Possible Transmission Flow of SARS-CoV-2 Based on ACE2 Features

 ,
,  , , , ,
, , , ,  , , ,
, , ,  , ,
, ,  , , ,
, , ,  , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
2. Results
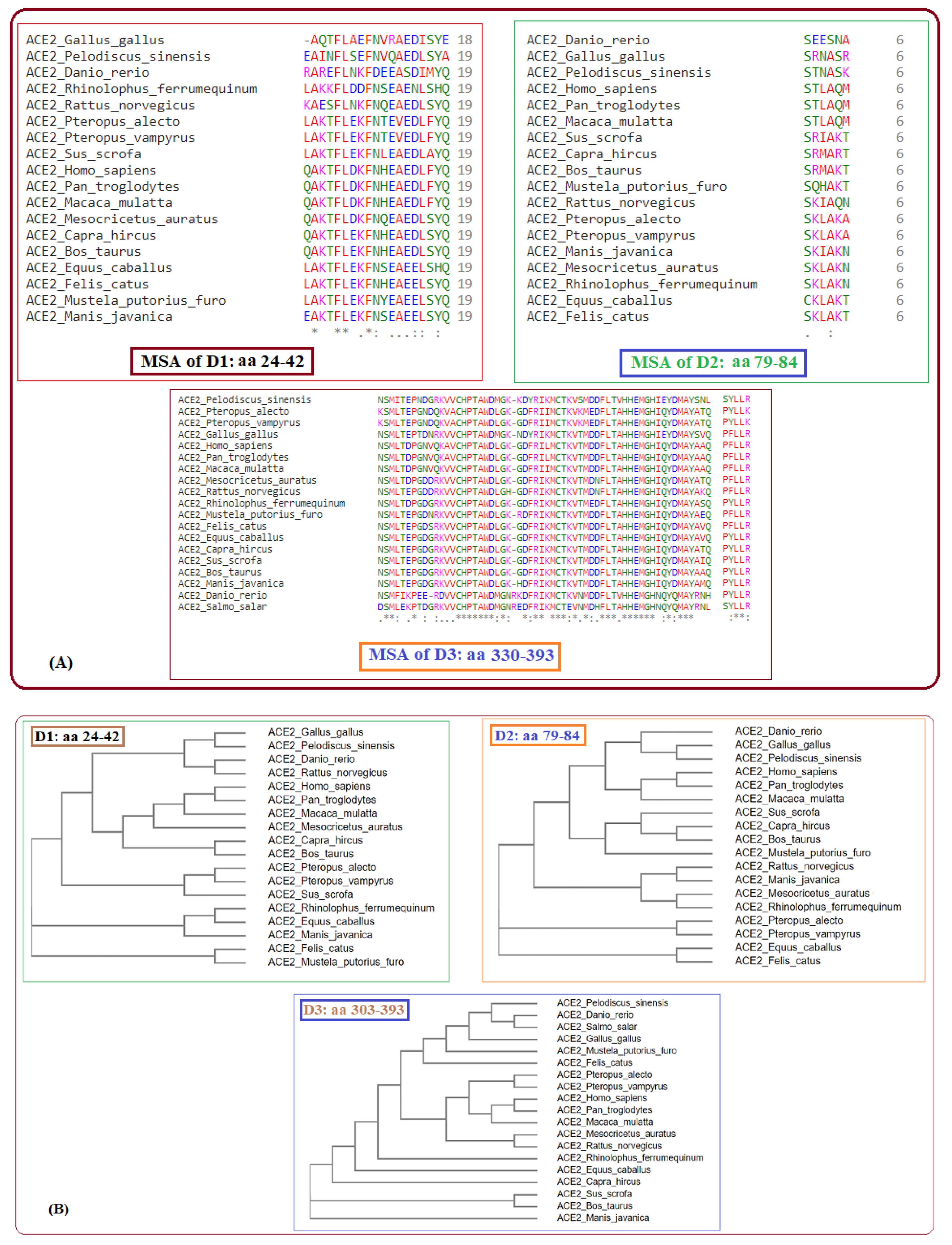
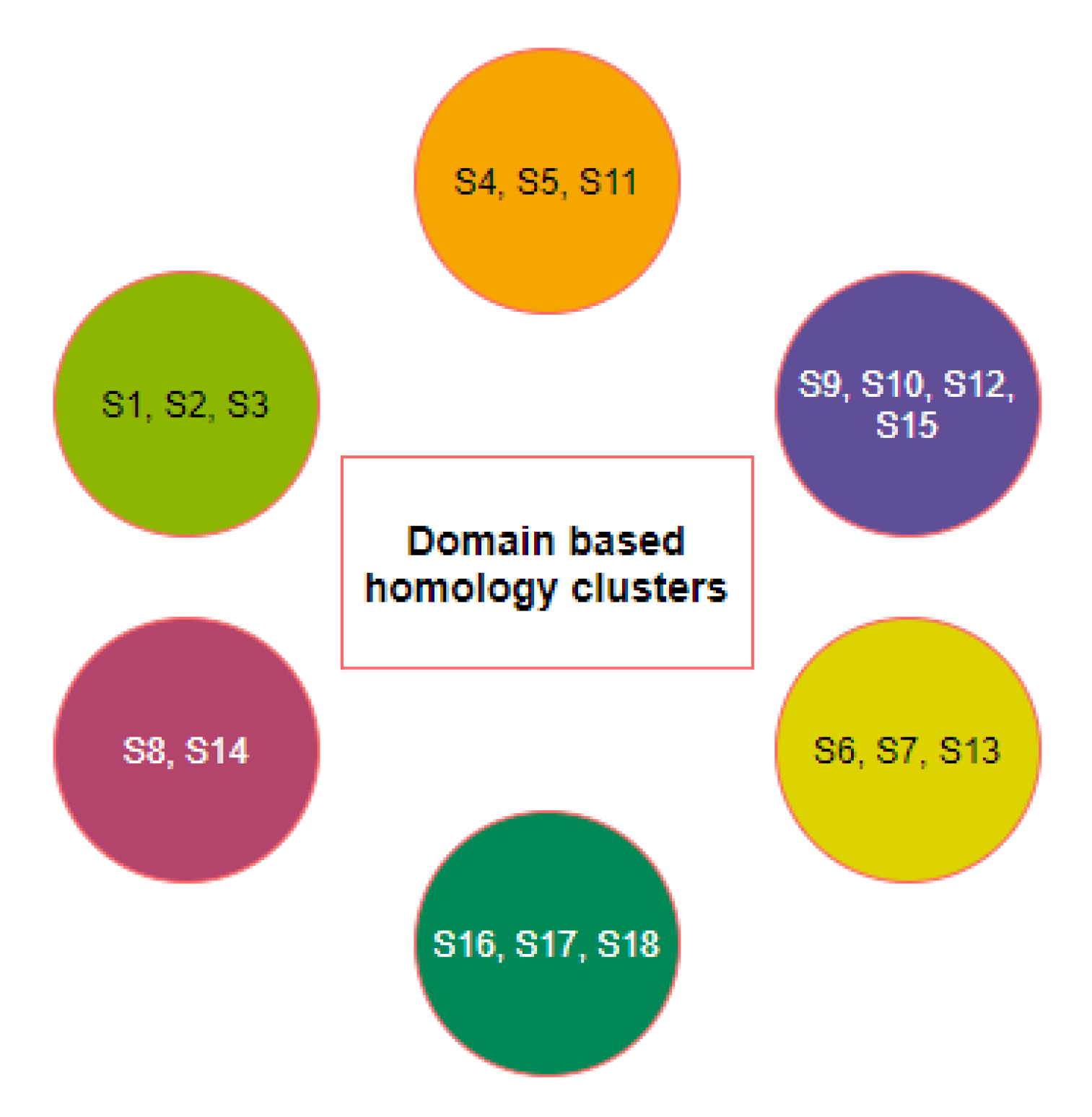
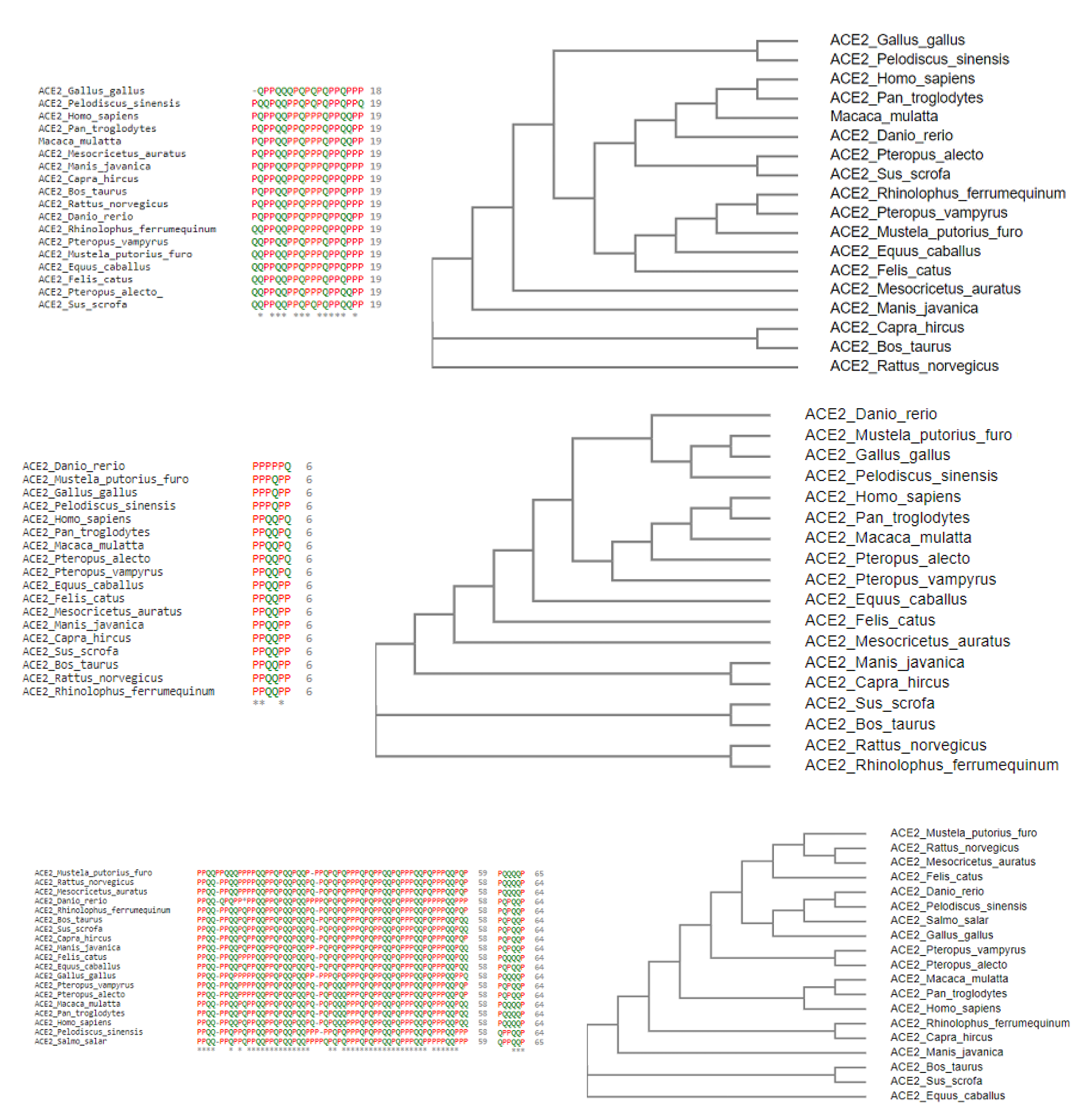
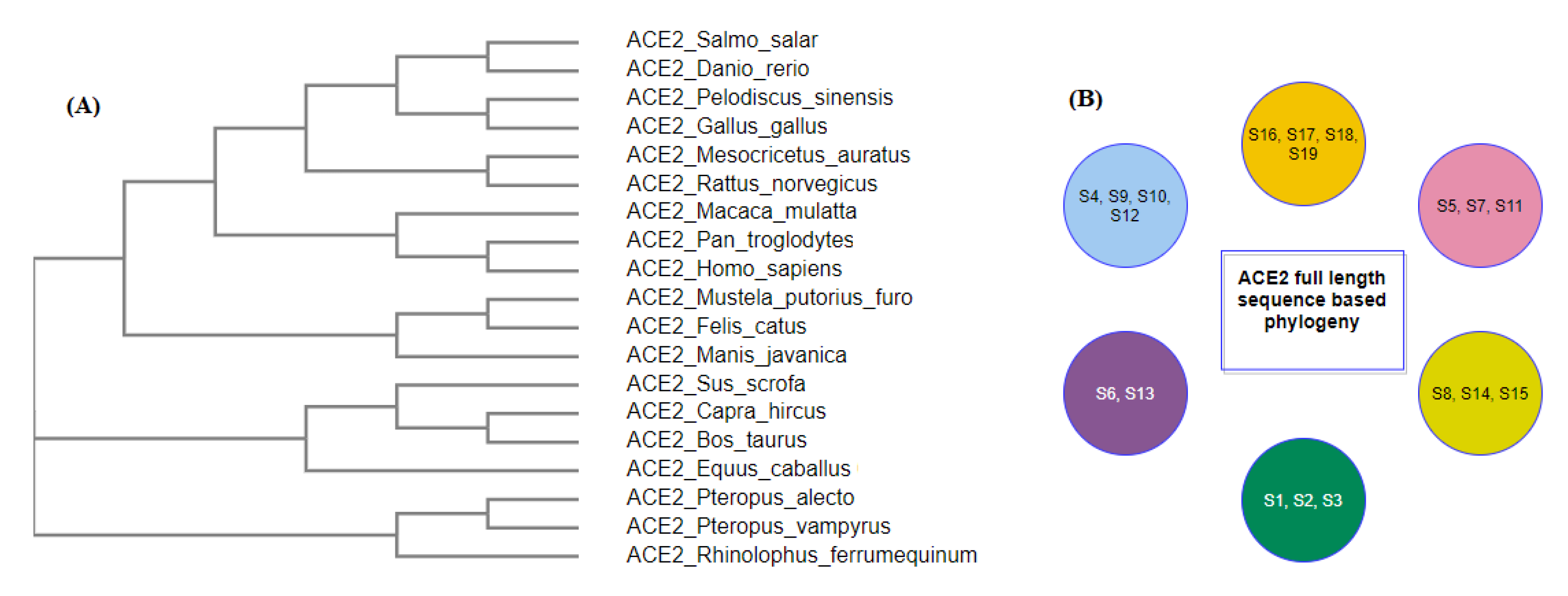
2.1. Phylogeny and Clustering Based on ACE2 Domain-Based Homology
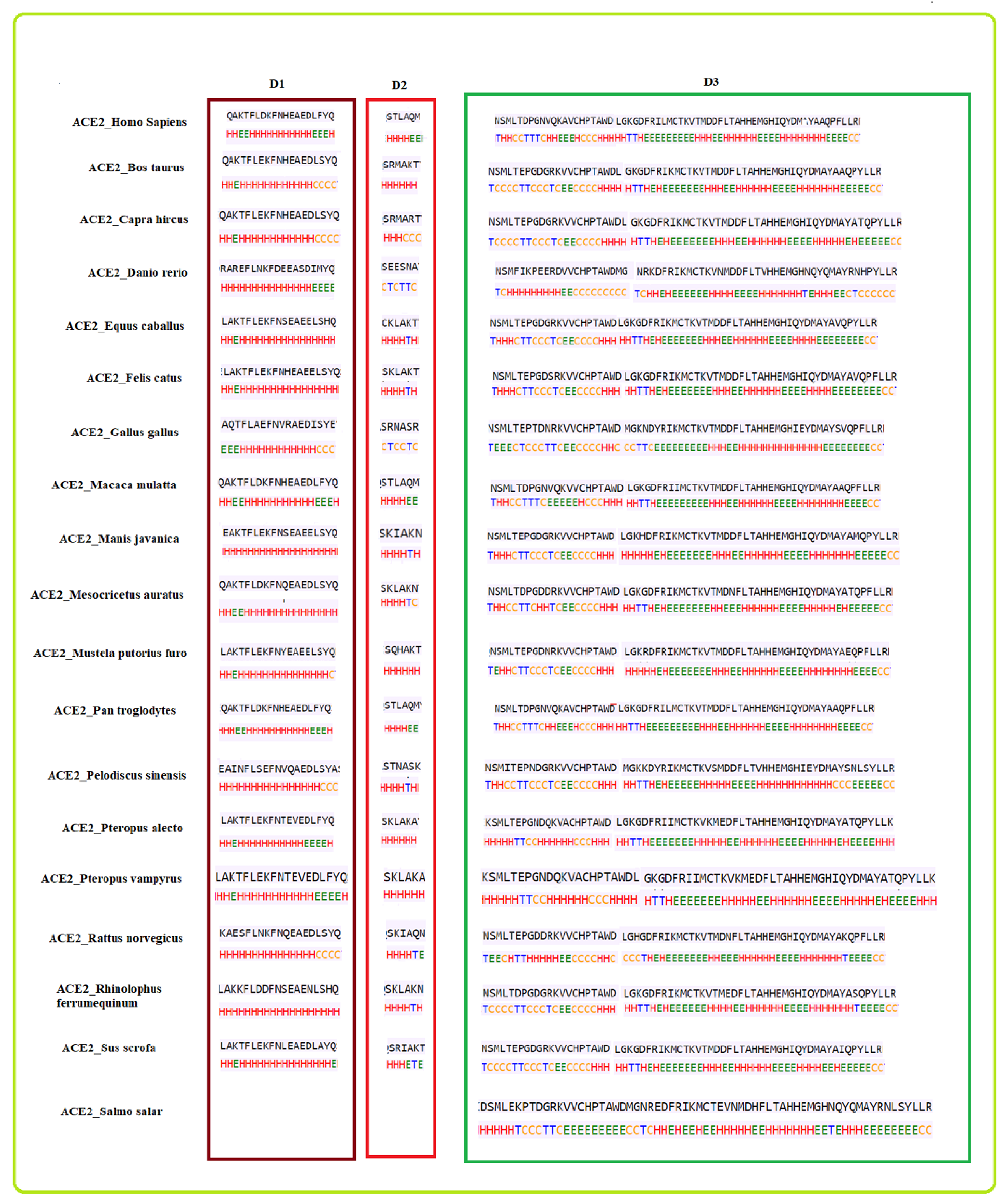
2.2. Clustering Based on Secondary Structures
- Bos taurus and Capra hircus
- Equus caballus and Felis catus
- Mustela putorius furo has a structure closer to Equus caballus and Felis catus as it has only one difference of a coil present at position 42. In addition, Mesocricetus auratus has a secondary structure similar to the above two, except an extended helix at position four. Similarly, Sus scrofa has an extended sheet instead of a helix at position 42. Thus, Mustela putorius furo, Mesocricetus auratus, and Sus scrofa can be put in the same cluster as Equus caballus and Felis catus.
- Homo sapiens, Macaca mulatta, and Pan troglodytes
- Manis javanica and Rhinolophus ferrumequinum
- Pteropus alecto and Pteropus Vampyrus
- Rattus norvegicus and Pelodiscus sinensis have similar structures differing by the presence of an extra coil at position 39 for Rattus norvegicus.
- Gallus gallus and Danio rerio have a unique secondary structure in comparison to the others.
