
The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite

 , , and
, , and
Abstract
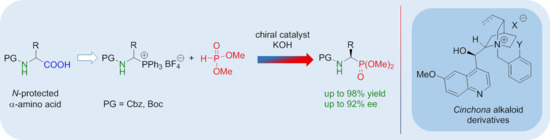
:
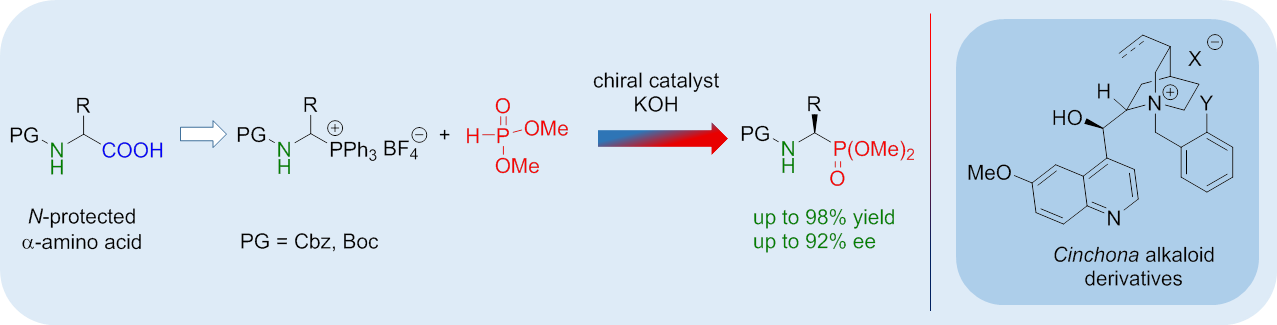
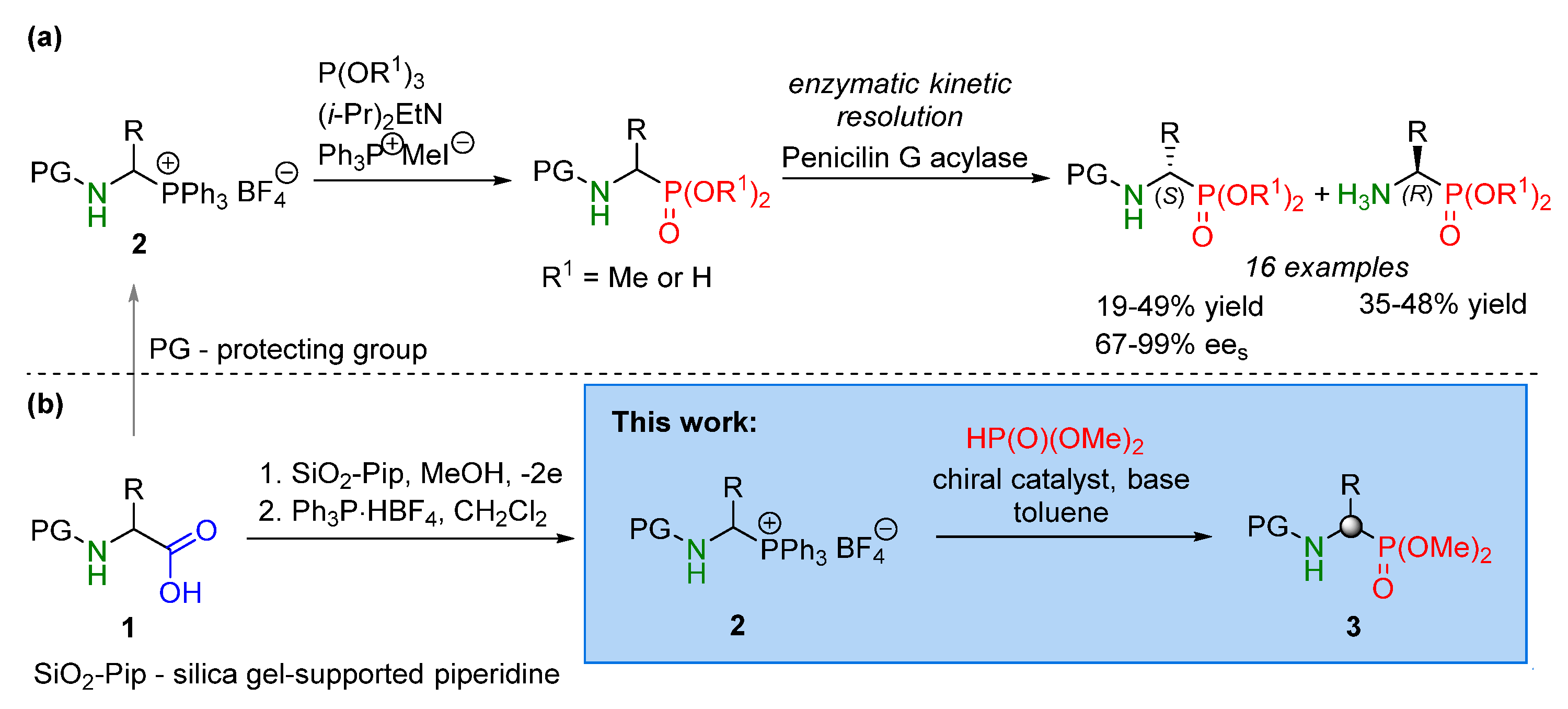
1. Introduction
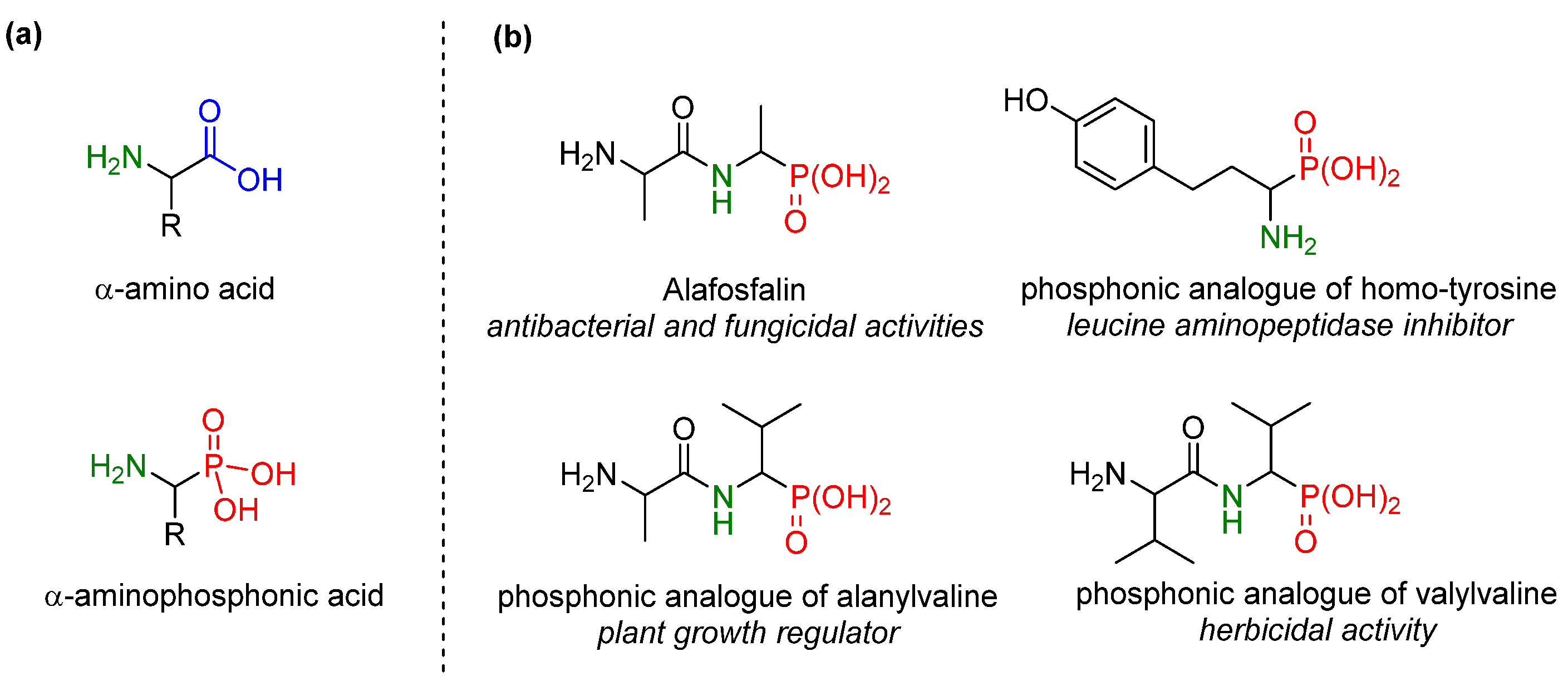
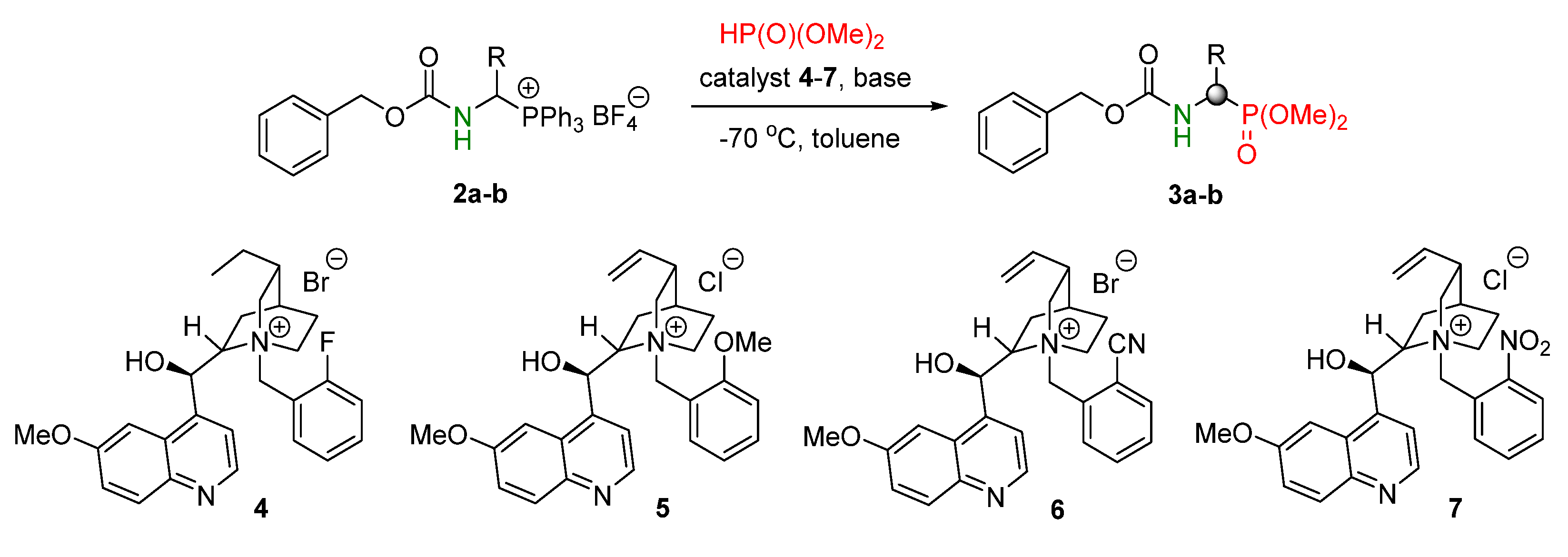
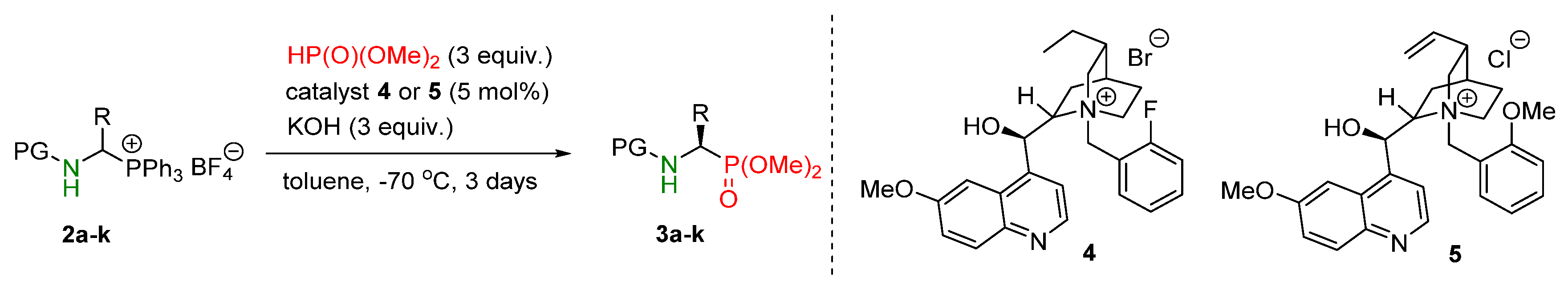
2. Result s and Discussion
3. Materials and Methods
3.1. General Information
3.2. Substrate Synthesis
General Procedure for the Synthesis of 1-(N-acylamino)alkyltriphenylphosphonium Tetrafluoroborates 2
3.3. General Procedure for the PT-Catalyzed Reaction of 1-(N-acylamino)alkyltriphenylphosphonium Salts 2 with Dimethyl Phosphite
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kafarski, P.; Lejczak, B. Biological Activity of Aminophosphonic Acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids. Chemistry and Biological Activity; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 2000. [Google Scholar]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P.; Lejczak, B. Aminophosphonic Acids of Potential Medical Importance. Curr. Med. Chem. Anticancer Agents 2001, 1, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef]
- Demkowicz, S.; Rachon, J.; Daśko, M.; Kozak, W. Selected Organophosphorus Compounds with Biological Activity. Applications in Medicine. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Lejczak, B.; Kafarski, P. Biological Activity of Aminophosphonic Acids and Their Short Peptides. In Top. Heterocycl. Chem.; Springer-Verlag: Berlin Heidelberg, Germany, 2009; pp. 31–63. [Google Scholar]
- Ordóñez, M.; Rojas-Cabrera, H.; Cativiela, C. An Overview of Stereoselective Synthesis of α-Aminophosphonic Acids and Derivatives. Tetrahedron 2009, 65, 17–49. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Romero-Estudillo, I. Amino Acid. In Amino Acid-New Insights and Roles in Plant and Animal; Toshiki Asao, Ed.; IntechOpen: London, UK, 2017; pp. 127–151. [Google Scholar]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An Update on the Stereoselective Synthesis of α-Aminophosphonic Acids and Derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Ganoub, N.A.; Sabry, E.; Abdou, W.M. Overview on the Phosphonation of the C=X Functional Groups Utilizing Alkyl Phosphites. Synth. Commun. 2017, 47, 1631–1660. [Google Scholar] [CrossRef]
