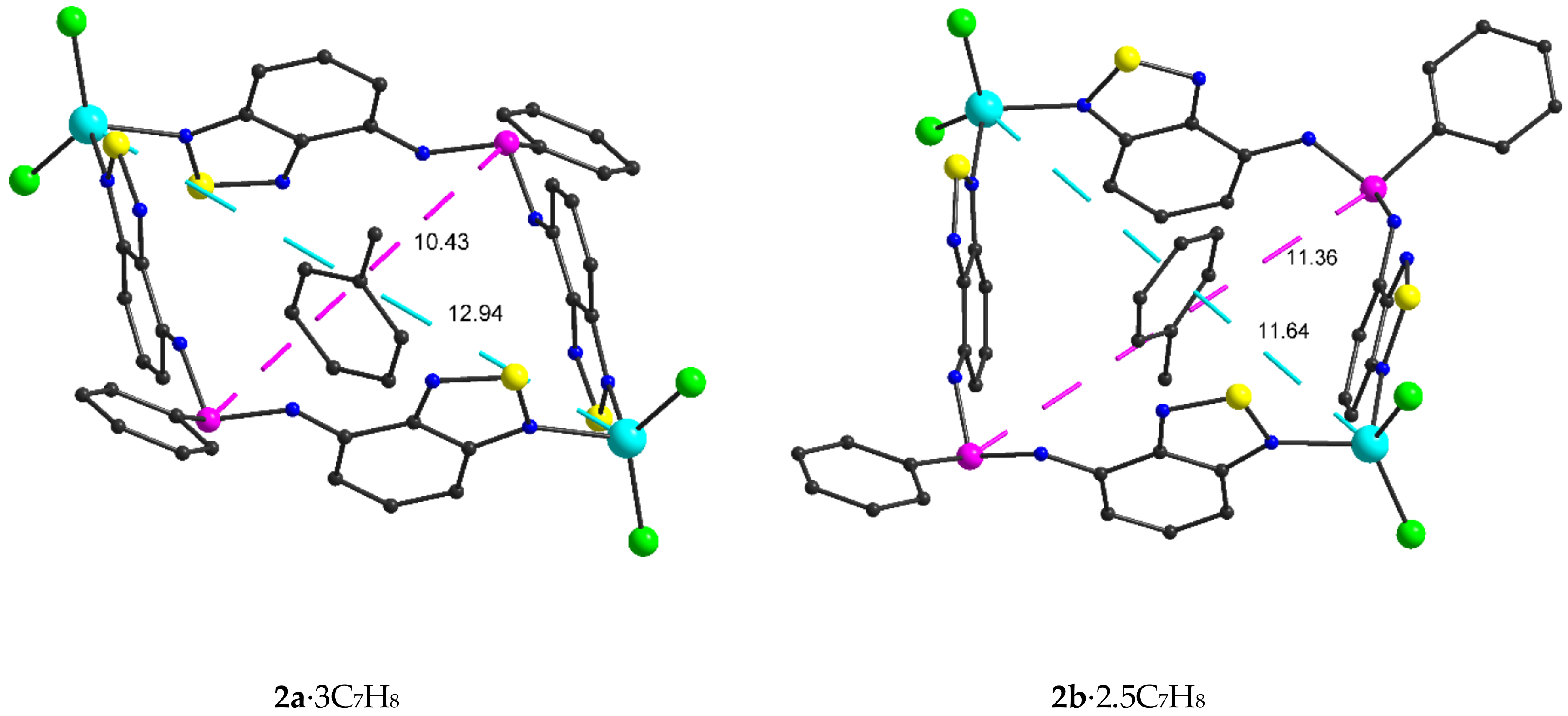
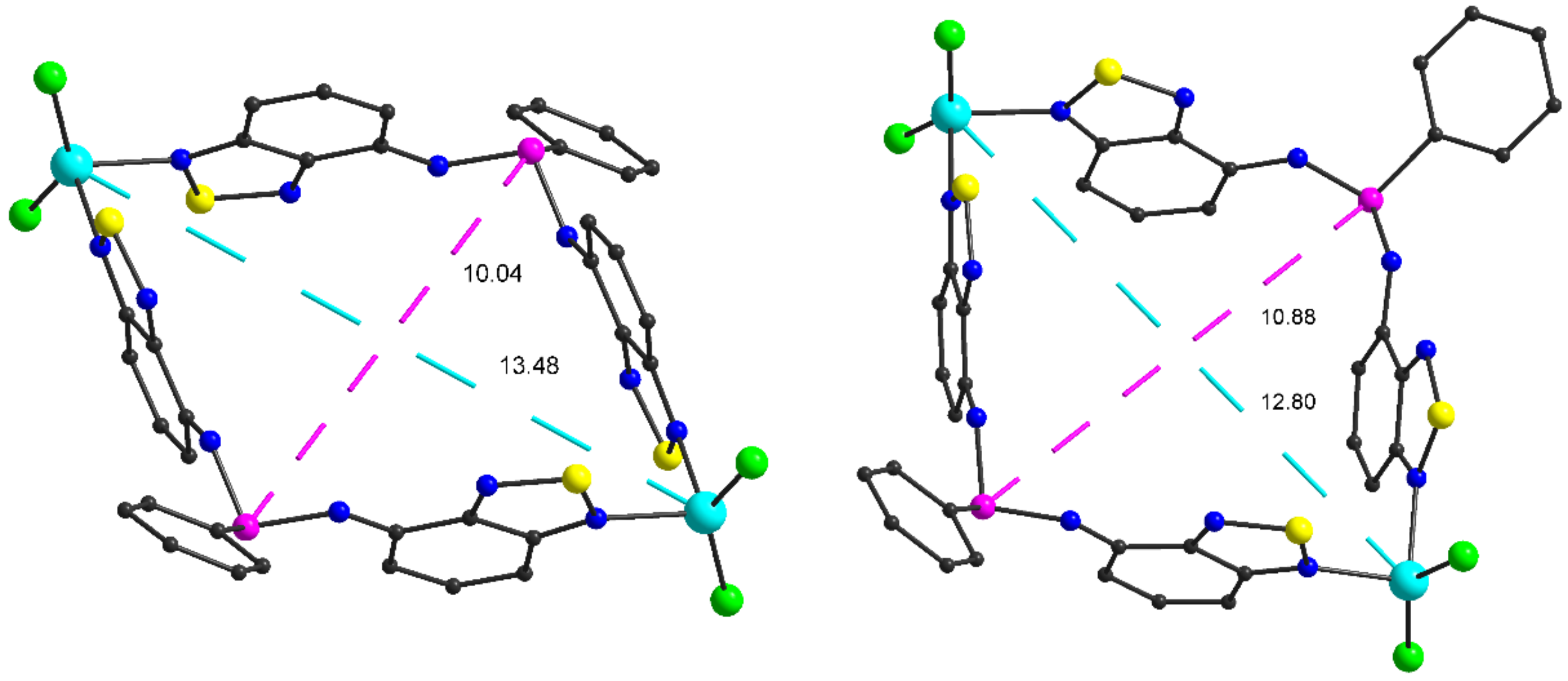
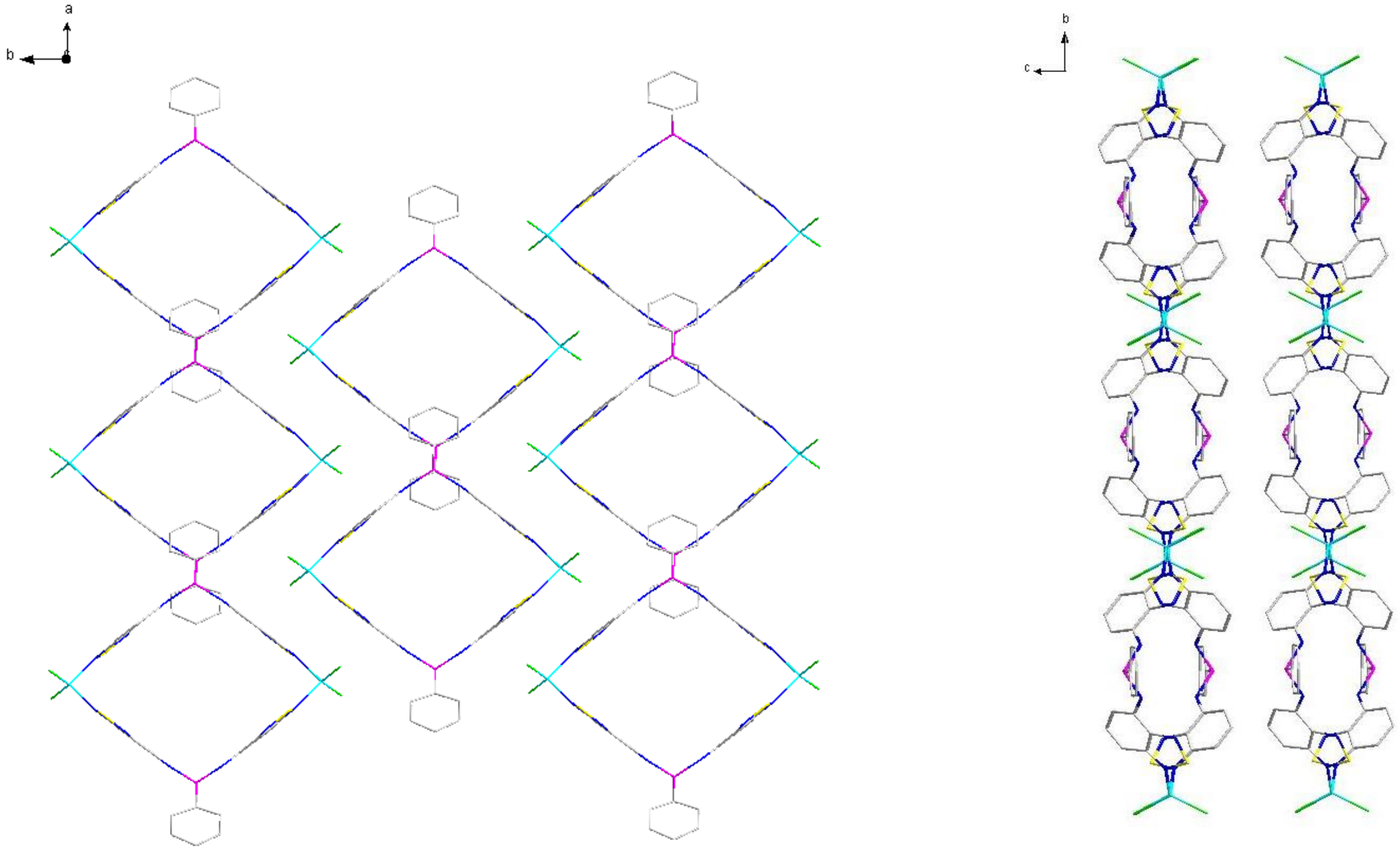
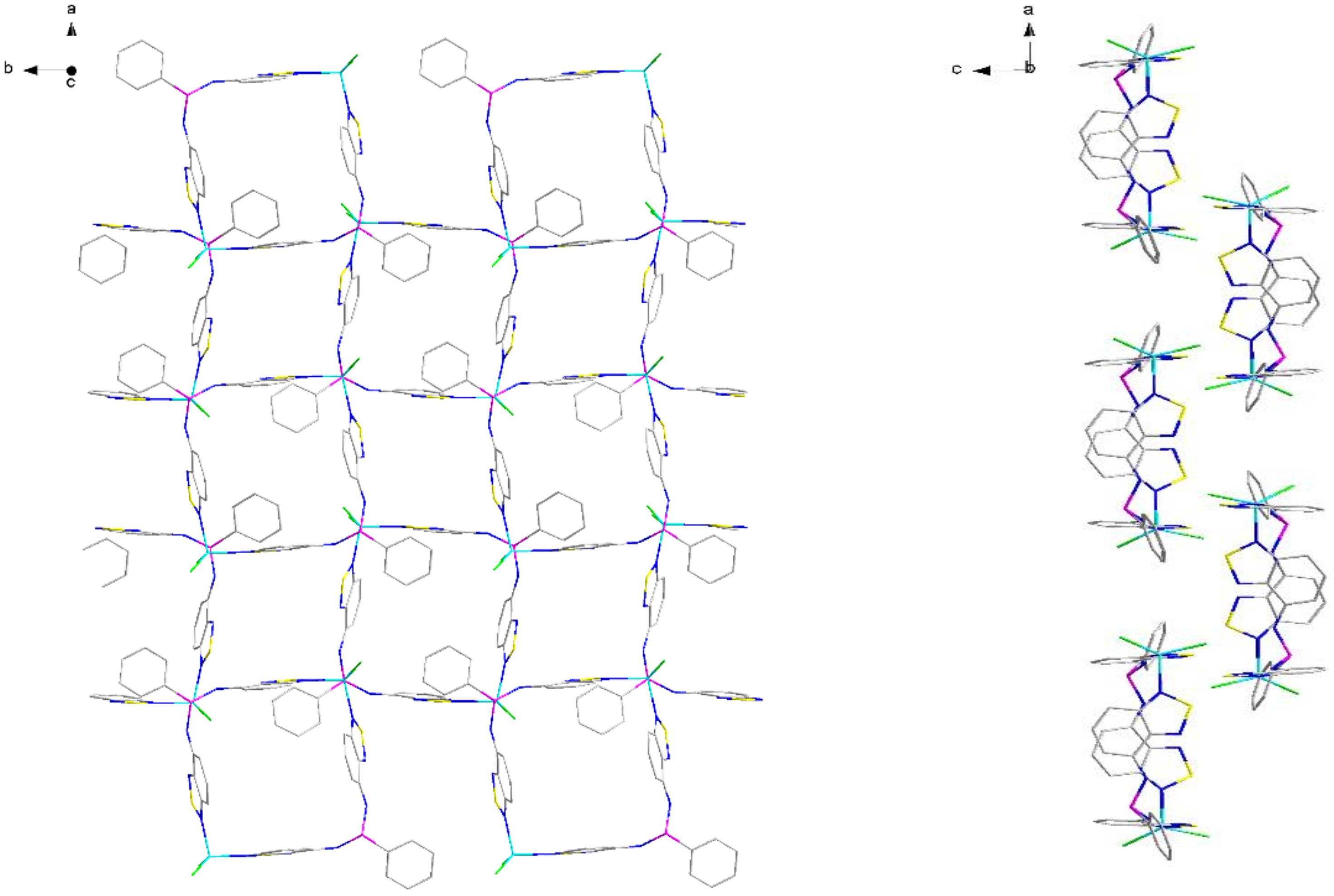
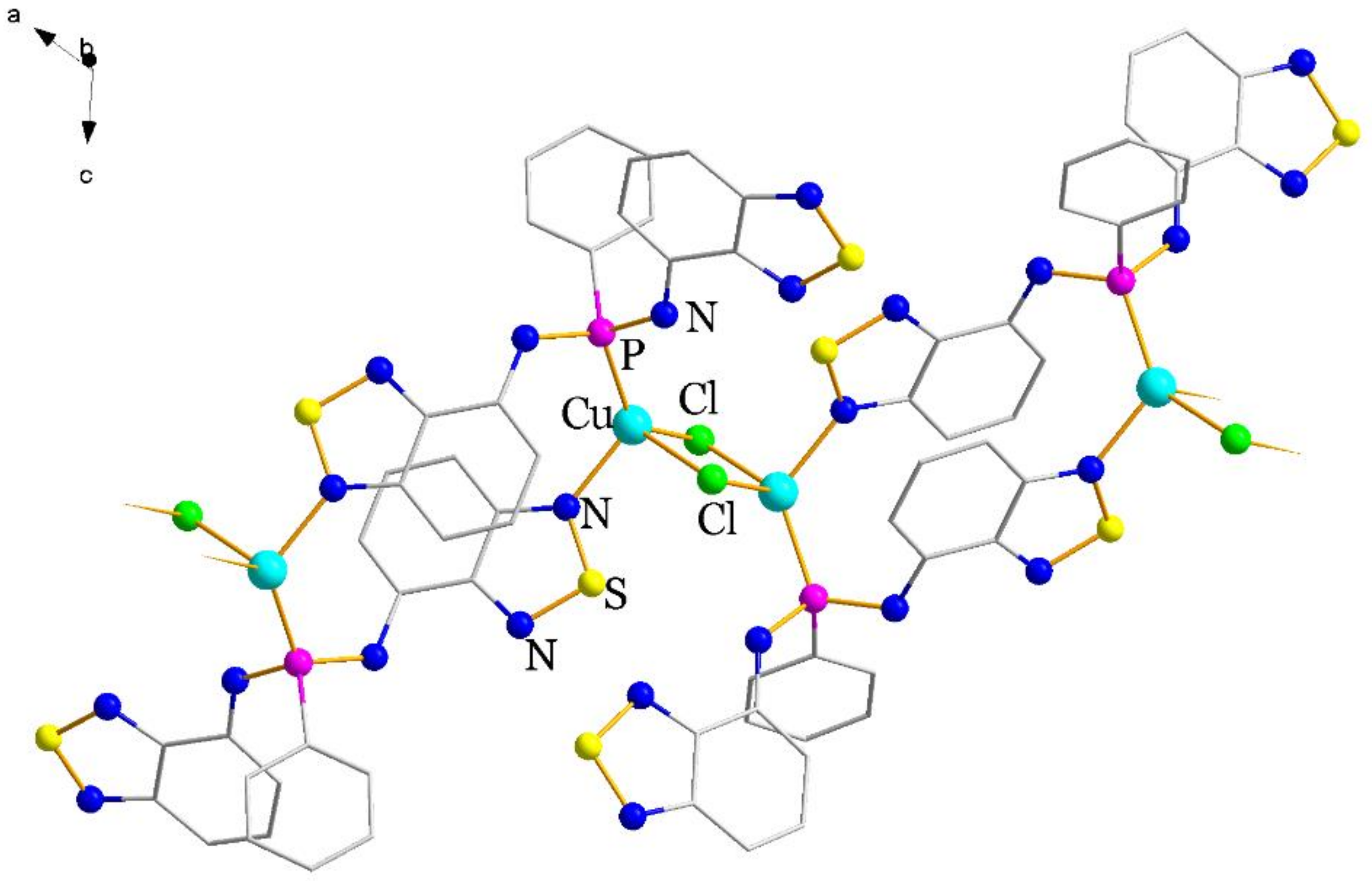
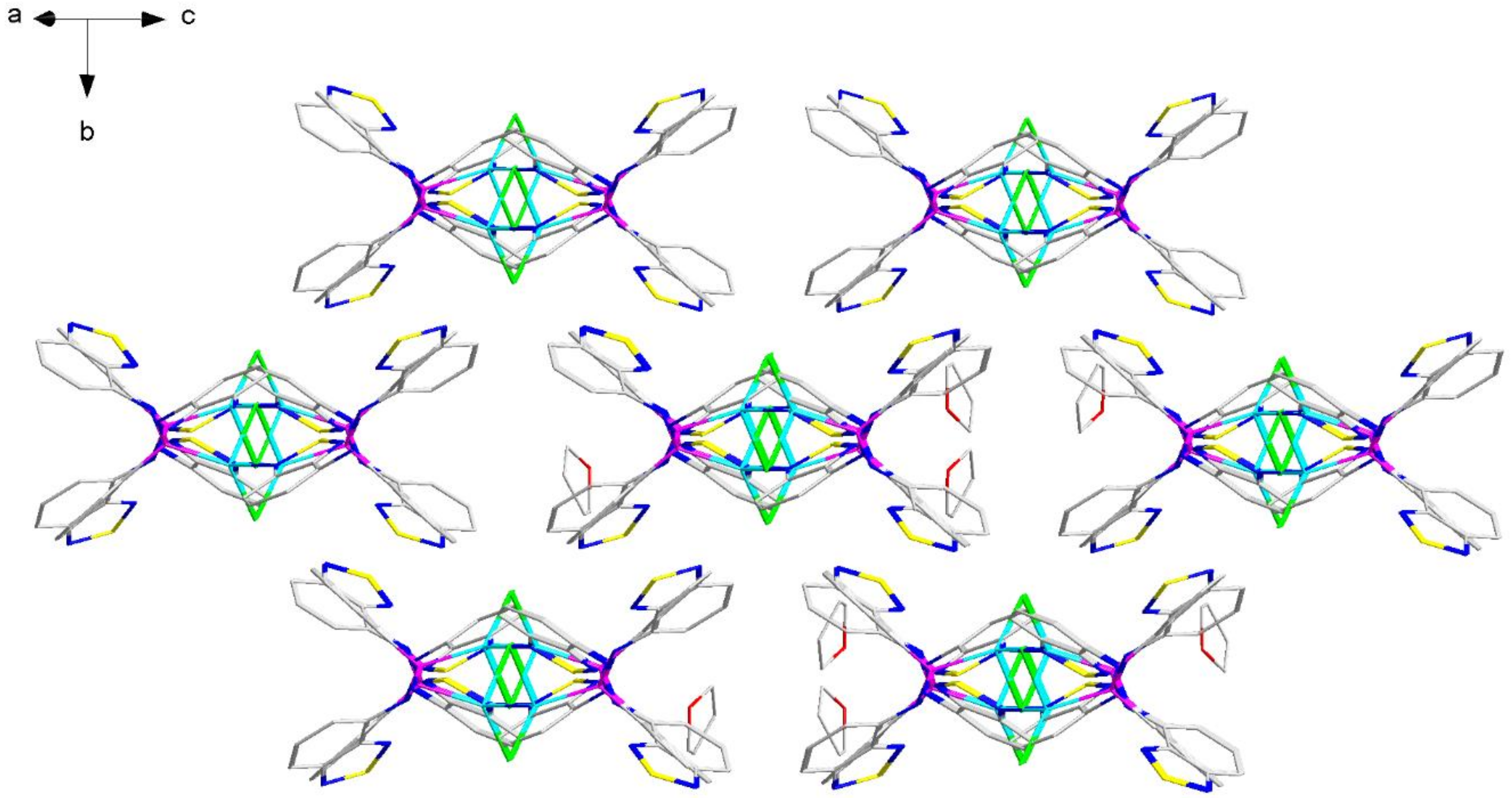
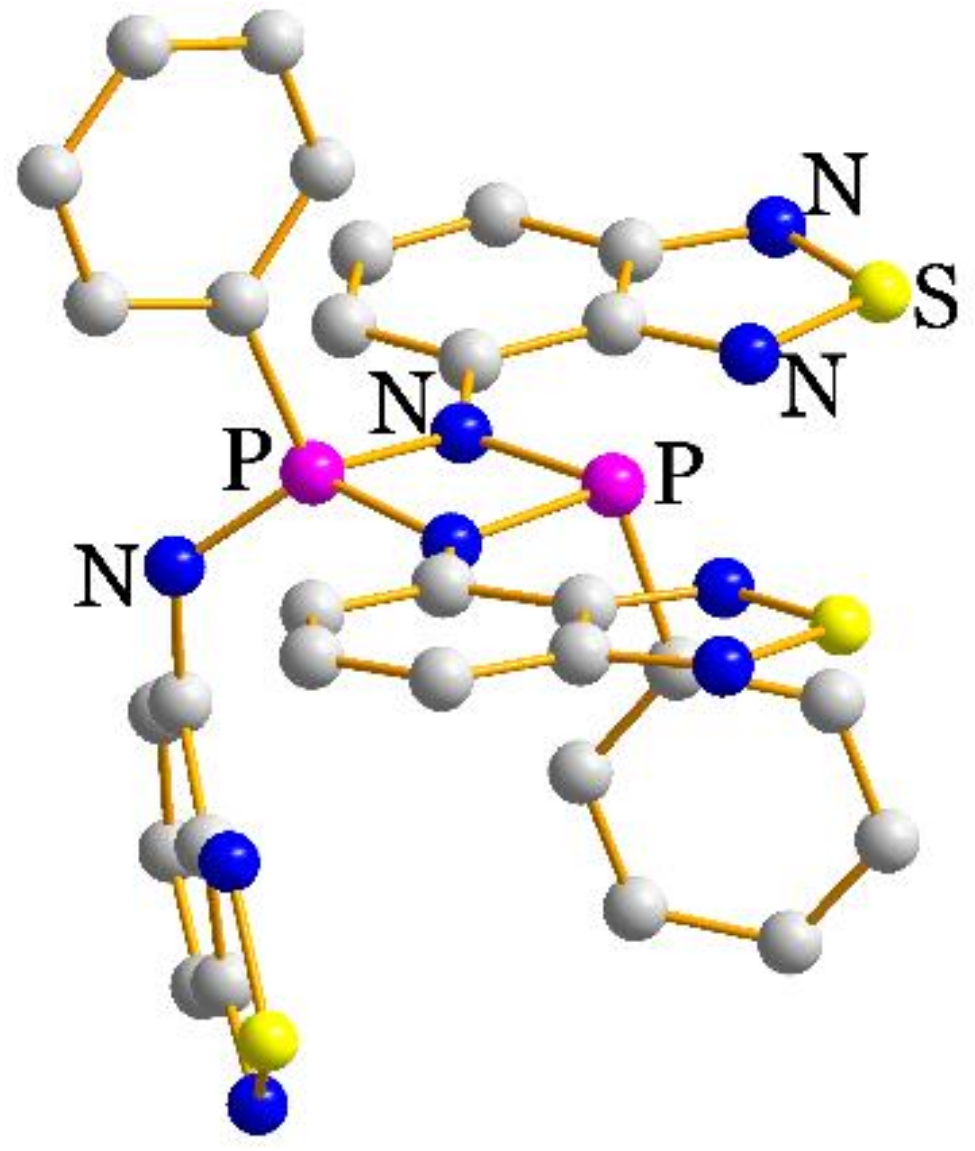
2.2. Structural Characterization
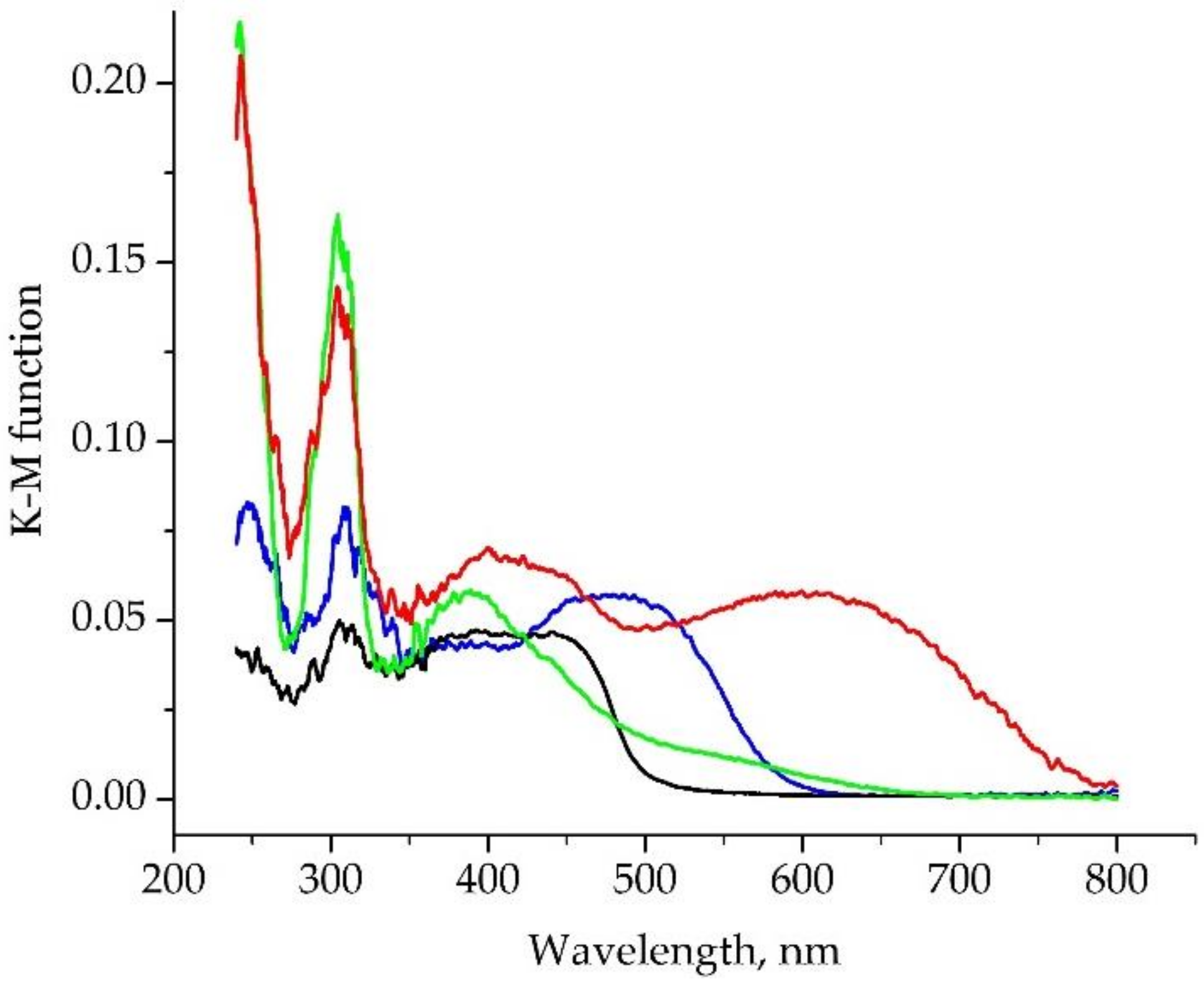
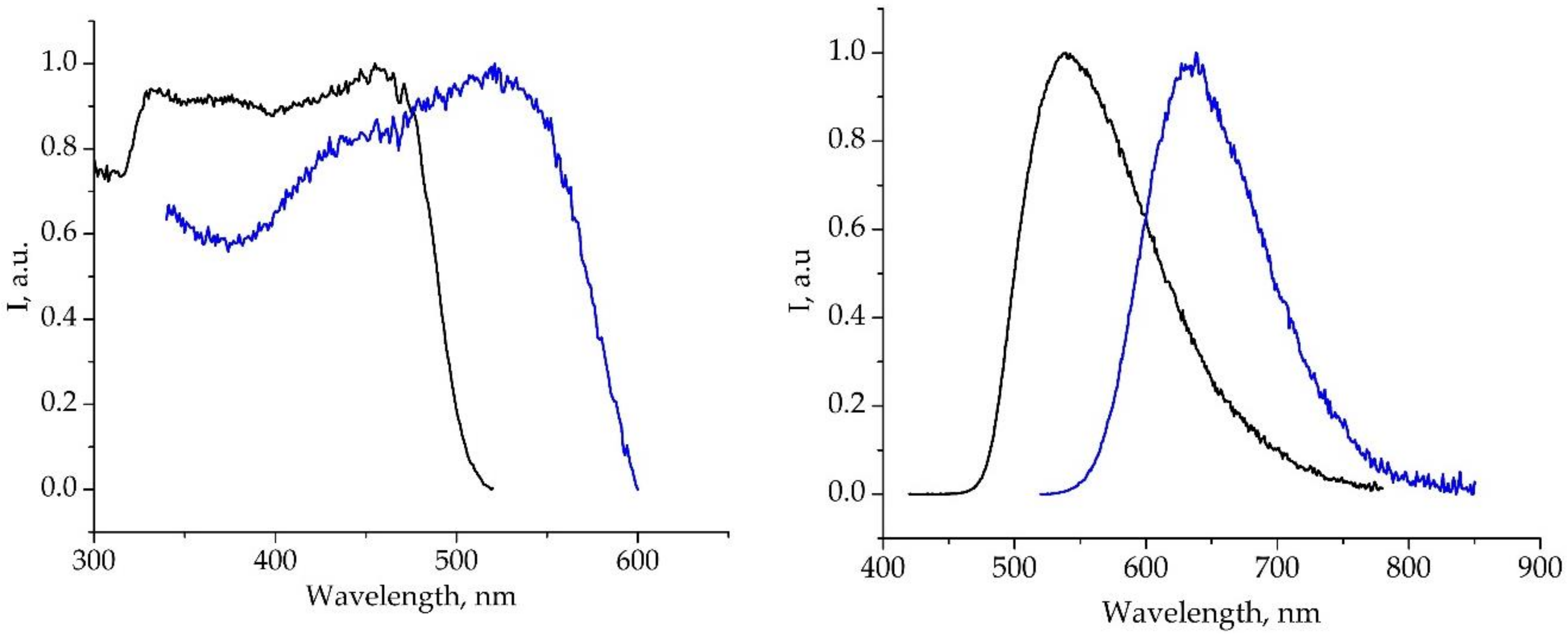
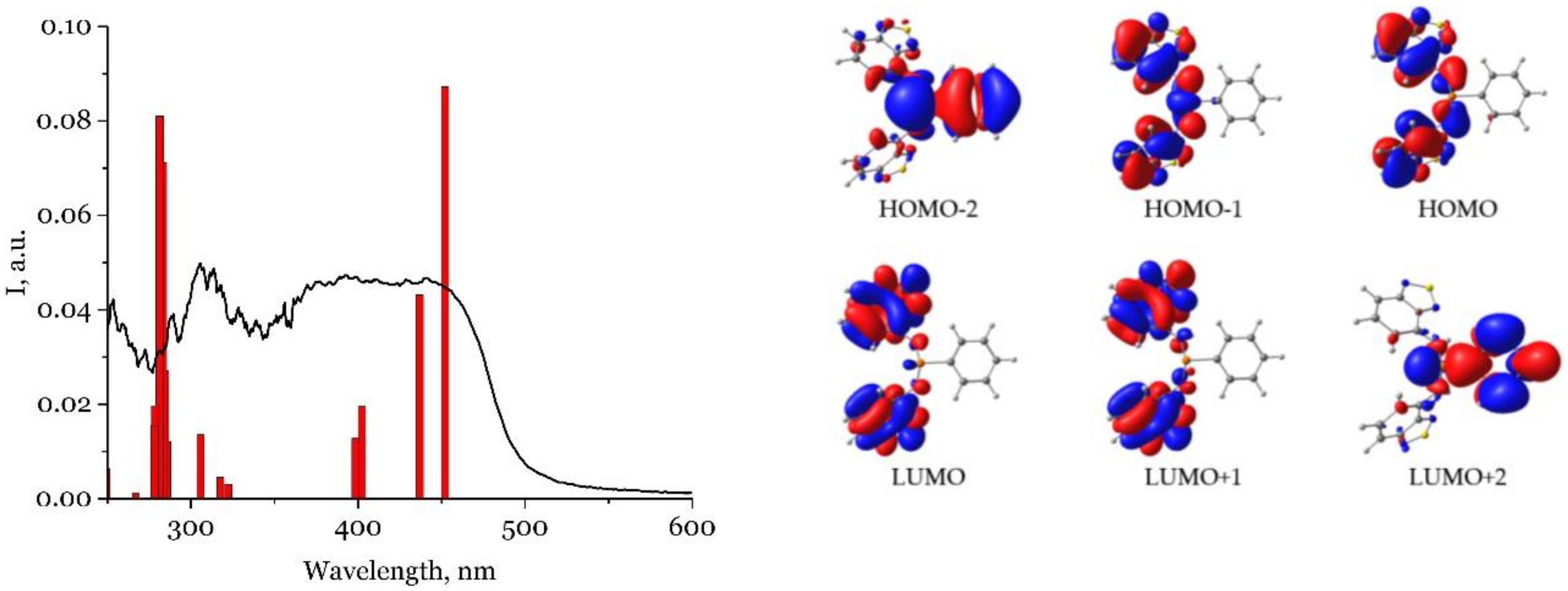
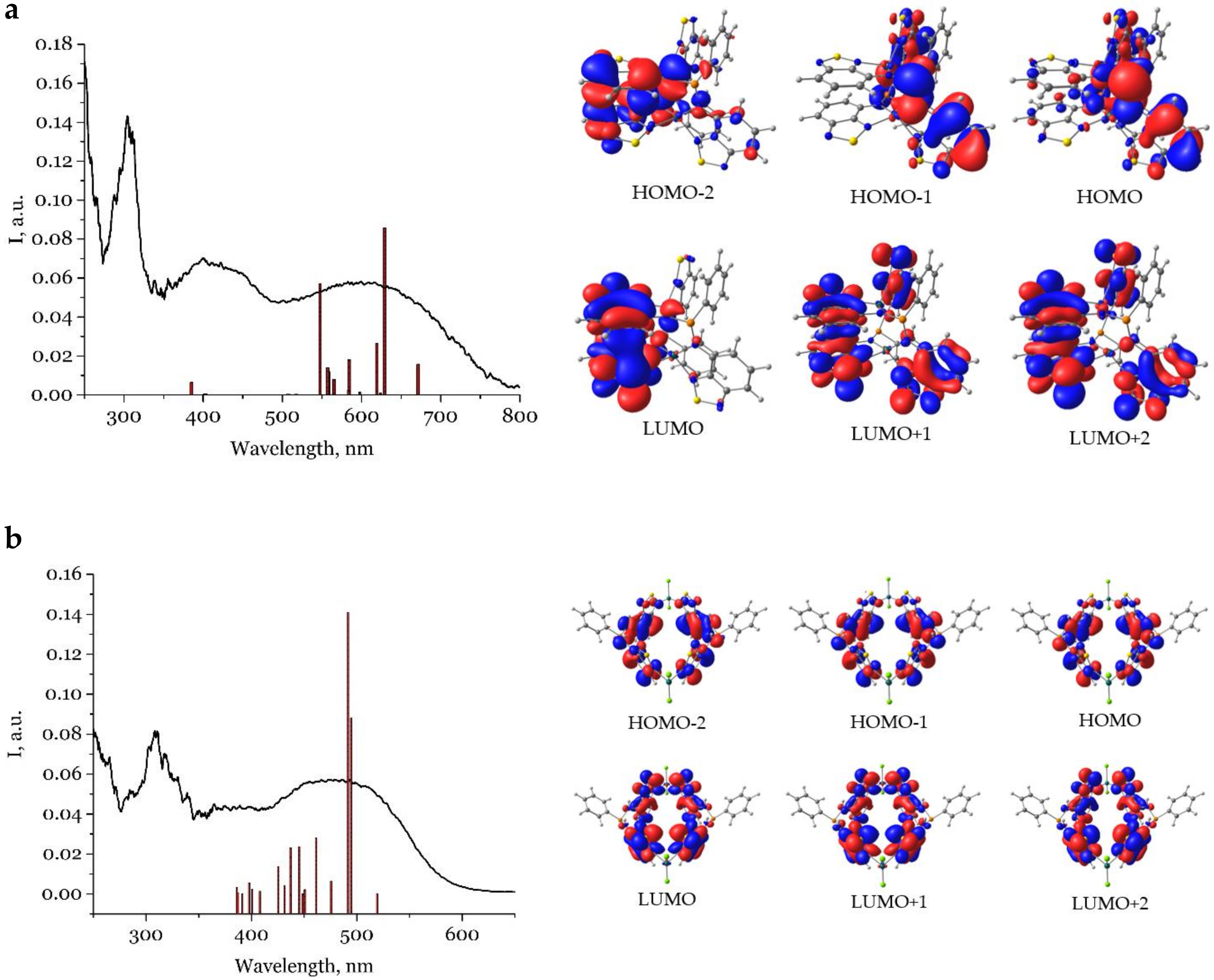
2.3. Photophysical Properties
3. Materials and Methods
3.1. General Methods
3.2. Quantum Chemical Calculations
3.3. X-ray Structure Determination
3.4. Syntheses
3.4.1. Synthesis of H2L
3.4.2. [Zn2L2]·nC7H8 (1)
3.4.3. [Zn2(H2L)2Cl4]·nC7H8 (2a·nC7H8 and 2b·nC7H8)
3.4.4. [Cu(H2L)Cl]n nTHF (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gutierrez, G.D.; Sazama, G.T.; Wu, T.; Baldo, M.A.; Swager, T.M. Red Phosphorescence from Benzo[2,1,3]thiadiazoles at Room Temperature. J. Org. Chem. 2016, 81, 4789–4796. [Google Scholar] [CrossRef] [Green Version]
- Paisley, N.R.; Tonge, C.M.; Mayder, D.M.; Thompson, K.A.; Hudson, Z.M. Tunable benzothiadiazole-based donor–acceptor materials for two-photon excited fluorescence. Mater. Chem. Front. 2020, 4, 555–566. