
The Effect of Enantiomer Elution Order on the Determination of Minor Enantiomeric Impurity in Ketoprofen and Enantiomeric Purity Evaluation of Commercially Available Dexketoprofen Formulations

 ,
,  ,
, 
 and
and
Abstract
:1. Introduction
2. Results
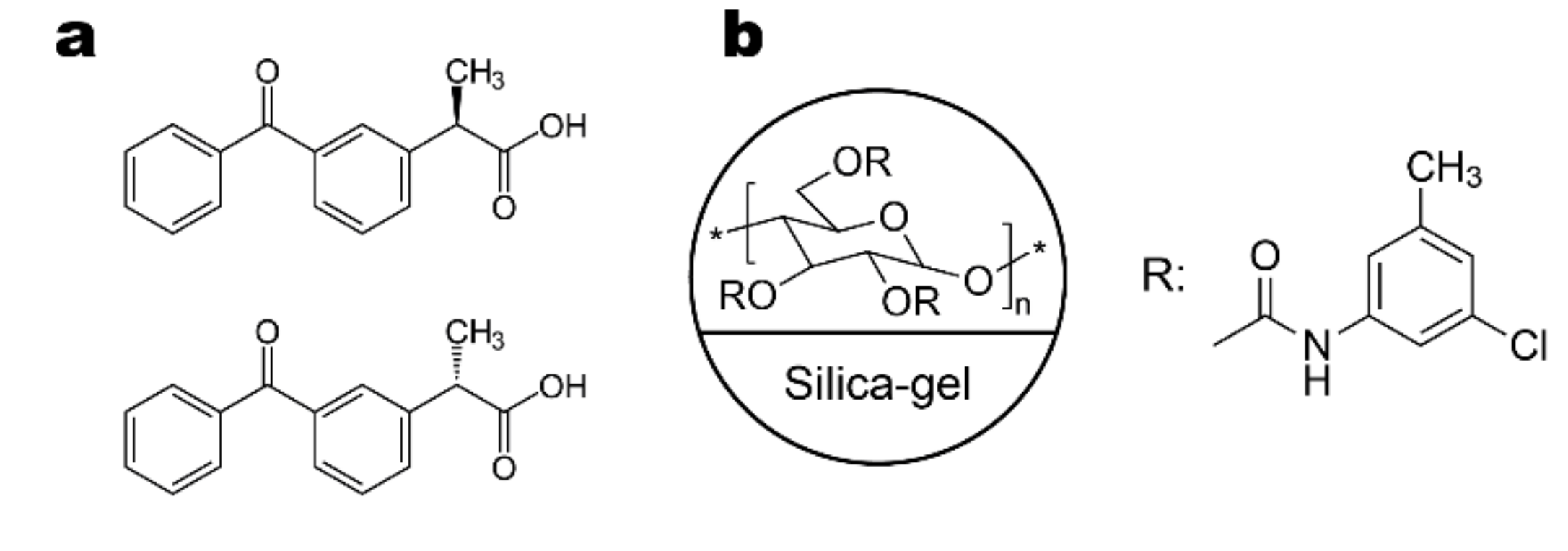
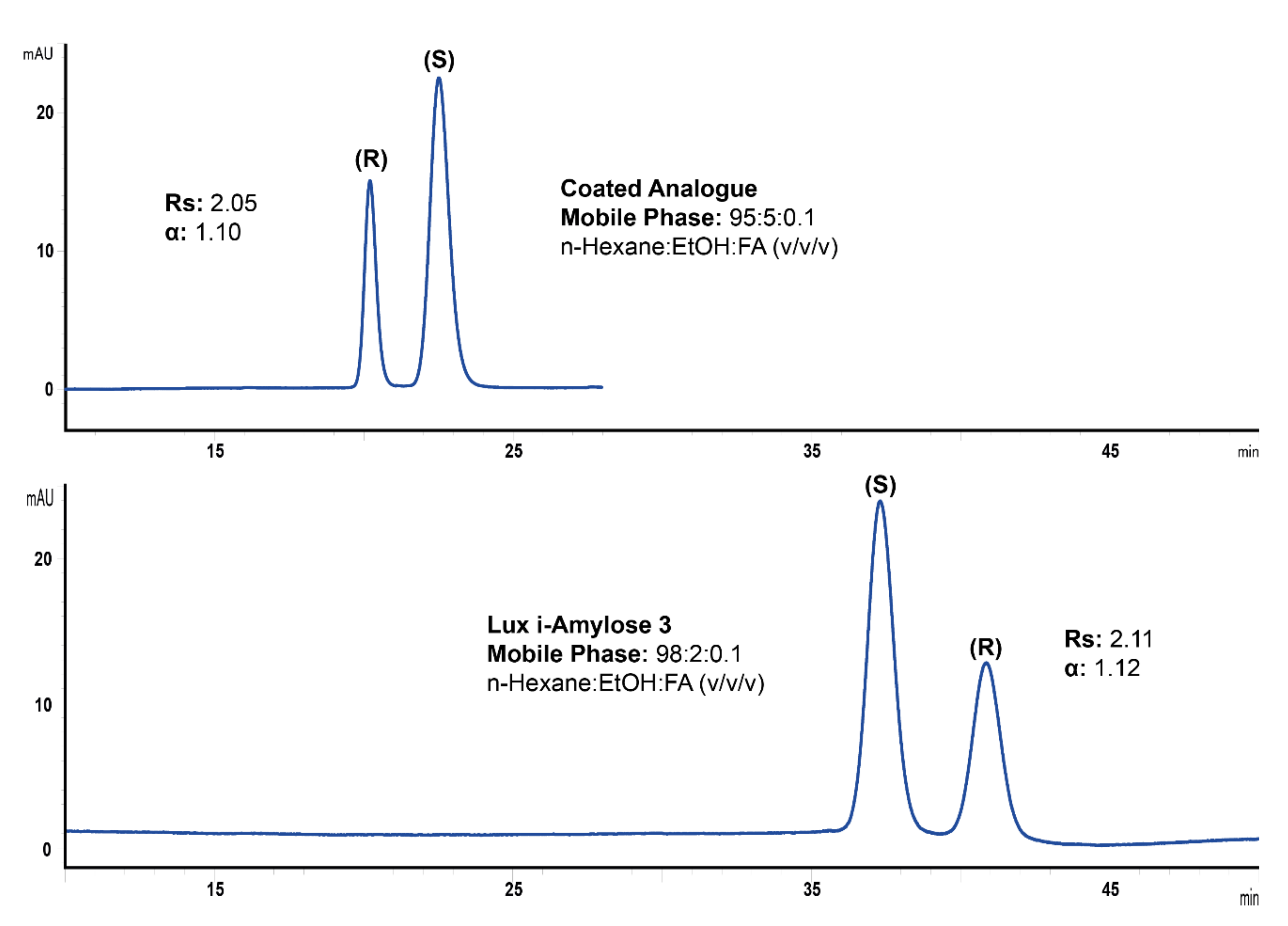
2.1. Method Development for the Separation of Ketoprofen Enantiomers on Lux i-Amylose-3 Column and Its Analogue with a Coated Chiral Selector
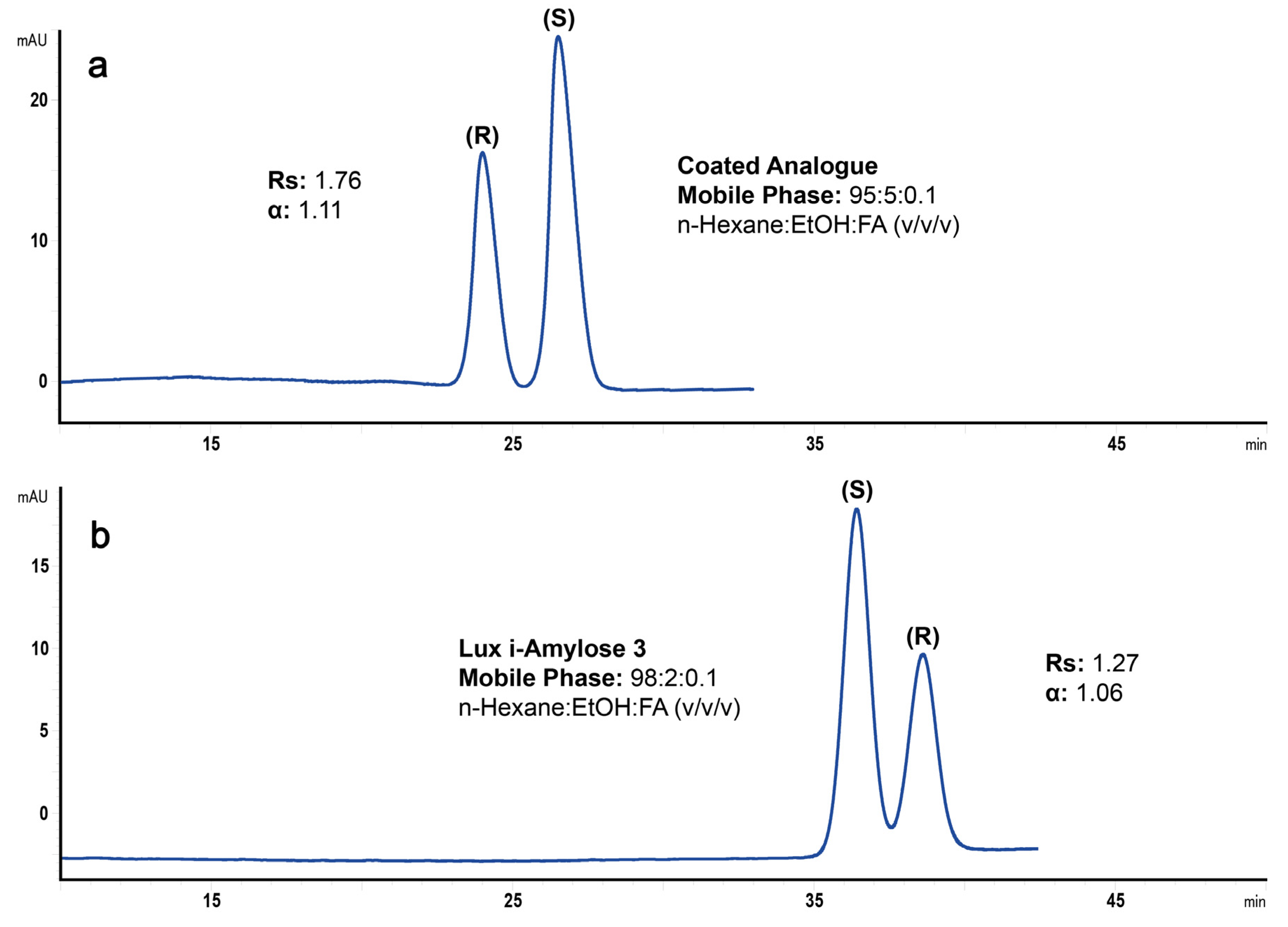
2.2. Method Validation Results and Application to Racemic Ketoprofen Formulation
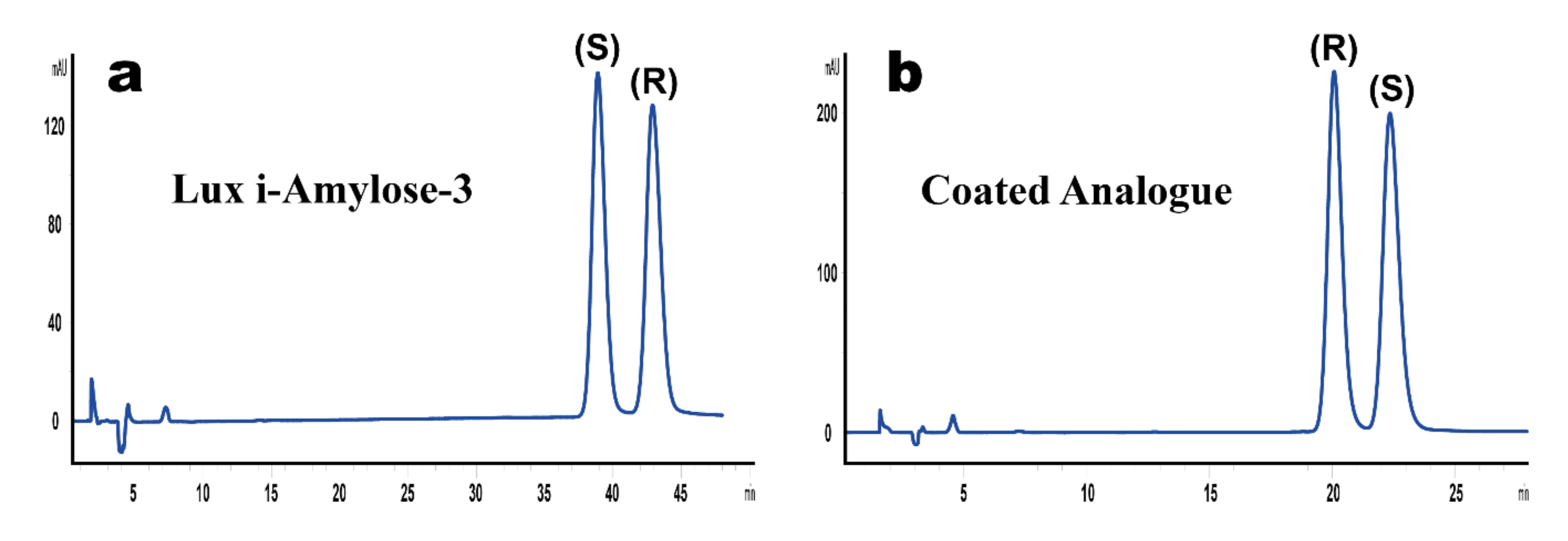
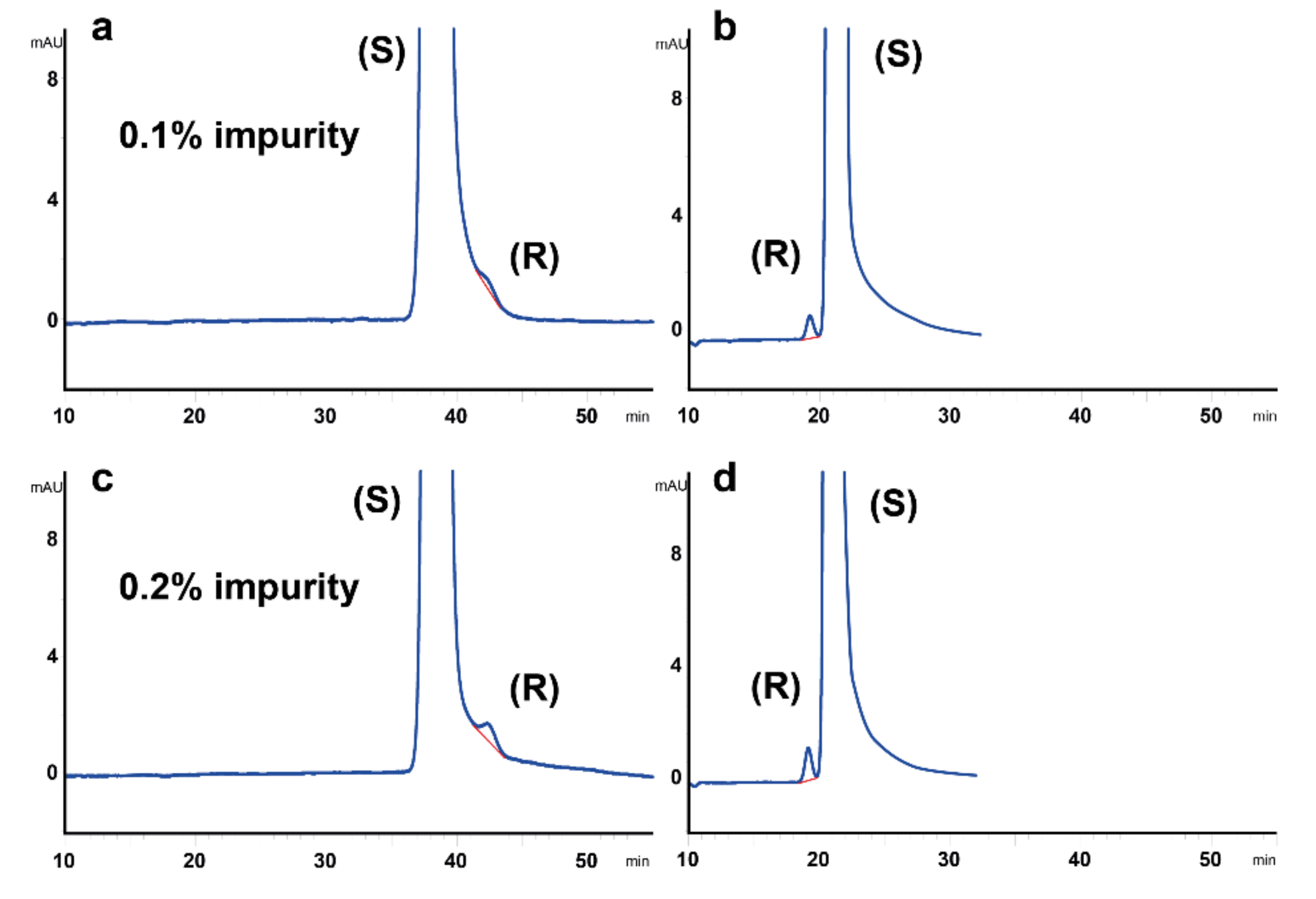
2.3. Determination of Minor Enantiomeric Impurity and Method Applicability to Dexketoprofen Formulations
3. Materials and Methods
3.1. Materials
3.2. Instrument
3.3. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maisuradze, M.; Sheklashvili, G.; Chokheli, A.; Matarashvili, I.; Gogatishvili, T.; Farkas, T.; Chankvetadze, B. Chromatographic and thermodynamic comparison of amylose tris(3-chloro-5-methylphenylcarbamate) coated or covalently immobilized on silica in high-performance liquid chromatographic separation of the enantiomers of select chiral weak acids. J. Chromatogr. A 2019, 1602, 228–236. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: It’s the same thing...only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef]
- Silva, B.; Fernandes, C.; Guedes de Pinho, P.; Remiao, F. Chiral resolution and enantioselectivity of synthetic cathinones: A Brief Review. J. Anal. Toxicol. 2018, 42, 17–24. [Google Scholar] [CrossRef]
- Rossi, D.; Tarantino, M.; Rossino, G.; Rui, M.; Juza, M.; Collina, S. Approaches for multi-gram scale isolation of enantiomers for drug discovery. Expert Opin. Drug Discov. 2017, 12, 1253–1269. [Google Scholar] [CrossRef]