- Renaud, P.; Seebach, D. Enantiomerenreine Pyrrolidin-Derivate Aus Trans-4-Hydroxy-L-Prolin Durch Elektrochemische Oxidative Decarboxylierung Und Titantetrachlorid-Vermittelte Umsetzung Mit Nukleophilen. Helv. Chim. Acta 1986, 69, 1704–1710. [Google Scholar] [CrossRef]
- Ösapay, G.; Szilagyi, I.; Seres, J. Conversion of Amino Acids and Dipeptides into Their Phosphonic Analogs: Aminoalkylphosphonic Acids and Peptides II. Tetrahedron 1987, 43, 2977–2983. [Google Scholar] [CrossRef]
- Seebach, D.; Charczuk, R.; Gerber, C.; Renaud, P.; Berner, H.; Schneider, H. Elektrochemische Decarboxylierung von L-Threonin-Und Oligopeptid-Derivaten Unter Bildung von N-Acyl-N,O-Acetalen: Herstellung von Oligopeptiden Mit Carboxamid-Oder Phosphonat-C-Terminus. Helv. Chim. Acta 1989, 72, 401–425. [Google Scholar] [CrossRef]
- Corcoran, R.C.; Green, J.M. Conversion of α-Aminocarboxylic Acids to α-Aminophosphonic Acids. Tetrahedron Lett. 1990, 31, 6827–6830. [Google Scholar] [CrossRef]
- Kaname, M.; Mashige, H.; Yoshifuji, S. Chemical Conversion of Cyclic α-Amino Acids to Cyclic α-Aminophosphonic Acids. Chem. Pharm. Bull. 2001, 49, 531–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pousset, C.; Larchevêque, M. An Efficient Synthesis of α- and β-Aminophosphonic Esters from α-Amino Acids. Tetrahedron Lett. 2002, 43, 5257–5260. [Google Scholar] [CrossRef]
- Matveeva, E.; Zefirov, N. Amino Acids in a Three-Component Synthesis of α-Aminophosphonates Derivatives. Russ. J. Org. Chem. 2006, 42, 1237–1238. [Google Scholar] [CrossRef]