- Homo sapiens, Macaca mulatta, and Pan troglodytes
- Bos taurus, Mustela putorius furo, Pteropus alecto, and Pteropus vampyrus
- Equus caballus, Felis catus, Manis javanica, Pelodiscus sinensis, and Rhinolophus ferrumequinum
- Danio rerio and Gallus gallus
- Homo sapiens and Pan troglodytes
- Bos Taurus, Rhinolophus ferrumequinum, Sus scrofa, and Capra hircus
- Equus caballus and Felis catus
- Pteropus alecto and Pteropus vampyrus
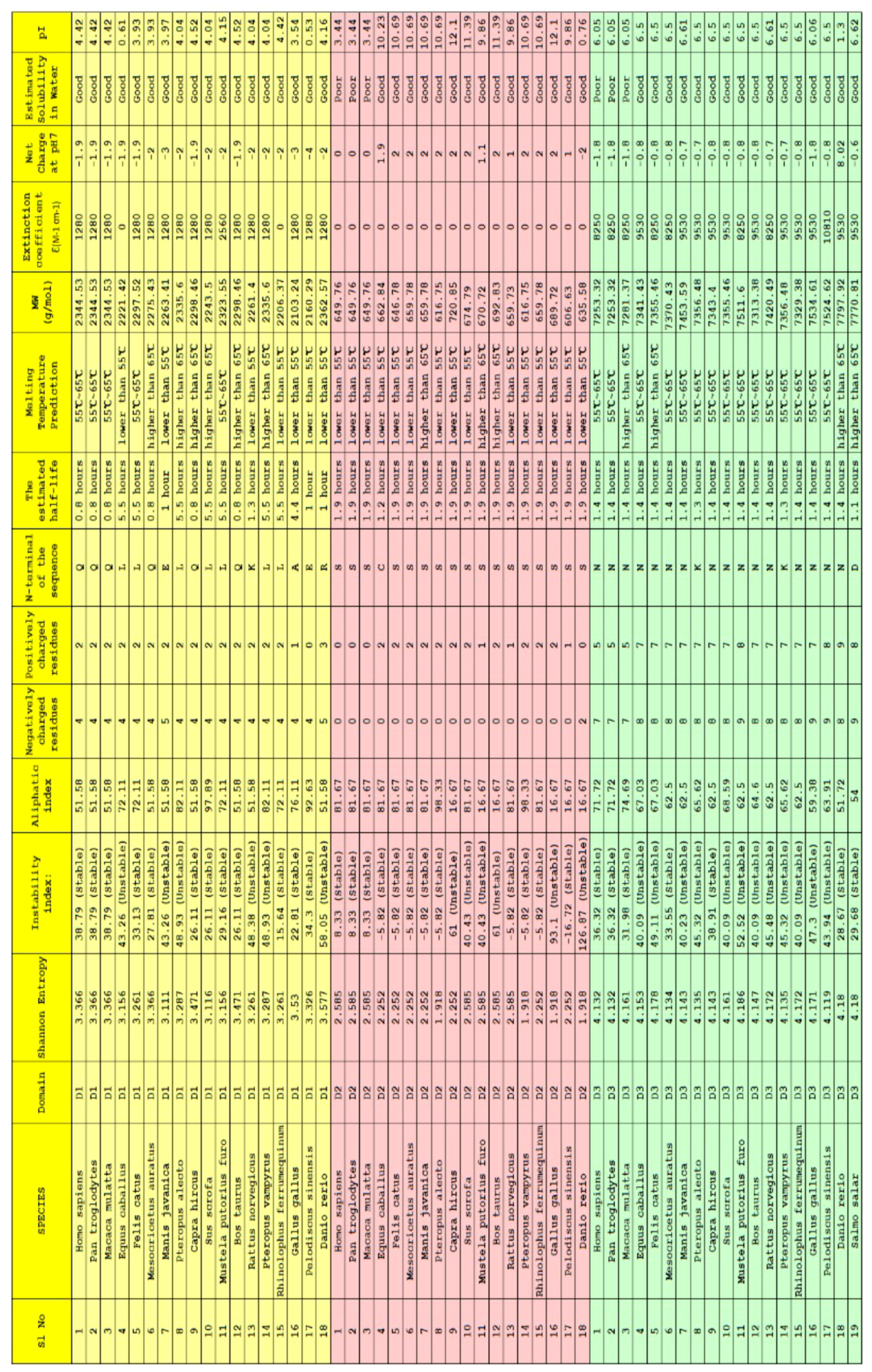
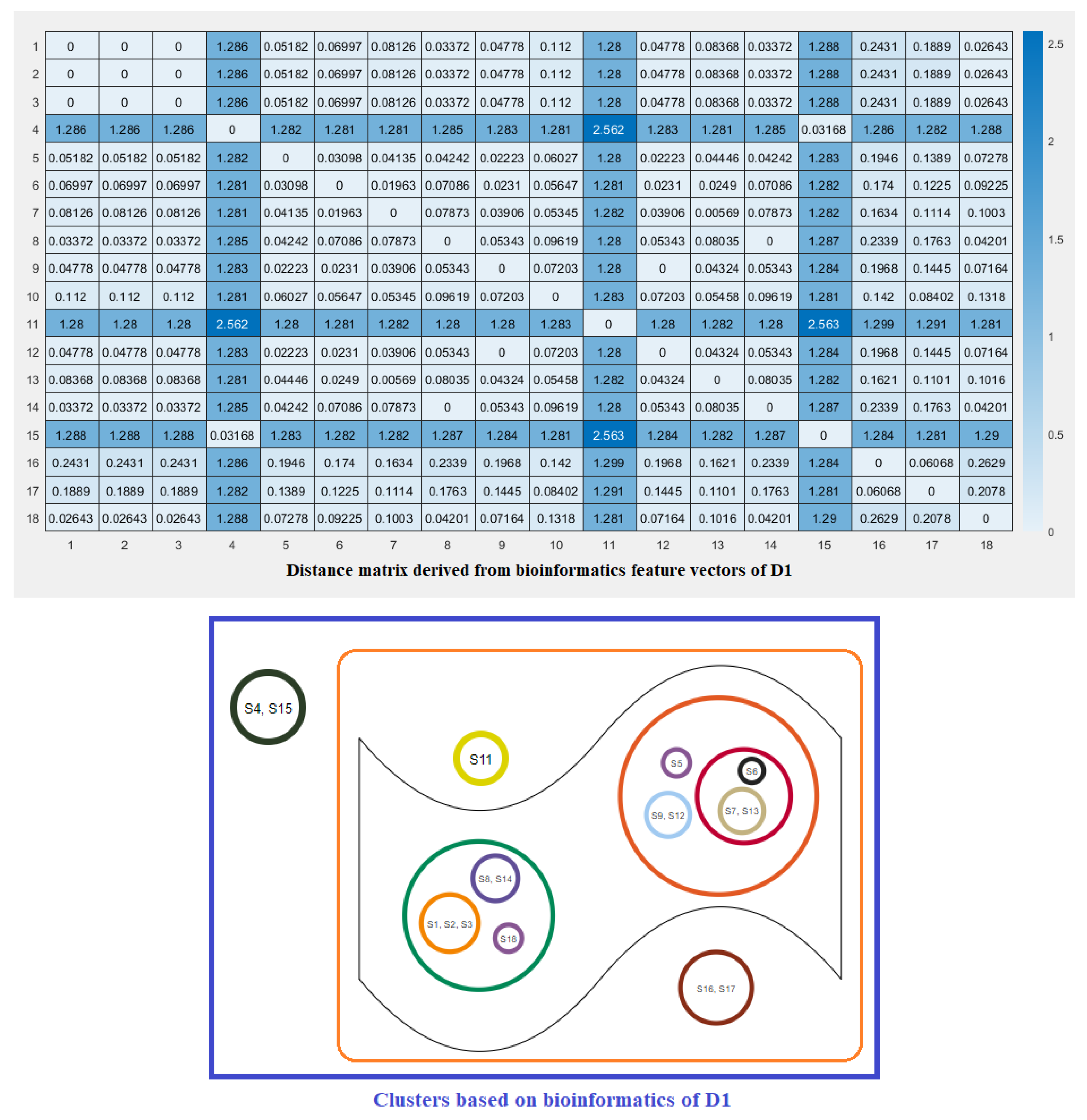
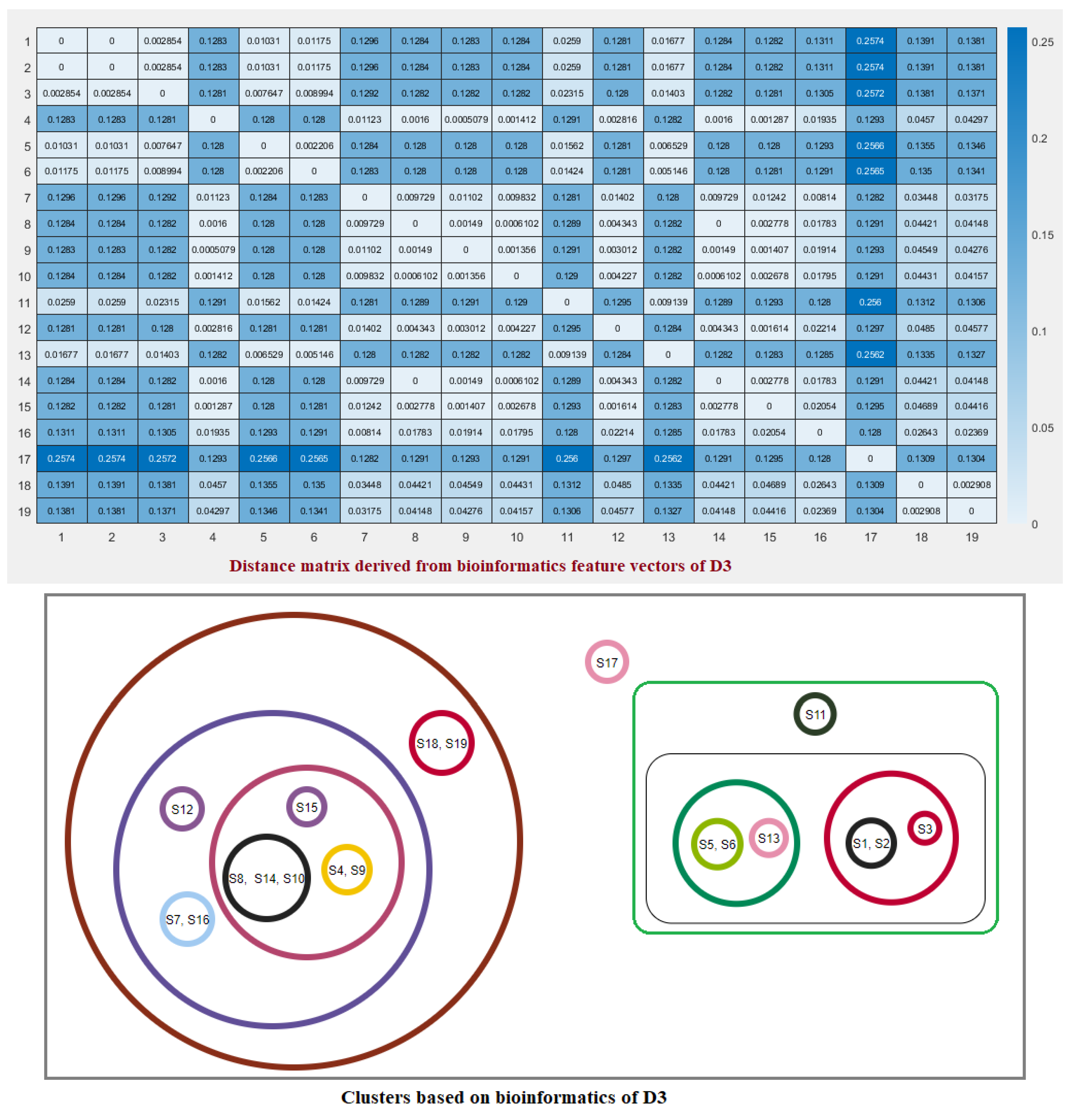
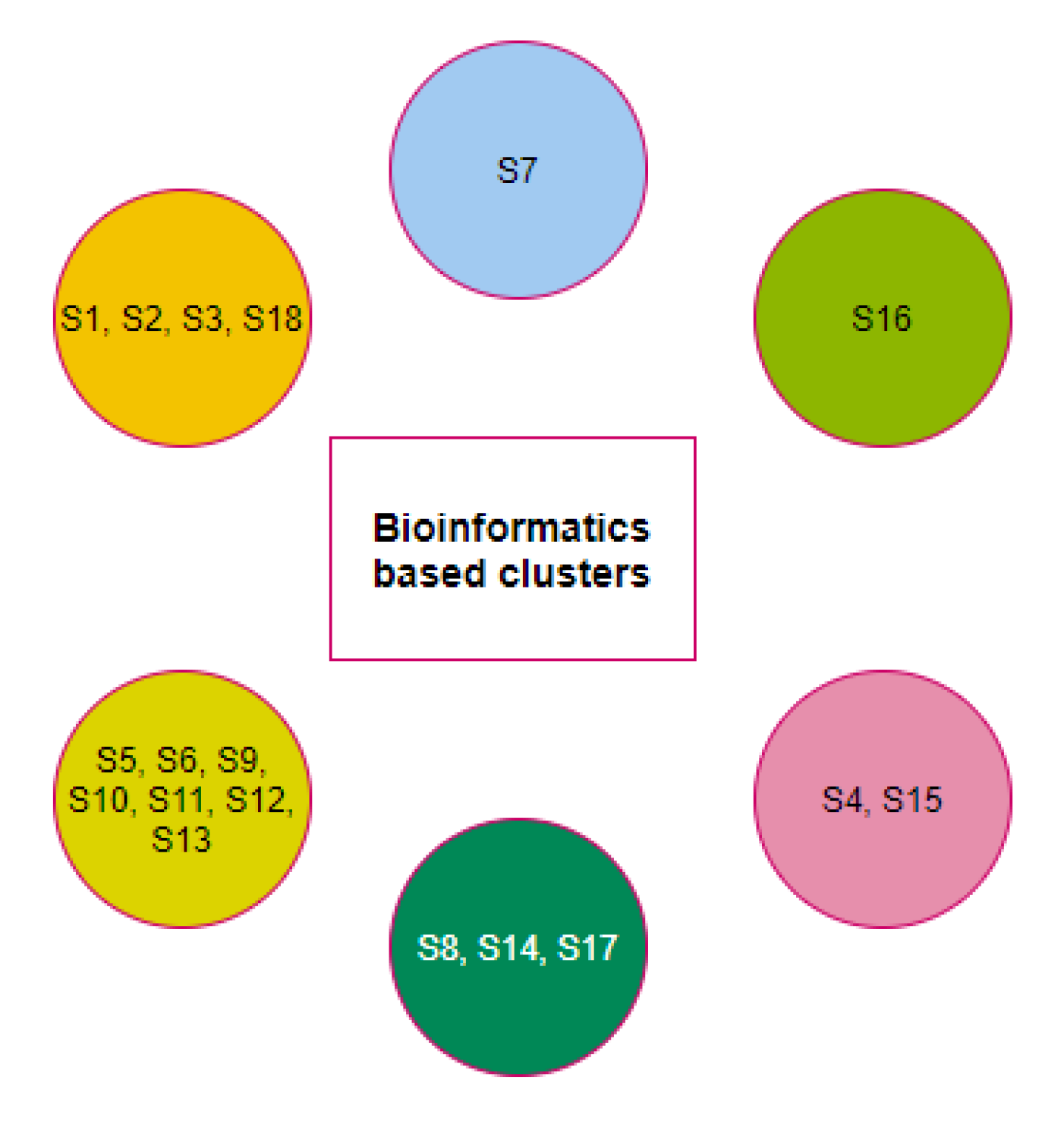
2.3. Clustering Based on Bioinformatics