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Carvalho, P.H.P.R.; Correa, J.R. Benzothiadiazole Derivatives as Fluorescence Imaging Probes: Beyond Classical Scaffolds. Acc. Chem. Res. 2015, 48, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Schill, J.; Ferrazzano, L.; Tolomelli, A.; Schenning, A.P.H.J.; Brunsveld, L. Fluorene benzothiadiazole co-oligomer based aqueous self-assembled nanoparticles. RSC Adv. 2020, 10, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Kini, G.P.; Choi, J.Y.; Jeon, S.J.; Suh, I.S.; Moon, D.K. Effect of mono alkoxy-carboxylate-functionalized benzothiadiazole-based donor polymers for non-fullerene solar cells. Dye. Pigment. 2019, 164, 62–71. [Google Scholar] [CrossRef]
- Prima, D.O.; Makarov, A.G.; Bagryanskaya, I.Y.; Kolesnikov, A.E.; Zargarova, L.V.; Baev, D.; Eliseeva, T.F.; Politanskaya, L.V.; Makarov, A.Y.; Slizhov, Y.G.; et al. Fluorine-Containing n-6 and Angular and Linear n-6-n′ (n, n′ = 5, 6, 7) Diaza-Heterocyclic Scaffolds Assembled on Benzene Core in Unified Way. Chemistry 2019, 4, 2383–2386. [Google Scholar] [CrossRef]
- Qian, G.; Wang, X.; Wang, S.; Zheng, Y.; Wang, S.; Zhu, W.; Wang, Y. Polymorphous Luminescent Materials Based on ’T’-Shaped Molecules Bearing 4,7-Diphenylbenzo[c][1,2,5]thiadiazole Skeletons: Effect of Substituents on the Photophysical Properties. Chem. A Eur. J. 2019, 25, 15401–15410. [Google Scholar] [CrossRef]
- Mikhailov, M.S.; Gudim, N.S.; Knyazeva, E.A.; Tanaka, E.; Zhang, L.; Mikhalchenko, L.V.; Robertson, N.; Rakitin, O.A. 9-(p-Tolyl)-2,3,4,4a,9,9a-hexahydro-1H-carbazole—A new donor building-block in the design of sensitizers for dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2020, 391, 112333. [Google Scholar] [CrossRef]