- Ambica; Kumar, S.; Taneja, S.C.; Hundal, M.S.; Kapoor, K.K. One-Pot Synthesis of α-Aminophosphonates Catalyzed by Antimony Trichloride Adsorbed on Alumina. Tetrahedron Lett. 2008, 49, 2208–2212. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, D.; Mao, L.; Wang, L.; Wang, R. Copper/DIPEA-Catalyzed, Aldehyde-Induced Tandem Decarboxylation-Coupling of Natural α-Amino Acids and Phosphites or Secondary Phosphine Oxides. J. Org. Chem. 2011, 76, 6426–6431. [Google Scholar] [CrossRef]
- Kaźmierczak, M.; Koroniak, H. DAST Mediated Preparation of β-Fluoro-α-Aminophosphonates. J. Fluor. Chem. 2012, 139, 23–27. [Google Scholar] [CrossRef]
- Kaboudin, B.; Karami, L.; Kato, J.; Aoyama, H.; Yokomatsu, T. A Catalyst-Free, Three-Component Decarboxylative Coupling of Amino Acids with Aldehydes and H-Dialkylphosphites for the Synthesis of α-Aminophosphonates. Tetrahedron Lett. 2013, 54, 4872–4875. [Google Scholar] [CrossRef]
- Zhu, Z.-Q.; Xiao, L.-J.; Guo, D.; Chen, X.; Ji, J.-J.; Zhu, X.; Xie, Z.-B.; Le, Z.-G. Cobalt-Catalyzed Oxidative Phosphonylation of α-Amino Acid Derivatives and α-Amino Ketones for α-Aminophosphonates. J. Org. Chem. 2019, 84, 435–442. [Google Scholar] [CrossRef]
- Miguélez-Ramos, J.; Batchu, V.R.; Boto, A. Tuning the Stereoselectivity in One-Pot Scission/Addition Processes: Synthesis of Azanucleotide Analogues from Proline Derivatives. Eur. J. Org. Chem. 2013, 846–852. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. α-Amidoalkylating Agents from N-Acyl-A-Amino Acids: 1-(N-Acylamino) Alkyltriphenylphosphonium Salts. J. Org. Chem. 2012, 77, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Kononienko, A. A Novel Synthesis of 1-Aminoalkanephosphonic Acid Derivatives from 1-(N-Acylamino)-Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon 2010, 185, 1986–1992. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl) Amides and 1-(N-Acylamino) Alkyltriphenylphosphonium Salts in the Michaelis-Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl) Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar]