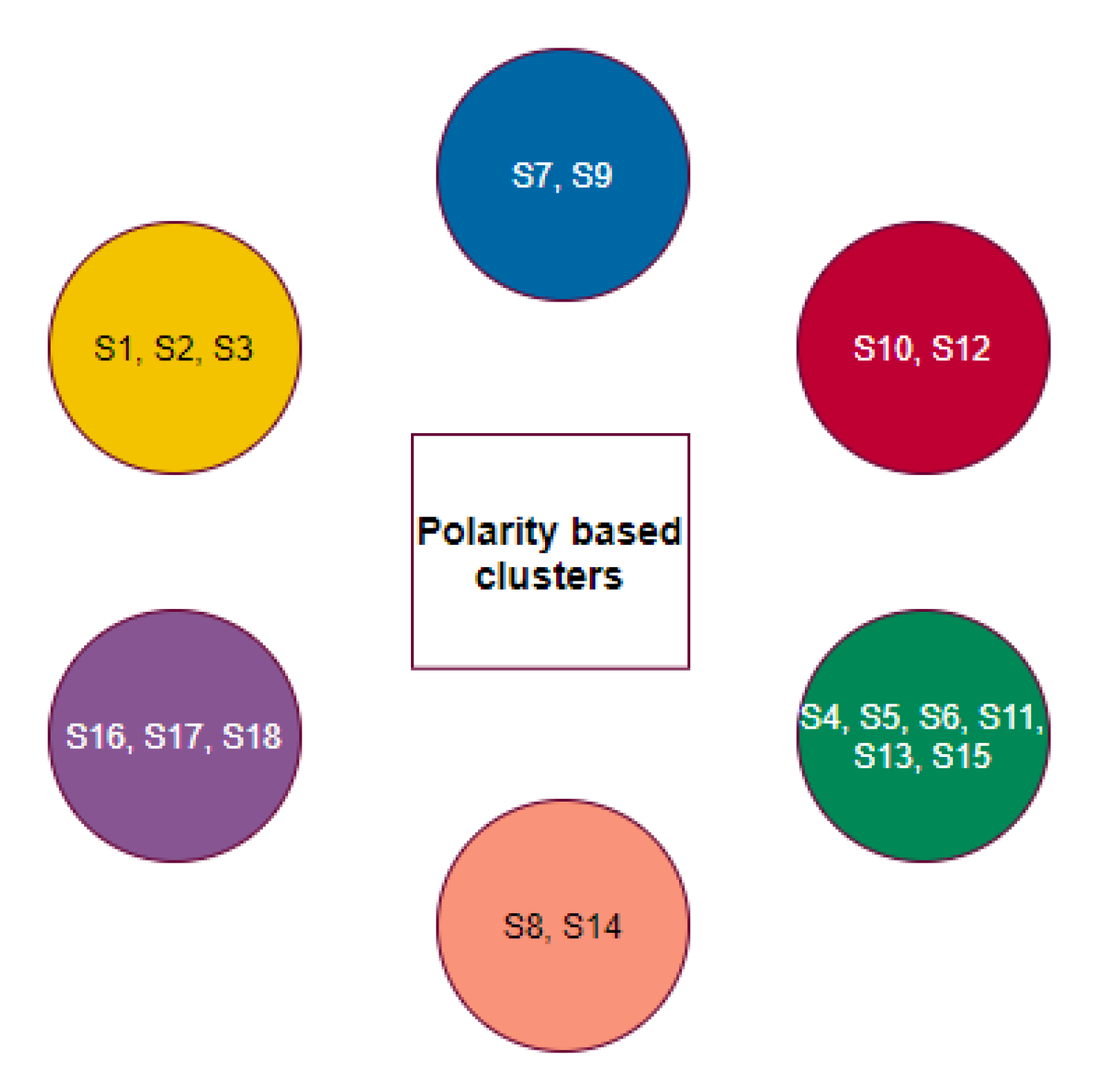
2.4. Phylogeny and Clustering Based on Polarity
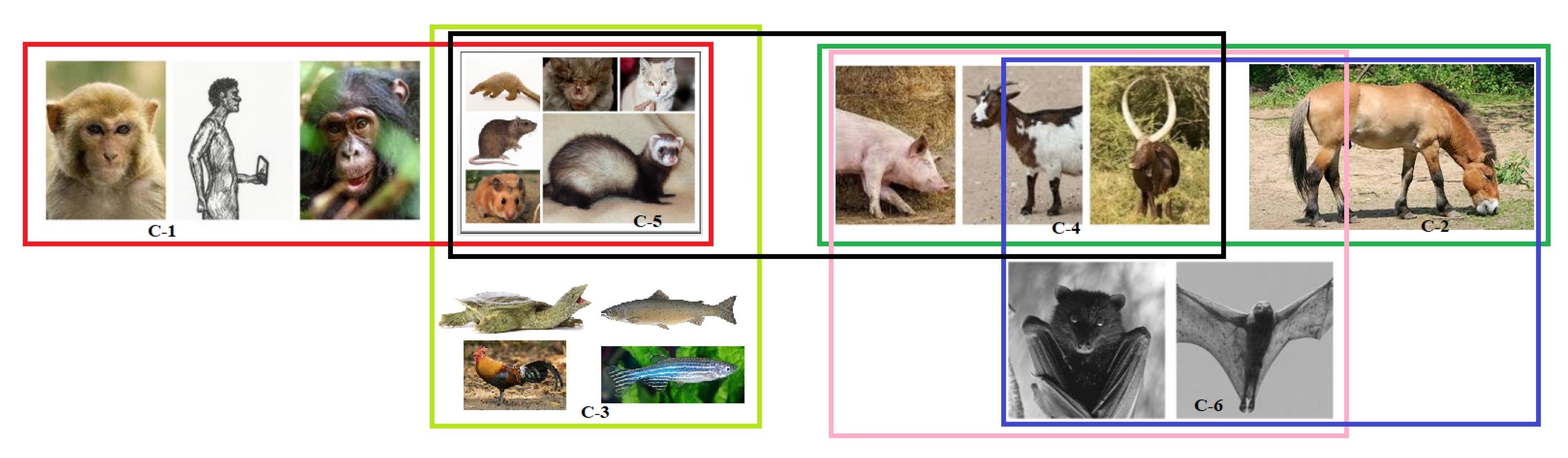
2.5. Possible Clusters of Transmission of SARS-CoV-2
- C-1: None of the species bear any mutation in the binding residues and are conserved, so viral transmission is immaculate.