- Leek, H.; Thunberg, L.; Jonson, A.C.; Ohlen, K.; Klarqvist, M. Strategy for large-scale isolation of enantiomers in drug discovery. Drug. Discov. Today 2017, 22, 133–139. [Google Scholar] [CrossRef]
- Farina, V.; Reeves, J.T.; Senanayake, C.H.; Song, J.J. Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev. 2006, 106, 2734–2793. [Google Scholar] [CrossRef]
- Etayo, P.; Vidal-Ferran, A. Rhodium-catalysed asymmetric hydrogenation as a valuable synthetic tool for the preparation of chiral drugs. Chem. Soc. Rev. 2013, 42, 728–754. [Google Scholar] [CrossRef]
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Analytical and preparative scale separation of enantiomers of chiral drugs by chromatography and related methods. Curr. Med. Chem. 2018, 25, 4152–4188. [Google Scholar] [CrossRef]
- Shen, J.; Okamoto, Y. Efficient separation of enantiomers using stereoregular chiral polymers. Chem. Rev. 2016, 116, 1094–1138. [Google Scholar] [CrossRef]
- Chankvetadze, B. Recent trends in preparation, investigation and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. Trac. Trends Anal. Chem. 2020, 122, 115709. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Conathy, J.M.; Owens, M.J. Stereochemistry in Drug Action. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 70–73. [Google Scholar] [CrossRef] [Green Version]
- Tucker, G.T. Chiral switches. Lancet 2000, 355, 1085–1087. [Google Scholar] [CrossRef]
- Perry, J.A.; Rateike, J.D.; Szczerba, T.J. Eluting trace components before major constituents: I. Sensitivity enhancement in analytical determinations of optical purity. J. Chromatogr. A 1987, 389, 57–64. [Google Scholar] [CrossRef]
- Wozniak, T.J.; Bopp, R.J.; Jensen, E.C. Chiral drugs: An industrial analytical perspective. J. Pharm. Biomed. Anal. 1991, 9, 363–382. [Google Scholar] [CrossRef]