- Ansell, J.; Wills, M. Enantioselective catalysis using phosphorus-donor ligands containing two or three P-N or P-O bonds. Chem. Soc. Rev. 2002, 31, 259–268. [Google Scholar] [CrossRef]
- Martín, R.; Prieto, P.; Carrillo, J.R.; Rodríguez, A.M.; De Cozar, A.; Boj, P.G.; Díaz-García, M.A.; Ramírez, M.G. Design, synthesis and amplified spontaneous emission of 1,2,5-benzothiadiazole derivatives. J. Mater. Chem. C 2019, 7, 9996–10007. [Google Scholar] [CrossRef]
- Sukhikh, T.S.; Ogienko, D.S.; Bashirov, D.A.; Konchenkoa, S.N. Luminescent complexes of 2,1,3-benzothiadiazole derivatives. Russ. Chem. Bull. 2019, 68, 651–661. [Google Scholar] [CrossRef]
- Ishi-I, T.; Tanaka, H.; Youfu, R.; Aizawa, N.; Yasuda, T.; Kato, S.-I.; Matsumoto, T. Mechanochromic fluorescence based on a combination of acceptor and bulky donor moieties: Tuning emission color and regulating emission change direction. N. J. Chem. 2019, 43, 4998–5010. [Google Scholar] [CrossRef]
- Rakitin, O.A.; Zibarev, A.V. Synthesis and Applications of 5-Membered Chalcogen-Nitrogen π-Heterocycles with Three Heteroatoms. Asian J. Org. Chem. 2018, 7, 2397–2416. [Google Scholar] [CrossRef]
