
Synthesis and In Vitro Cytotoxicity and Antibacterial Activity of Novel 1,2,3-Triazol-5-yl-Phosphonates

 ,
, 
Abstract
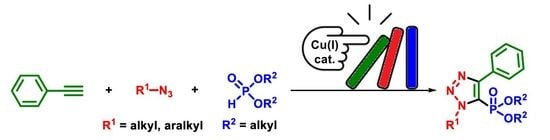
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Organic Azides
3.2.1. Method A
3.2.2. Method B
3.3. General Procedure for the Synthesis of Dipropyl and Dipentyl Phosphite
3.4. General Procedure for the Synthesis of 1,2,3-Triazol-5-yl-Phosphonates
3.5. Biological Evaluation
3.5.1. Cell Culture
3.5.2. Antibacterial Activity
3.5.3. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moonen, K.; Laureyn, I.; Stevens, C.V. Synthetic methods for azaheterocyclic phosphonates and their biological activity. Chem. Rev. 2004, 104, 6177–6216. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.K. (Ed.) Phosphorus Heterocycles II. In Topics in Heterocyclic Chemistry; Springer: Berlin, Germany, 2010; Volume 21, ISBN 978-3-642-12253-8. [Google Scholar]
- Tappe, F.M.J.; Trepohl, V.T.; Oestreich, M. Transition-metal-catalyzed CP cross-coupling reactions. Synthesis 2010, 18, 3037–3062. [Google Scholar] [CrossRef]
- Ali, T.E.; Abdel-Kariem, S.M. Methods for the synthesis of α-heterocyclic/heteroaryl-α-aminophosphonic acids and their esters. Arkivoc 2015, 2015, 246–287. [Google Scholar] [CrossRef]
- Haji, M. Multicomponent Reactions: A simple and efficient route to heterocyclic phosphonates. Beilstein J. Org. Chem. 2016, 12, 1269–1301. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.J.J. (Ed.) Multicomponent Reactions 1. In Science of Synthesis; Thieme: Stuttgart, Germany, 2014; ISBN 978-3-131-66881-3. [Google Scholar]
- Katritzky, A.R.; Rees, C.W.; Scriven, C.W.V. (Eds.) Comprehensive Heterocyclic Chemistry; Elsevier Science: Oxford, UK, 1996; Volume 4, pp. 1–126. [Google Scholar]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef]
- Lauria, A.; Delisi, R.; Mingoia, F.; Terenzi, A.; Martorana, A.; Barone, G.; Almerico, A.M. 1,2,3-Triazole in heterocyclic compounds, endowed with biological activity, through 1,3-dipolar cycloadditions. Eur. J. Org. Chem. 2014, 2014, 3289–3306. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1,2,3-triazole-linked isosteres as pharmacophores in drug discovery: Review. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef]
- Lal, K.; Yadav, P.; Kumar, A.; Kumar, A.; Paul, A.K. Design, synthesis, characterization, antimicrobial evaluation and molecular modeling studies of some dehydroacetic acid-chalcone-1,2,3-triazole hybrids. Bioorg. Chem. 2018, 77, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Kategaonkar, A.H.; Shinde, P.V.; Kategaonkar, A.H.; Pasale, S.K.; Shingate, B.B.; Shingare, M.S. Synthesis and biological evaluation of new 2-chloro-3-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)quinoline derivatives via click chemistry approach. Eur. J. Med. Chem. 2010, 45, 3142–3146. [Google Scholar] [CrossRef] [PubMed]