- Cirilli, R.; Ferretti, R.; Gallinella, B.; Zanitti, L.; La Torre, F. A new application of stopped-flow chiral HPLC: Inversion of enantiomer elution order. J. Chromatogr. A 2004, 1061, 27–34. [Google Scholar] [CrossRef]
- Dossou, K.S.; Edorh, P.A.; Chiap, P.; Chankvetadze, B.; Servais, A.C.; Fillet, M.; Crommen, J. Determination of enantiomeric purity of S-amlodipine by chiral LC with emphasis on reversal of enantiomer elution order. J. Sep. Sci. 2011, 34, 1772–1780. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Finn, J.M.; Schreiner, J.L.; Hamper, B.C. A widely useful chiral stationary phase for the high-performance liquid chromatography separation of enantiomers. J. Am. Chem. Soc. 1981, 103, 3964–3966. [Google Scholar] [CrossRef]
- Ilisz, I.; Bajtai, A.; Lindner, W.; Peter, A. Liquid chromatographic enantiomer separations applying chiral ion-exchangers based on Cinchona alkaloids. J. Pharm. Biomed. Anal. 2018, 159, 127–152. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.W.; DeMond, W. Cyclodextrin bonded phases for the liquid chromatographic separation of optical, geometrical, and structural isomers. J. Chromatogr. Sci. 1984, 22, 411–415. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Tang, Y.; Chen, S.; Zhou, Y.; Bagwill, C.; Chen, J.-R. Macrocyclic antibiotics as a new class of chiral selectors for liquid chromatography. Anal. Chem. 1994, 66, 1473–1484. [Google Scholar] [CrossRef]
- Haginaka, J. Protein-based chiral stationary phases for high-performance liquid chromatography enantioseparations. J. Chromatogr. A 2001, 906, 253–273. [Google Scholar] [CrossRef]