- Islam, A.; Akhtaruzzaman, M.; Chowdhury, T.H.; Qin, C.; Han, L.; Bedja, I.M.; Stalder, R.; Schanze, K.; Reynolds, J.R. Enhanced Photovoltaic Performances of Dye-Sensitized Solar Cells by Co-Sensitization of Benzothiadiazole and Squaraine-Based Dyes. ACS Appl. Mater. Interfaces 2016, 8, 4616–4623. [Google Scholar] [CrossRef] [PubMed]
- Page, Z.A.; Liu, Y.; Puodziukynaite, E.; Russell, T.P.; Emrick, T. Hydrophilic Conjugated Polymers Prepared by Aqueous Horner–Wadsworth–Emmons Coupling. Macromolecules 2016, 49, 2526–2532. [Google Scholar] [CrossRef]
- Boucard, J.; Boudjemaa, R.; Steenkeste, K.; Jacqueline, C.; Stephant, N.; Lefèvre, F.-X.; Laurent, A.D.; Lartigue, L.; Hulin, P.; Nedellec, S.; et al. Phosphonic Acid Fluorescent Organic Nanoparticles for High-Contrast and Selective Staining of Gram-Positive Bacteria. ACS Omega 2018, 3, 17392–17402. [Google Scholar] [CrossRef]
- Ren, Y.; Sezen, M.; Guo, F.; Jäkle, F.; Loo, Y.-L. [d]-Carbon–carbon double bond engineering in diazaphosphepines: A pathway to modulate the chemical and electronic structures of heteropines. Chem. Sci. 2016, 7, 4211–4219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jin, Z.; Fu, W.; Shi, M.; Chen, H. Phosphate ester side-chain-modified conjugated polymer for hybrid solar cells. J. Appl. Polym. Sci. 2017, 134, 134. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.-Y.; Zhao, Y.-D.; Cao, Y.-C. Recent advancements of high efficient donor–acceptor type blue small molecule applied for OLEDs. Mater. Today 2017, 20, 258–266. [Google Scholar] [CrossRef]