- Garudachari, B.; Isloor, A.M.; Satyanarayana, M.N.; Fun, H.K.; Hegde, G. Click chemistry approach: Regioselective one-pot synthesis of some new 8-trifluoromethylquinoline based 1,2,3-triazoles as potent antimicrobial agents. Eur. J. Med. Chem. 2014, 74, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza-Fagundes, E.M.; Delp, J.; Prazeres, P.D.M.; Marques, B.; Maria, A.; Carmo, L.; Henrique, P.; Stroppa, F.; Glanzmann, N.; Kisitu, J.; et al. Correlation of structural features of novel 1,2,3-triazoles with their neurotoxic and tumoricidal properties. Chem.-Biol. Interact. 2018, 291, 253–263. [Google Scholar] [CrossRef]
- Wu, M.J.; Wu, D.M.; Chen, J.B.; Zhao, J.F.; Gong, L.; Gong, Y.X.; Li, Y.; Yang, X.D.; Zhang, H. Synthesis and anti-proliferative activity of allogibberic acid derivatives containing 1,2,3-triazole pharmacophore. Bioorg. Med. Chem. Lett. 2018, 28, 2543–2549. [Google Scholar] [CrossRef]
- Praveena, K.S.S.; Shivaji Ramarao, E.V.V.; Murthy, N.Y.S.; Akkenapally, S.; Ganesh Kumar, C.; Kapavarapu, R.; Pal, S. Design of new hybrid template by linking quinoline, triazole and dihydroquinoline pharmacophoric groups: A greener approach to novel polyazaheterocycles as cytotoxic agents. Bioorg. Med. Chem. Lett. 2015, 25, 1057–1063. [Google Scholar] [CrossRef]
- Li, L.; Hao, G.; Zhu, A.; Fan, X.; Zhang, G.; Zhang, L. A copper(I)-catalyzed three-component domino process: Assembly of complex 1,2,3-triazolyl-5-phosphonates from azides, alkynes, and H-phosphates. Chem. Eur. J. 2013, 19, 14403–14406. [Google Scholar] [CrossRef]
- Radi, S.; Lazrek, H.B. Synthesis and biological activity of new 1,2,3-triazole acyclonucleosides analogues of ACV. J. Chem. Res. Synop. 2002, 264–266. [Google Scholar] [CrossRef]
- Costa, C.P.C.; Boechat, N.; Bastos, M.M.; da Silva, F.D.C.; Marttorelli, A.; Souza, M.L.T.; Baptista, S.M.; Hoelz, V.B.L.; Cafffarena, R.E. New Efavirenz derivatives and 1,2,3-triazolyl-phosphonates as inhibitors of reverse transcriptase of HIV-1. Curr. Top. Med. Chem. 2018, 2018, 1494–1505. [Google Scholar] [CrossRef]
- McAllister, T.E.; Horner, K.A.; Webb, M.E. Evaluation of the interaction between phosphohistidine analogues and phosphotyrosine binding domains. Chem. Bio. Chem. 2014, 15, 1088–1091. [Google Scholar] [CrossRef] [Green Version]
- Kee, J.M.; Villani, B.; Carpenter, L.R.; Muir, T.W. Development of stable phosphohistidine analogues. J. Am. Chem. Soc. 2010, 132, 14327–14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, S.; Flematti, G.R.; Byrne, L.T.; Besant, P.G.; Attwood, P.V.; Piggott, M.J. Stable triazolylphosphonate analogues of phosphohistidine. Amino Acids. 2012, 43, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. 1963, 2, 565–632. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew. Chem. Int. Ed. 1963, 2, 633–696. [Google Scholar] [CrossRef]
- Hall, R.G.; Trippet, S. The preparation and Diels-Alder reactivity of ethyl (diethoxyphosphinyl)propynoate. Tetrahedron Lett. 1982, 23, 2603–2604. [Google Scholar] [CrossRef]