- Matarashvili, I.; Chankvetadze, L.; Fanali, S.; Farkas, T.; Chankvetadze, B. HPLC separation of enantiomers of chiral arylpropionic acid derivatives using polysaccharide-based chiral columns and normal-phase eluents with emphasis on elution order. J. Sep. Sci. 2013, 36, 140–147. [Google Scholar] [CrossRef]
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Separation and elution order of the enantiomers of some beta-agonists using polysaccharide-based chiral columns and normal phase eluents by high-performance liquid chromatography. J. Chromatogr. A 2016, 1467, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Jibuti, G.; Mskhiladze, A.; Takaishvili, N.; Karchkhadze, M.; Chankvetadze, L.; Farkas, T.; Chankvetadze, B. HPLC separation of dihydropyridine derivatives enantiomers with emphasis on elution order using polysaccharide-based chiral columns. J. Sep. Sci. 2012, 35, 2529–2537. [Google Scholar] [CrossRef]
- Balmér, K.; Persson, B.-A.; Lagerström, P.-O. Stereoselective effects in the separation of enantiomers of omeprazole and other substituted benzimidazoles on different chiral stationary phases. J. Chromatogr. A 1994, 660, 269–273. [Google Scholar] [CrossRef]
- Balmér, K.; Lagerström, P.-O.; Persson, B.-A.; Schill, G. Reversed retention order and other stereoselective effects in the separation of amino alcohols on Chiralcel OD. J. Chromatogr. 1992, 592, 331–337. [Google Scholar] [CrossRef]
- Matarashvili, I.; Ghughunishvili, D.; Chankvetadze, L.; Takaishvili, N.; Khatiashvili, T.; Tsintsadze, M.; Farkas, T.; Chankvetadze, B. Separation of enantiomers of chiral weak acids with polysaccharide-based chiral columns and aqueous-organic mobile phases in high-performance liquid chromatography: Typical reversed-phase behavior? J. Chromatogr. A 2017, 1483, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Gyllenhaal, O.; Stefansson, M. Reversal of elution order for profen acid enantiomers in normal phase LC on Chiralpak AD. J. Pharm. Biomed. Anal. 2008, 46, 860–863. [Google Scholar] [CrossRef]