- Duan, C.; Li, J.; Han, C.; Ding, D.; Yang, H.; Wei, Y.; Xu, H. Multi-dipolar Chromophores Featuring Phosphine Oxide as Joint Acceptor: A New Strategy toward High-Efficiency Blue Thermally Activated Delayed Fluorescence Dyes. Chem. Mater. 2016, 28, 5667–5679. [Google Scholar] [CrossRef]
- Li, H.; Hong, M.; Scarpaci, A.; He, X.; Risko, C.; Sears, J.S.; Barlow, S.; Winget, P.; Marder, S.R.; Kim, D.; et al. Chemical Stabilities of the Lowest Triplet State in Aryl Sulfones and Aryl Phosphine Oxides Relevant to OLED Applications. Chem. Mater. 2019, 31, 1507–1519. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Wang, Q.; Shi, H.; An, Z.; Huang, W. Manipulating the Ultralong Organic Phosphorescence of Small Molecular Crystals. Chem. A Eur. J. 2020, 26, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Joly, D.; Bouit, P.-A.; Hissler, M. Organophosphorus derivatives for electronic devices. J. Mater. Chem. C 2016, 4, 3686–3698. [Google Scholar] [CrossRef] [Green Version]
- Sukhikh, T.S.; Khisamov, R.M.; Bashirov, D.A.; Komarov, V.Y.; Molokeev, M.S.; Ryadun, A.A.; Benassi, E.; Konchenko, S.N. Tuning of the Coordination and Emission Properties of 4-Amino-2,1,3-Benzothiadiazole by Introduction of Diphenylphosphine Group. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]