- Zielińska, K.; Mazurkiewicz, R.; Szymańska, K.; Jarzębski, A.; Magiera, S.; Erfurt, K. Penicillin G Acylase-Mediated Kinetic Resolution of Racemic 1-(N-Acylamino) Alkylphosphonic and 1-(N-Acylamino) Alkylphosphinic Acids and Their Esters. J. Mol. Catal. B: Enzym. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Song, E.C. Cinchona Alkaloids in Synthesis & Catalysis. Ligands, Immobilization and Organocatalysis; Song, E.C., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, G.S. Recent Application of Cinchona Alkaloids and Their Derivatives as Catyst in Metal-Free Asymmetric Synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Fini, F.; Bernardi, L.; Herrera, R.P.; Pettersen, D.; Ricci, A.; Sgarzani, V. Phase-Transfer-Catalyzed Enantioselective Mannich Reaction of Malonates with α-Amido Sulfones. Adv. Synth. Catal. 2006, 348, 2043–2046. [Google Scholar] [CrossRef]
- Marianacci, O.; Micheletti, G.; Bernardi, L.; Fini, F.; Fochi, M.; Pettersen, D.; Sgarzani, V.; Ricci, A. Organocatalytic Asymmetric Mannich Reactions with N-Boc and N-Cbz Protected α-Amido Sulfones (Boc: Tert-Butoxycarbonyl, Cbz: Benzyloxycarbonyl). Chem. Eur. J. 2007, 13, 8338–8351. [Google Scholar] [CrossRef]
- González, P.B.; Lopez, R.; Palomo, C. Catalytic Enantioselective Mannich-Type Reaction with β-Phenyl Sulfonyl Acetonitrile. J. Org. Chem. 2010, 75, 3920–3922. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N. Asymmetric Synthesis of Quaternary α-Amino Acids and Their Phosphonate Analogues. Asian J. Org. Chem. 2014, 3, 1234–1260. [Google Scholar] [CrossRef]
- Pettersen, D.; Marcolini, M.; Bernardi, L.; Fini, F.; Herrera, R.P.; Sgarzani, V.; Ricci, A. Direct Access to Enantiomerically Enriched α-Amino Phosphonic Acid Derivatives by Organocatalytic Asymmetric Hydrophosphonylation of Imines. J. Org. Chem. 2006, 71, 6269–6272. [Google Scholar] [CrossRef] [PubMed]
- Fini, F.; Micheletti, G.; Bernardi, L.; Pettersen, D.; Fochi, M.; Ricci, A. An Easy Entry to Optically Active α-Amino Phosphonic Acid Derivatives Using Phase-Transfer Catalysis (PTC). Chem. Commun. 2008, 36, 4345–4347. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Song, A.; Zhang, X.; Chen, X.; Li, H.; Sheng, C.; Wang, W. Quinine-Thiourea Catalyzed Enantioselective Hydrophosphonylation of Trifluoromethyl 2(1H)-Quinazolinones. Chem. Commun. 