- Xiang, C.; Liu, G.; Kang, S.; Guo, X.; Yao, B.; Weng, W.; Zeng, Q. Unusual chromatographic enantioseparation behavior of naproxen on an immobilized polysaccharide-based chiral stationary phase. J. Chromatogr. A 2011, 1218, 8718–8721. [Google Scholar] [CrossRef]
- Gaffney, M.H.; Stiffin, R.M.; Wainer, I.W. The effect of alcoholic mobile phase modifiers on retention and stereoselectivity on a commercially available cellulose-based HPLC chiral stationary phase: An unexpected reversal in enantiometric elution order. Chromatographia 1989, 27, 15–18. [Google Scholar] [CrossRef]
- Okamoto, M.; Nakazawa, H. Reversal of elution order during direct enantiomeric separation of pyriproxyfen on a cellulose-based chiral stationary phase. J. Chromatogr. A 1991, 588, 177–180. [Google Scholar] [CrossRef]
- Chankvetadze, L.; Ghibradze, N.; Karchkhadze, M.; Peng, L.; Farkas, T.; Chankvetadze, B. Enantiomer elution order reversal of fluorenylmethoxycarbonyl-isoleucine in high-performance liquid chromatography by changing the mobile phase temperature and composition. J. Chromatogr. A 2011, 1218, 6554–6560. [Google Scholar] [CrossRef]
- Mosiashvili, L.; Chankvetadze, L.; Farkas, T.; Chankvetadze, B. On the effect of basic and acidic additives on the separation of the enantiomers of some basic drugs with polysaccharide-based chiral selectors and polar organic mobile phases. J. Chromatogr. A 2013, 1317, 167–174. [Google Scholar] [CrossRef]
- Dossou, K.S.S.; Chiap, P.; Chankvetadze, B.; Servais, A.-C.; Fillet, M.; Crommen, J. Enantioresolution of basic pharmaceuticals using cellulose tris(4-chloro-3-methylphenylcarbamate) as chiral stationary phase and polar organic mobile phases. J. Chromatogr. A 2009, 1216, 7450–7455. [Google Scholar] [CrossRef]