- Burford, N.; Cameron, T.S.; Conroy, K.D.; Ellis, B.; Lumsden, M.; Macdonald, C.L.B.; McDonald, R.; Phillips, A.D.; Ragogna, P.J.; Schurko, R.W.; et al. Transformations between Monomeric, Dimeric, and Trimeric Phosphazanes: Oligomerizing NP Analogues of Olefins. J. Am. Chem. Soc. 2002, 124, 14012–14013. [Google Scholar] [CrossRef]
- Ams, M.; Trapp, N.; Schwab, A.; Milić, J.V.; Diederich, F. Chalcogen Bonding “2S–2N Squares” versus Competing Interactions: Exploring the Recognition Properties of Sulfur. Chem. A Eur. J. 2018, 25, 323–333. [Google Scholar] [CrossRef]
- Sukhikh, T.S.; Bashirov, D.; Shuvaev, S.; Komarov, V.; Kuratieva, N.; Konchenko, S.; Benassi, E. Noncovalent interactions and photophysical properties of new Ag(I) complexes with 4-amino-2,1,3-benzothiadiazole. Polyhedron 2018, 141, 77–86. [Google Scholar] [CrossRef]
- CrystalExplorer, version 17; University of Western: Perth, Australia, 2017.
- Bond, A.D.; Doyle, E.L.; García, F.; Kowenicki, R.A.; Moncrieff, D.; McPartlin, M.; Riera, L.; Woods, A.D.; Wright, D.S. Thermodynamic/Kinetic Control in the Isomerization of the[{tBuNP(μ-NtBu)}2]2− Ion. Chem. A Eur. J. 2004, 10, 2271–2276. [Google Scholar] [CrossRef]
- Tirreé, J.; Gudat, D.; Nieger, M.; Niecke, E. Reversible Tautomeric Transformation between a Bis(amino)cyclodiphosph(V)azene and a Bis(imino)cyclodiphosph(V)azane. Angew. Chem. Int. Ed. 2001, 40, 3025–3028. [Google Scholar] [CrossRef]
- Moser, D.F.; Carrow, C.J.; Stahl, L.; Staples, R.J. Titanium complexes of bis(1°-amido)cyclodiphosph(III)azanes and bis(1°-amido)cyclodiphosph(V)azanes: Facial versus lateral coordination. J. Chem. Soc. Dalton Trans. 2001, 1246–1252. [Google Scholar] [CrossRef]