2013, 49, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Y.; Xu, D. Asymmetric Synthesis of α-Amino Phosphonates by Using Cinchona Alkaloid-Based Chiral Phase Transfer Catalyst. Eur. J. Org. Chem. 2018, 5422–5426. [Google Scholar] [CrossRef]
- Nakamura, S.; Nakashima, H.; Yamamura, A.; Shibata, N.; Toru, T. Organocatalytic Enantioselective Hydrophosphonylation of Sulfonylimines Having a Heteroarenesulfonyl Group as a Novel Stereocontroller. Adv. Synth. Catal. 2008, 350, 1209–1212. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Adamek, J.; Mazurkiewicz, R.; Zielińska, K. Amidoalkylating Properties of 1-(N-Acylamino) Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 205–212. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Walęcka-Kurczyk, A.; Musioł, S.; Stecko, S. Catalyst-Free Mannich-Type Reaction of 1-(N-Acylamino) Alkyltriphenylphosphonium Salts with Silyl Enolates. Tetrahedron 2019, 75, 732–742. [Google Scholar] [CrossRef]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The Self-Disproportionation of Enantiomers (SDE): A Menace or an Opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, T.; Imai, T.; Wzorek, A.; Marcinkowska, M.; Kolbus, A.; Kitagawa, O.; Soloshonok, V.A.; Klika, K.D. The Self-Disproportionation of Enantiomers (SDE) of α-Amino Acid Derivatives: Facets of Steric and Electronic Properties. Amino Acids 2019, 51, 283–294. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Remarkable Magnitude of the Self-Disproportionation of Enantiomers (SDE) via Achiral Chromatography: Application to the Practical-Scale Enantiopurification of β-Amino Acid Esters. Amino Acids 2016, 48, 605–613. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Marcinkowska, M.; Wzorek, A.; Pajkert, R.; Han, J.; Klika, K.D.; Soloshonok, V.A.; Rӧschenthaler, G.