- Matarashvili, I.; Kobidze, G.; Chelidze, A.; Dolidze, G.; Beridze, N.; Jibuti, G.; Farkas, T.; Chankvetadze, B. The effect of temperature on the separation of enantiomers with coated and covalently immobilized polysaccharide-based chiral stationary phases. J. Chromatogr. A 2019, 1599, 172–179. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Chankvetadze, L.; Sidamonidze, S.; Kasashima, E.; Yashima, E.; Okamoto, Y. 3-Fluoro-, 3-chloro- and 3-bromo-5-methylphenylcarbamates of cellulose and amylose as chiral stationary phases for high-performance liquid chromatographic enantioseparation. J. Chromatogr. A 1997, 787, 67–77. [Google Scholar] [CrossRef]
- CPMP/ICH/381/95–ICH Harmonised Tripartite Guideline–Validation of Analytical Procedures: Text and Methodology Q2(R1). 2014. Available online: https://www.ema.europa.eu/en/ich-q2-r1-validation-analytical-procedures-text-methodology (accessed on 4 October 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Lux i-Amylose-3 | Coated Analogue | ||
|---|---|---|---|---|
| S-Enantiomer | R-Enantiomer | R-Enantiomer | S-Enantiomer | |
| Retention time (min) | 37.31 | 40.86 | 20.23 | 22.54 |
| Capacity factor (k) | 8.33 | 9.22 | 4.07 | 4.64 |
| Theoretical plates numbers (N) | 8101 | 8337 | 5666 | 5499 |
| Separation factor (α) | - | 1.12 | - | 1.10 |
| Resolution (Rs) | - | 2.11 | - | 2.05 |
| Symmetry | 0.94 | 0.92 | 0.85 | 0.85 |
| Tailing factor (Tf) | 1.06 | 1.00 | 1.16 | 1.17 |
| Parameters | Lux i-Amylose-3 | Coated Analogue | ||
|---|---|---|---|---|
| S-Enantiomer | R-Enantiomer | R-Enantiomer | S-Enantiomer | |
| Linearity range (mg/mL) | 0.1–100 | 0.01–2.5% | 0.01–2.5% | 0.1–100 |
| Slope | 19.72 × 103 | 0.0011 | 0.0012 | 19.64 × 103 |