- Shen, Y.; Zheng, J.; Xin, Y.; Lin, Y.; Qi, M. Synthesis of perfluoroalkylated heterocyclic phosphonates. J. Chem. Soc. Perkin Trans. 1 1995, 8, 997–999. [Google Scholar] [CrossRef]
- Huang, P.; Su, Q.; Dong, W.; Zhang, Y.; An, D. BrØnsted/Lewis acids-promoted selective preparations of 3-hetero quinolines or 4/5-hetero triazoles from azides and hetero-alkynes. Tetrahedron 2017, 73, 4275–4284. [Google Scholar] [CrossRef]
- Artyushin, O.I.; Matveeva, E.V.; Bushmarinov, I.S.; Odinets, I.L. Water as a promoting media for 1,3-dipolar cycloaddition of phosphorylated azides to internal alkynes. Arkivoc 2012, 4, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Louërat, F.; Bougrin, K.; Loupy, A.; de Retana, A.M.O.; Pagalday, J.; Palacios, F. Cycloaddition reactions of azidomethyl phosphonate with acetylenes and enamines. Synthesis of triazoles. Heterocycles 1998, 48, 161–170. [Google Scholar] [CrossRef]
- Thomas, J.; Jubi, J.; Parekh, N.; Dehaen, W. A metal-free three-component reaction for the regioselective synthesis of 1,4,5-trisubstituted 1,2,3-triazoles. Angew. Chem. Int. Ed. 2014, 53, 10155–10160. [Google Scholar] [CrossRef]
- Ahamad, S.; Kant, R.; Mohanan, K. Metal-free three-component domino approach to phosphonylated triazolines and triazoles. Org. Lett. 2016, 18, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Tripolszky, A.; Tóth, E.; Bálint, E. Synthesis of 1,2,3-triazolyl-5-diethylphosphonate by domino reaction. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 377–378. [Google Scholar] [CrossRef]
- Gann, A.W.; Amoroso, J.W.; Einck, V.J.; Rice, W.P.; Chambers, J.J.; Schnarr, N.A. A photoinduced, benzyne click reaction. Org. Lett. 2014, 16, 2003–2005. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Lu, W.; Su, H.; Qiu, L.; Xu, X. A convergent formal [4 + 2] cycloaddition of 1,6-diynes and benzyl azides: Construction of spiro-polyheterocycles. Org. Biomol. Chem. 2018, 16, 3258–3265. [Google Scholar] [CrossRef]
- Wang, X.; Mei, T.S.; Yu, J.Q. Versatile Pd(OTf)2 ∙ 2H2O-catalyzed ortho-fluorination using NMP as a promoter. J. Am. Chem. Soc. 2009, 131, 7520–7521. [Google Scholar] [CrossRef]
- Colombano, G.; Albani, C.; Ottonello, G.; Ribeiro, A.; Scarpelli, R.; Tarozzo, G.; Daglian, J.; Jung, K.M.; Piomelli, D.; Bandiera, T. O-(Triazolyl)methyl carbamates as a novel and potent class of fatty acid amide hydrolase (FAAH) inhibitors. Chem. Med. Chem. 2014, 10, 380–395. [Google Scholar] [CrossRef] [Green Version]
- Tesch, M.; Kudruk, S.; Letzel, M.; Studer, A. Orthogonal click postfunctionalization of alternating copolymers prepared by nitroxide-mediated polymerization. Chem. Eur. J. 2017, 23, 5915–5919. [Google Scholar] [CrossRef]
- Colombano, G.; Travelli, C.; Galli, U.; Caldarelli, A.; Chini, M.G.; Canonico, P.L.; Sorba, G.; Bifulco, G.; Tron, G.C.; Genazzani, A.A. A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 2010, 53, 616–623. [Google Scholar] [CrossRef]