- Sukhikh, T.S.; Ogienko, D.S.; Bashirov, D.A.; Kurat’Eva, N.V.; Smolentsev, A.I.; Konchenko, S.N. Samarium, Europium, and Gadolinium Complexes with 4-(2,1,3-Benzothiadiazol-4-ylamino)pent-3-en-2-onate. Russ. J. Coord. Chem. 2019, 45, 30–35. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Prashad, M.; Repič, O.; Blacklock, T.J. A Practical and Chemoselective Reduction of Nitroarenes to Anilines Using Activated Iron. Adv. Synth. Catal. 2005, 347, 217–219. [Google Scholar] [CrossRef]
- Kubelka, P. New contributions to the optics intensely light scattering materials. J. Opt. Soc. Am. 1948, 38, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Bruker Apex3 Software Suite: Apex3, SADABS-2017/2 and SAINT, version 2018.7-2; Bruker AXS Inc.: Madison, WI, USA, 2017.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. Struct. Science 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples used are available from the authors. |



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P–N–C | N–P–N | C–P–N–C | P–N–C–C(N) | N–Zn–N | |
|---|---|---|---|---|---|
| H2L | 125.7, 127.3, | 107.4 | 151.4, 145.7 | 176.0, 171.8 | |
| H2L (optimized) | 125.6, 126.0 | 105.5 | 154.9, 156.4 | 170.2, 164.9 | |
| 1·C7H8 | 122.0, 122.1 | 97.9 | 80.0, 145.9 | 166.7, 177.8 | 84.3, 84.7 |