| Determination coefficient (R2) | 0.9999 | 0.9997 | 0.9999 | 0.9999 |
| Intercept | 6.3 × 103 | 8 × 10−5 | 2 × 10−4 | 4.7 × 103 |
| Limit of detection (µg/mL) | 0.033 | 0.033 | 0.01 | 0.01 |
| Limit of quantification (µg/mL) | 0.10 | 0.10 | 0.03 | 0.03 |
| Within-day precision (RSD, %; n = 5) | 2.80 | 3.29 | 1.69 | 0.70 |
| Between-day precision (RSD, %; n = 5) | 3.13 | 3.65 | 2.59 | 1.12 |
| Lux i-Amylose-3 | Coated Analogue | |||
|---|---|---|---|---|
| S-Enantiomer | R-Enantiomer | R-Enantiomer | S-Enantiomer | |
| Labeled Amount (mg/mL) | 50 | 50 | ||
| Found Amount (mg/mL) | 23.89 | 23.78 | 24.18 | 23.92 |
| Total Amount (mg/mL) | 47.67 | 48.10 | ||
| RSD (%) * | 0.46 | 0.59 | 0.12 | 0.13 |
| Bias (%) * | 4.46 | 4.91 | 3.28 | 4.32 |
| Total Bias (%) * | 4.66 | 3.80 | ||
| Formulation | Lux i-Amylose-3 | Coated Analogue | ||||||
|---|---|---|---|---|---|---|---|---|
| Added Amount (%) | Found Amount (%) | RSD (%) * | Recovery (%) * | Added Amount (%) | Found Amount (%) | RSD (%) * | Recovery (%) * | |
| Formulation 1 | 0.5 | 0.493 | 4.20 | 98.51 | 0.5 | 0.509 | 3.70 | 101.80 |
| Formulation 2 | 0.5 | 0.510 | 2.35 | 101.99 | 0.5 | 0.501 | 0.77 | 100.20 |
| Formulation 6 | 0.5 | 0.477 | 3.30 | 95.36 | 0.5 | 0.503 | 2.90 | 100.68 |
| Dexketoprofen Formulation | F1 | F2 | F3 | F4 | F5 | F6 | |
|---|---|---|---|---|---|---|---|
| Lux i-Amylose-3 | Content of S-Enantiomer, mg/mL | 23.44 | 23.74 | 24.64 | 24.43 | 24.73 | 23.42 |
| Content of R-Enantiomer, mg/mL | 0.09 | 0.49 | 0.14 | 0.02 | 0.06 | 0.04 | |
| Enantiomeric impurity of dexketoprofen, % (w/w) | 0.34 | 1.95 | 0.57 | 0.07 | 0.24 | 0.16 | |
| RSD (%) * | 3.09 | 4.42 | 3.69 | 2.66 | 4.19 | 3.86 | |
| Total content of ketoprofen, mg/mL | 23.53 | 24.23 | 24.78 | 24.45 | 24.79 | 23.46 | |
| Declared content of dexketoprofen, mg/mL | 25 | 25 | 25 | 25 | 25 | 25 | |