-V. The Self-Disproportionation of Enantiomers (SDE) via Column Chromatography of β-Amino-α, α-Difluorophosphonic Acid Derivatives. Amino Acids 2019, 51, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Stamper, C.; Bennett, B.; Edwards, T.; Holz, R.C.; Ringe, D.; Petsko, G. Inhibition of the Aminopeptidase from Aeromonas Proteolytica by L-Leucinephosphonic Acid. Spectroscopic and Crystallographic Characterization of the Transition State of Peptide Hydrolysis. Biochemistry 2001, 40, 7035–7046. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.E.; Kaplan, A.P.; Bartlett, P.A. Phosphonate Analogs of Carboxypeptidase A Substrates Are Potent Transition-State Analog Inhibitors. Biochemistry 1989, 28, 6294–6305. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-Y.; Huang, J.-D.; Hu, X.-P.; Deng, J.; Yu, S.-B.; Duan, Z.-C.; Zheng, Z. Readily Available Chiral Phosphine- Aminophosphine Ligands for Highly Efficient Rh-Catalyzed Asymmetric Hydrogenation of α-Enol Ester Phosphonates and α-Enamido Phosphonates. J. Org. Chem. 2008, 73, 2011–2014. [Google Scholar] [CrossRef]
- Szewczyk, J.; Lejczak, B.; Kafarski, P. Transesterification of Diphenyl Phosphonates Using the Potassium Fluoride/Crown Ether/Alcohol System; Part 1. Transesterification of Diphenyl 1-(Benzyloxycarbonylamino)-Alkanephosphonates. Synthesis 1982, 409–412. [Google Scholar] [CrossRef]
- Hoffmann, M. Application of t-Butoxycarbonylazide for Preparation of 1-Aminoalkylphosphonic Acid Derivatives. Pol. J. Chem. 1979, 53, 1153–1158. [Google Scholar]
- Bartlett, P.A.; Hanson, J.E.; Giannousis, P.P. Potent Inhibition of Pepsin and Penicillopepsin by Phosphorus-Containing Peptide Analogs. J. Org. Chem. 1990, 55, 6268–6274. [Google Scholar] [CrossRef]
- Pícha, J.; Buděšínský, M.; Hančlová, I.; Šanda, M.; Fiedler, P.; Vaněk, V.; Jiráček, J. Efficient Synthesis of Phosphonodepsipeptides Derived from Norleucine. Tetrahedron 2009, 65, 6090–6103. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 2, 3 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Salt | R | Catalyst (5 mol%) | Base (equiv.) | Time [days] | Yield 2 [%] | ee 3 [%] |
|---|---|---|---|---|---|---|---|
| 1 | 2a | Me | 4 | K2CO3 (3) | 3 | 62 | 49 |