| 2a·3C7H8 | 124.1 | 106.2 | 150.0 | 177.4 | 96.2 |
| 2a (optimized) | 126.4, 126.4 | 104.8 | 152.0, 152.0 | 176.5 | 92.6 |
| 2b·2.5C7H8 | 122.2, 124.5 | 105.2 | 156.3, 154.9 | 160.0, 165.9 | 101.5 |
| 2b (optimized) | 125.4, 126.2 | 104.5 | 156.6, 151.2 | 171.9, 178.5 | 94.6 |
| 3·THF | 120.9, 129.9 | 109.1 | 156.9, 49.2 | 137.9, 169.2 |
| Compound | λabs, nm | λEm, nm | τ | QY, % |
|---|---|---|---|---|
| H2L | 305, 390-440 | 540 | 4.3 ns | 8 |
| 1·nC7H8 | 304, 400, 600 | |||
| 2·nC7H8 | 309, 480 | 635 | 3.1 ns | 3 |
| 3·THF | 304, 385, 550(sh) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khisamov, R.; Sukhikh, T.; Bashirov, D.; Ryadun, A.; Konchenko, S. Structural and Photophysical Properties of 2,1,3-Benzothiadiazole-Based Phosph(III)azane and Its Complexes. Molecules 2020, 25, 2428. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25102428
Khisamov R, Sukhikh T, Bashirov D, Ryadun A, Konchenko S. Structural and Photophysical Properties of 2,1,3-Benzothiadiazole-Based Phosph(III)azane and Its Complexes. Molecules. 2020; 25(10):2428. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25102428
Chicago/Turabian StyleKhisamov, Radmir, Taisiya Sukhikh, Denis Bashirov, Alexey Ryadun, and Sergey Konchenko. 2020. "Structural and Photophysical Properties of 2,1,3-Benzothiadiazole-Based Phosph(III)azane and Its Complexes" Molecules 25, no. 10: 2428. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25102428