| Bias (%) | 5.88 | 3.08 | 0.88 | 2.20 | 0.84 | 6.16 | |
| Coated Analogue | Content of S-Enantiomer, mg/mL | 23.66 | 24.38 | 24.95 | 25.01 | 24.91 | 24.36 |
| Content of R-Enantiomer, mg/mL | 0.09 | 0.42 | 0.13 | 0.03 | 0.07 | 0.04 | |
| Enantiomeric impurity of dexketoprofen, % (w/w) | 0.34 | 1.66 | 0.50 | 0.10 | 0.26 | 0.17 | |
| RSD (%) * | 0.39 | 0.22 | 0.58 | 1.90 | 2.47 | 3.18 | |
| Total content of ketoprofen, mg/mL | 23.75 | 24.80 | 25.08 | 25.04 | 24.98 | 24.40 | |
| Stated content of dexketoprofen, mg/mL | 25 | 25 | 25 | 25 | 25 | 25 | |
| Bias (%) * | 5.00 | 0.80 | −0.32 | −0.16 | 0.08 | 2.40 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tok, K.C.; Gumustas, M.; Jibuti, G.; Suzen, H.S.; Ozkan, S.A.; Chankvetadze, B. The Effect of Enantiomer Elution Order on the Determination of Minor Enantiomeric Impurity in Ketoprofen and Enantiomeric Purity Evaluation of Commercially Available Dexketoprofen Formulations. Molecules 2020, 25, 5865. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245865
Tok KC, Gumustas M, Jibuti G, Suzen HS, Ozkan SA, Chankvetadze B. The Effect of Enantiomer Elution Order on the Determination of Minor Enantiomeric Impurity in Ketoprofen and Enantiomeric Purity Evaluation of Commercially Available Dexketoprofen Formulations. Molecules. 2020; 25(24):5865. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245865
Chicago/Turabian StyleTok, Kenan Can, Mehmet Gumustas, Giorgi Jibuti, Halit Sinan Suzen, Sibel A. Ozkan, and Bezhan Chankvetadze. 2020. "The Effect of Enantiomer Elution Order on the Determination of Minor Enantiomeric Impurity in Ketoprofen and Enantiomeric Purity Evaluation of Commercially Available Dexketoprofen Formulations" Molecules 25, no. 24: 5865. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245865