- C-2: This cluster has an equal number of transmission affecting and transmission non-affecting types of substitutions. Therefore, both have an equal probability of getting infected from each other.
- C-3: Here, again, Gallus gallus, Pelodiscus sinensis, and Danio rerio have a similar ratio of S1 to S2, signifying possible flow of viral transmission within these three species. However, Salmo salar is unique and distant, and therefore, the probability of viral transmission is unlikely.
- C-4: The species in this cluster have a similar number of transmission-affecting and transmission non-affecting types of substitutions show that the flow of viral transmission would be continuous among these three species.
- C-5: Transmission between Felis catus and Mesocrietus auratus is highly likely, which is the same for Manis javanica, Mustela putorius furo, and Rattus norvegicus as indicated by their similar number of substitutions. Therefore, the inter-transmission between these species is highly plausible. While Rhinolophus ferrumequinum has a relatively high value of transmission affecting substitutions from all of the above, its susceptibility to getting infected from other species is uncertain.
- C-6: A total of five transmission affecting substitutions in the three domains for Homo sapiens were observed.
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Findings
4.2. Methods
- Assign the number of desired clusters (K) (in the present study, ).
- Find centroids by first shuffling the dataset and then randomly selecting K data points for the centroids without replacement.
- Keep iterating until there is no change to the centroids.
- Find the sum of the squared distance between data points and all centroids.
- Assign each data point to the closest cluster (centroid).
- Compute the centroids for the clusters by taking the average of the all data points that belong to each cluster.
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Severe acute respiratory syndrome (SARS). Wkly. Epidemiol. Rec. 2003, 78, 89. [Google Scholar]
- World Health Organization. Outbreak news: Severe acute respiratory syndrome (SARS). Wkly. Epidemiol. Rec. 2003, 78, 81–83. [Google Scholar]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ning, Z.; Chen, Y.; Guo, M.; Liu, Y.; Gali, N.K.; Sun, L.; Duan, Y.; Cai, J.; Westerdahl, D.; et al. Aerodynamic analysis of SARS-CoV-2 in two Wuhan hospitals. Nature 2020, 582, 557–560. [Google Scholar] [CrossRef]
- Sun, J.; He, W.T.; Wang, L.; Lai, A.; Ji, X.; Zhai, X.; Li, G.; Suchard, M.A.; Tian, J.; Zhou, J.; et al. COVID-19: Epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol. Med. 2020, 26, 483–495. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Coronavirus Disease (COVID-19): Weekly Epidemiological, Update 1; World Health Organization: Washington, DC, USA, 2020. [Google Scholar]
- Zhang, Y.Z.; Holmes, E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Zheng, M.; Song, L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell. Mol. Immunol. 2020, 17, 536–538. [Google Scholar] [CrossRef] [Green Version]
- Minakshi, R.; Jan, A.T.; Rahman, S.; Kim, J. A testimony of the surgent SARS-CoV-2 in the immunological panorama of the human host. Front. Cell. Infect. Microbiol. 2020, 10, 539. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; Yu, T.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and multi-organ response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the era of SARS-CoV-2: Controversies and novel perspectives. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Han, J.; Zhang, A.; Han, Y.; Chen, M.; Liu, Z.; Shao, M.; Cao, W. Exploring the Demographics and Clinical Characteristics Related to the Expression of Angiotensin-Converting Enzyme 2, a Receptor of SARS-CoV-2. Front. Med. 2020, 7, 530. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Zhao, Y.B.; Wang, Q.; Li, J.Y.; Zhou, Z.J.; Liao, C.H.; Ge, X.Y. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes Infect. 2020, 22, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, P.; Uhal, B.D. COVID-19–A theory of autoimmunity to ACE-2. MOJ Immunol. 2020, 7, 17. [Google Scholar]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 562, 165–169. [Google Scholar] [CrossRef]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-COV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Thunuguntla, V.; Bobbillapati, J.; Bondili, J.S. Structural and simulation analysis of hotspot residues interactions of SARS-CoV 2 with human ACE2 receptor. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, K.; Susumu, K.; Chen, J.; Xu, M.; Pradhan, M.; Zhu, W.; Hu, X.; Breger, J.C.; Wolak, M.; Oh, E. Quantum Dot-Conjugated SARS-CoV-2 Spike Pseudo-Virions Enable Tracking of Angiotensin Converting Enzyme 2 Binding and Endocytosis. ACS Nano 2020, 14, 12234–12247. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Elrashdy, F.; Aljadawi, A.; Redwan, E.M. Household pets and SARS-CoV2 transmissibility in the light of the ACE2 intrinsic disorder status. J. Biomol. Struct. Dyn. 2020, 1–4. [Google Scholar] [CrossRef]