| 2 | 2a | Me | 4 | KOH (3) | 3 | 84 | 84 |
| 3 4 | 2a | Me | 4 | KOH (3) | 3 | 71 | 59 |
| 4 | 2a | Me | 4 | KOH (1.5) | 3 | 64 | 78 |
| 5 | 2a | Me | 5 | KOH (3) | 3 | 84 | 83 |
| 6 | 2a | Me | 6 | KOH (3) | 3 | 75 | 81 |
| 7 | 2a | Me | 7 | KOH (3) | 3 | 82 | 72 |
| 8 | 2b | PhCH2 | 4 | KOH (3) | 3 | 87 | 65 |
| 9 | 2b | PhCH2 | 4 | KOH (3) | 1 | 78 | 68 |
| 10 | 2b | PhCH2 | 4 | KOH (1.5) | 3 | 47 5 | 78 |
| 11 | 2b | PhCH2 | 4 | KOH (2) | 1 | 70 | 73 |
| 12 | 2b | PhCH2 | 4 | KOH (1.5) | 1 | 63 | 70 |
| 13 | 2b | PhCH2 | 5 | KOH (3) | 3 | 84 | 73 |
| 14 | 2b | PhCH2 | 5 | KOH (3) | 2 | 79 | 70 |
| 15 | 2b | PhCH2 | 5 | KOH (3) | 1 | 71 | 74 |
| Entry | Product 3 | Catalyst | Yield 2 [%] | ee 3 [%] | |
|---|---|---|---|---|---|
| 1 |  | 3a | 4 | 84 | 84 |
| 5 | 84 | 83 | |||
| 2 |  | 3b | 4 | 84 | 73 |
| 3 |  | 3c | 4 | 93 | 72 |
| 5 | 73 | 61 | |||
| 4 |  | 3d | 4 | 82 | 56 |
| 5 |  | 3e | 4 | 88 | 23 4 |
| 6 |  | 3f | 4 | 82 | 80 |
| 5 | 60 | 79 | |||
| 7 |  | 3g | 4 | 85 | 80 |
| 8 |  | 3h | 4 | 80 | 92 |
| 9 |  | 3i | 4 | 79 | 65 |
| 5 | 69 | 55 | |||
| 10 |  | 3j | 4 | 98 | 53 |
| 5 | 88 | 54 | |||
| 11 |  | 3k | 4 | 69 | 42 |
| 5 | 84 | 53 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walęcka-Kurczyk, A.; Walczak, K.; Kuźnik, A.; Stecko, S.; Październiok-Holewa, A. The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite. Molecules 2020, 25, 405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020405
Walęcka-Kurczyk A, Walczak K, Kuźnik A, Stecko S, Październiok-Holewa A. The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite. Molecules. 2020; 25(2):405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020405
Chicago/Turabian StyleWalęcka-Kurczyk, Alicja, Krzysztof Walczak, Anna Kuźnik, Sebastian Stecko, and Agnieszka Październiok-Holewa. 2020. "The Synthesis of α-Aminophosphonates via Enantioselective Organocatalytic Reaction of 1-(N-Acylamino)alkylphosphonium Salts with Dimethyl Phosphite" Molecules 25, no. 2: 405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25020405