- Swetha, M.; Ramana, P.V.; Shirodkar, S.G. Simple and efficient method for the synthesis of azides in water- THF solvent system. Org. Prep. Proced. Int. 2011, 43, 348–353. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Tóth, N.; Keglevich, G. Continuous flow alcoholysis of dialkyl H-phosphonates with aliphatic alcohols. Molecules 2018, 23, 1618. [Google Scholar] [CrossRef] [Green Version]
- Csörgő, B.; Fehér, T.; Tímár, E.; Blattner, F.R.; Pósfai, G. Low-mutation-rate, reduced-genome Escherichia coli: An improved host for faithful maintenance of engineered genetic constructs. Microb. Cell Fact. 2012, 11, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.T.; Tran, L.T.; Schumann, W.; Nguyen, H.D. Development of Pgrac100-based expression vectors allowing high protein production levels in Bacillus subtilis and relatively low basal expression in Escherichia coli. Microb. Cell Fact. 2015, 14, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szebeni, G.J.; Balázs, A.; Madarász, I.; Pocz, G.; Ayaydin, F.; Kanizsai, I.; Fajka-Boja, R.; Alföldi, R.; Hackler, L., Jr.; Puskás, L.G. Achiral mannich-base curcumin analogs induce unfolded protein response and mitochondrial membrane depolarization in panc-1 cells. Int. J. Mol. Sci. 2017, 18, 2105. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 8 and 11–26 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Y1 | Y2 | R | Cat. | Solvent | T, t | Product Ratio (%) | Yield (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | |||||||||
| 1 | CH2COOMe | COOEt | Et | - | toluene | 110 °C | 64 | 36 | - | [28] |
| 2 | CH2COOtBu | CF3 | iPr | - | DEE | 25 °C, 20 h | 75 | 25 | 90 | [29] |
| 3 | Bn | Ph, 4-MeC6H4, 4-FC6H4, etc. | Et | CuSO4·5H2O/NaAsc | DMF | 170 °C, 12 h | 100 | 0 | 83–92 | [30] |
| 4 | (EtO)2P(O)(CH2)2 | (MeO)2P(O) | Me | - | water | 25 °C, 36 h or 60 °C, 2 h | 100 | 95 | [31] | |
| 5 | (EtO)2P(O)CH2 | Me | Et | - | - | MW, 90 °C, 20 min | 34 | 66 | 78 | [32] |

| Entry | DBP (Equiv.) | Condition | Catalyst (10 mol%) | Base (2 Equiv.) | Solvent | Product Composition (%) a | ||
|---|---|---|---|---|---|---|---|---|
| 8 | 9 | 10 | ||||||
| 1 | 1.1 | N2 bubbling | CuCl | TEA | MeCN | 22 | 77 | 1 |
| 2 | 1.1 | air bubbling | CuCl | TEA | MeCN | 55 | 42 | 3 |
| 3 | 1.1 | O2 bubbling | CuCl | TEA | MeCN | 24 | 70 | 6 |
| 4 | 1.1 | air bubbling | CuCl | DPA | MeCN | 46 | 49 | 5 |
| 5 | 1.1 | air bubbling | CuCl | DIPA | MeCN | 54 | 44 | 2 |
| 6 | 1.1 | air bubbling | CuCl | DIPEA | MeCN | 35 | 63 | 2 |
| 7 | 1.1 | air bubbling | CuCl | TEA | DMF | 41 | 57 | 2 |
| 8 | 1.1 | air bubbling | CuCl | TEA | DCM | 43 | 56 | 1 |
| 9 | 1.1 | air bubbling | CuCl | TEA | THF | 21 | 78 | 1 |
| 10 | 1.1 | air bubbling | CuCl | TEA | EtOH | 39 | 59 | 2 |
| 11 | 1.1 | air bubbling | CuBr | TEA | MeCN | 42 | 56 | 2 |
| 12 | 1.1 | air bubbling | CuI | TEA | MeCN | 26 | 72 | 2 |
| 13 | 1.1 | air bubbling | CuSO4·5H2O/NaAsc | TEA | MeCN | 0 | 100 | 0 |
| 14 | 2 | air bubbling | CuCl | TEA | MeCN | 59 | 37 | 4 |
| 15 | 3 | air bubbling | CuCl | TEA | MeCN | 51 | 45 | 4 |
| 16 b | 2 | air bubbling | CuCl | TEA | MeCN | 49 | 49 | 2 |