- Sang, E.R.; Tian, Y.; Gong, Y.; Miller, L.C.; Sang, Y. Integrate structural analysis, isoform diversity, and interferon-inductive propensity of ACE2 to predict SARS-CoV2 susceptibility in vertebrates. Heliyon 2020, 6, e04818. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic interactions in protein structure, folding, binding, and condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Flocco, M.M.; Mowbray, S.L. Strange bedfellows: Interactions between acidic side-chains in proteins. J. Mol. Biol. 1995, 254, 96–105. [Google Scholar] [CrossRef]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Kinjo, A.R.; Nishikawa, K. Eigenvalue analysis of amino acid substitution matrices reveals a sharp transition of the mode of sequence conservation in proteins. Bioinformatics 2004, 20, 2504–2508. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E. On the relationship between sequence and structure similarities in proteomics. Bioinformatics 2007, 23, 717–723. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Liu, C.; Xu, R.; Ruan, Z.; Zhao, S.; Zhang, H.; Wang, W.; Huang, X.; Yang, L.; Tang, Y.; et al. SARS-CoV-2 Infection of Cats and Dogs? Preprints 2020, 2020, 2020040116. [Google Scholar]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sit, T.H.; Brackman, C.J.; Ip, S.M.; Tam, K.W.; Law, P.Y.; To, E.M.; Yu, V.Y.; Sims, L.D.; Tsang, D.N.; Chu, D.K.; et al. Infection of dogs with SARS-CoV-2. Nature 2020, 586, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federhen, S. The NCBI taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Jenuth, J.P. The NCBI. In Bioinformatics Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2000; pp. 301–312. [Google Scholar]
- Likas, A.; Vlassis, N.; Verbeek, J.J. The global k-means clustering algorithm. Pattern Recognit. 2003, 36, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Lu, S.; Fotouhi, F.; Deng, Y.; Brown, S.J. FGKA: A fast genetic k-means clustering algorithm. In Proceedings of the 2004 ACM Symposium on Applied Computing, Nicosia, Cyprus, 14–17 March 2004; pp. 622–623. [Google Scholar]
- Kumar, T.A. CFSSP: Chou and Fasman secondary structure prediction server. Wide Spectr. 2013, 1, 15–19. [Google Scholar]
- Pande, A.; Patiyal, S.; Lathwal, A.; Arora, C.; Kaur, D.; Dhall, A.; Mishra, G.; Kaur, H.; Sharma, N.; Jain, S.; et al. Computing wide range of protein/peptide features from their sequence and structure. bioRxiv 2019, 599126. [Google Scholar]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI–multi FASTA ProtParam interface. Bioinformation 2016, 12, 74. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins Struct. Funct. Bioinform. 2005, 61, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.L.; Kurgan, L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012, 13, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Species | DI (M1, M2) | D2 (M1, M2) | D3 (M1, M2) | Total (M1, M2) |
|---|---|---|---|---|---|
| 1 | Human ACE2 | 0 | 0 | 0 | 0 |
| 1 | Pan troglodytes | 0 | 0 | 0 | 0 |
| 1 | Macaca mulatta | 0 | 0 | 0 | 0 |
| 6 | Equus caballus | (4,1) | (1,0) | (0,0) | (5,1) |
| 5 | Felis catus | (1,2) | (1,0) | (0,0) | (2,2) |
| 5 | Mesocrietus auratus | (1,0) | (1,0) | (0,0) | (2,0) |
| 5 | Manis javanica | (2,2) | (1,1) | (1,0) | (4,3) |
| 5 | Mustela putorius furo | (2,2) | (2,0) | (1,0) | (5,2) |
| 5 | Rattus norvegicus | (3,1) | (3,1) | (0,1) | (6,3) |
| 5 | Rhinolophus ferrumequinum | (6,0) | (2,0) | (0,0) | (8,0) |
| 4 | Capra hircus | (0,1) | (2,0) | (0,0) | (2,1) |
| 4 | Bos taurus | (0,1) | (2,0) | (0,0) | (2,1) |
| 4 | Sus scrofa | (1,2) | (1,1) | (0,1) | (2,4) |
| 2 | Pteropus vampyrus | (2,1) | (1,0) | (1,1) | (4,2) |
| 2 | Pteropus alecto | (2,1) | (1,0) | (1,1) | (4,2) |
| 3 | Gallus gallus | (6,0) | (3,0) | (1,0) | (10,0) |
| 3 | Pelodiscus sinensis | (7,0) | (2,0) | (1,0) | (10,0) |
| 3 | Danio rerio | (5,0) | (2,0) | (2,1) | (9,1) |
| 3 | Salmo salar | NA | NA | (3,1) | (3,1) |
| Name | Species | ACE2 Accession ID | Length |
|---|---|---|---|
| S1 | Homo sapiens | NP_001358344.1 | 805 |
| S2 | Pan troglodytes | PNI38577.1 | 805 |
| S3 | Macaca mulatta | XP_028697658.1 | 805 |
| S4 | Equus caballus | XP_001490241.1 | 805 |
| S5 | Felis catus | NP_001034545.1 | 805 |
| S6 | Mesocricetus auratus | XP_005074266.1 | 805 |
| S7 | Manis javanica | XP_017505752.1 | 805 |
| S8 | Pteropus alecto | XP_006911709.1 | 805 |
| S9 | Capra hircus | AHI85757.1 | 804 |
| S10 | Sus scrofa | NP_001116542.1 | 805 |
| S11 | Mustela putorius furo | XP_004758943.1 | 805 |
| S12 | Bos taurus | NP_001019673.2 | 804 |
| S13 | Rattus norvegicus | NP_001012006.1 | 805 |
| S14 | Pteropus vampyrus | XP_011361275.1 | 804 |
| S15 | Rhinolophus ferrumequinum | XP_032963186.1 | 805 |
| S16 | Gallus gallus | XP_416822.2 | 808 |
| S17 | Pelodiscus sinensis | XP_006122891.1 | 808 |
| S18 | Danio rerio | XP_005169417.1 | 807 |
| S19 | Salmo salar | XP_014062928.1 | 695 |
| SARS-CoV-2 RBD | ACE2 (Homo Sapiens) | SARS-CoV-2 RBD | ACE2 (Homo Sapiens) |
|---|---|---|---|
| K417 | Q24 | Q493 | Q42 |
| G446 | T27 | G496 | L79 |
| Y449 | F28 | Q498 | M82 |
| Y453 | D30 | T500 | Y83 |
| L455 | K31 | N501 | N330 |
| F456 | H34 | G502 | K353 |
| A475 | E35 | Y505 | G354 |
| F486 | E37 | D355 | |
| N487 | D38 | R357 | |
| Y489 | Y41 | R393 |
Sample Availability: Data used in this manuscript is available in the NCBI public depository. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.S.; Ghosh, S.; Attrish, D.; Choudhury, P.P.; Aljabali, A.A.A.; Uhal, B.D.; Lundstrom, K.; Rezaei, N.; Uversky, V.N.; Seyran, M.; et al. Possible Transmission Flow of SARS-CoV-2 Based on ACE2 Features. Molecules 2020, 25, 5906. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245906
Hassan SS, Ghosh S, Attrish D, Choudhury PP, Aljabali AAA, Uhal BD, Lundstrom K, Rezaei N, Uversky VN, Seyran M, et al. Possible Transmission Flow of SARS-CoV-2 Based on ACE2 Features. Molecules. 2020; 25(24):5906. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245906
Chicago/Turabian StyleHassan, Sk. Sarif, Shinjini Ghosh, Diksha Attrish, Pabitra Pal Choudhury, Alaa A. A. Aljabali, Bruce D. Uhal, Kenneth Lundstrom, Nima Rezaei, Vladimir N. Uversky, Murat Seyran, and et al. 2020. "Possible Transmission Flow of SARS-CoV-2 Based on ACE2 Features" Molecules 25, no. 24: 5906. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245906