| 17 c | 2 | air bubbling | CuCl | TEA | MeCN | 36 | 62 | 2 |
| 18 d | 2 | air bubbling | CuCl | TEA | MeCN | 23 | 75 | 2 |

| Compound | R1 | R2 | Antibacterial Activity (IC50, µM) | In Vitro Cytotoxicity (IC50, µM) | |||
|---|---|---|---|---|---|---|---|
| B. subtilis | E. coli | A549 | NIH/3T3 | HL-60 | |||
| 8 | Bn | Bu | >30 | >30 | >30 | 27.5 ± 1.4 | >30 |
| 11 | Bn | Me | >30 | >30 | >30 | >30 | >30 |
| 12 | Bn | Et | >30 | >30 | >30 | >30 | >30 |
| 13 | Bn | Pr | 29.3 ± 1.2 | >30 | >30 | >30 | 12.6 ± 1.7 |
| 14 | Bn | iPr | >30 | >30 | >30 | >30 | >30 |
| 15 | Bn | Pn | 23.9 ± 1.0 | >30 | >30 | 26.2 ± 1.1 | 15.4 ± 1.2 |
| 16 | 4-MeC6H4 | Me | >30 | >30 | >30 | 19.8 ± 1.2 | 11.0 ± 1.2 |
| 17 | 4-MeC6H4 | Et | >30 | >30 | >30 | >30 | >30 |
| 18 | 4-MeC6H4 | Bu | 18.2 ± 1.0 | >30 | >30 | >30 | >30 |
| 19 | 2-FC6H4 | Bu | 19.3 ± 1.0 | >30 | >30 | 27.5 ± 1.1 | 11.7 ± 1.2 |
| 20 | 3-FC6H4 | Bu | >30 | >30 | >30 | >30 | 15.4 ± 1.2 |
| 21 | 4-FC6H4 | Bu | 23.2 ± 1.1 | >30 | >30 | >30 | 16.6 ± 1.3 |
| 22 | 4-CF3C6H4 | Bu | >30 | >30 | >30 | 23.1 ± 1.2 | 9.7 ± 1.1 |
| 23 | Oct | Me | >30 | >30 | >30 | >30 | >30 |
| 24 | Oct | Et | >30 | >30 | >30 | >30 | >30 |
| 25 | Oct | Bu | >30 | >30 | >30 | >30 | >30 |
| 26 | iOct | Bu | 29.6 ± 1.0 | >30 | >30 | >30 | 26.7 ± 1.0 |
| Doxycycline | 0.04 ± 0.01 | 0.10 ± 0.02 | – | – | – | ||
| Gentamicin | 0.49 ± 0.14 | 4.23 ± 0.99 | – | – | – | ||
| Doxorubicin | – | – | 0.31 ± 0.24 | 5.65 ± 0.81 | – | ||
| Bortezomib | – | – | – | – | 7.42 × 10−3 2.60 × 10−3 | ||
| R1N3 | Yield | [M + H]+found | [M + H]+requires |
|---|---|---|---|
| BnN3 [36] | 93% (1.24 g) | 134.0725 | 134.0718 |
| 4-Me-BnN3 [37] | 80% (1.18 g) | 148.0880 | 148.0874 |
| 2-F-BnN3 [38] | 68% (1.02 g) | 152.0632 | 152.0624 |
| 3-F-BnN3 [39] | 76% (1.14 g) | 152.0633 | 152.0624 |
| 4-F-BnN3 [37] | 83% (1.25 g) | 152.0632 | 152.0624 |
| 4-CF3-BnN3 [40] | 86% (1.72 g) | 202.0601 | 202.0592 |
| OctN3 [41] | 67% (1.04 g) | 156.1514 | 156.1501 |
| iOctN3 [42] | 55% (0.85 g) | 156.1514 | 156.1501 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripolszky, A.; Tóth, E.; Szabó, P.T.; Hackler, L., Jr.; Kari, B.; Puskás, L.G.; Bálint, E. Synthesis and In Vitro Cytotoxicity and Antibacterial Activity of Novel 1,2,3-Triazol-5-yl-Phosphonates. Molecules 2020, 25, 2643. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112643
Tripolszky A, Tóth E, Szabó PT, Hackler L Jr., Kari B, Puskás LG, Bálint E. Synthesis and In Vitro Cytotoxicity and Antibacterial Activity of Novel 1,2,3-Triazol-5-yl-Phosphonates. Molecules. 2020; 25(11):2643. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112643
Chicago/Turabian StyleTripolszky, Anna, Emese Tóth, Pál Tamás Szabó, László Hackler, Jr., Beáta Kari, László G. Puskás, and Erika Bálint. 2020. "Synthesis and In Vitro Cytotoxicity and Antibacterial Activity of Novel 1,2,3-Triazol-5-yl-Phosphonates" Molecules 25, no. 11: 2643. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112643