
Recent Advances in the Synthesis of Selenophenes and Their Derivatives
by
 , , , and
, , , and
Paola S. Hellwig
1 ,
,
Thiago J. Peglow
1,
Filipe Penteado
1, 
Luana Bagnoli
2,
Gelson Perin
1,* and
Eder J. Lenardão
1,*
1
Laboratório de Síntese Orgânica Limpa-LASOL-CCQFA, Universidade Federal de Pelotas-UFPel, P.O. Box 354, 96010-900 Pelotas, RS, Brazil
2
Group of Catalysis, Synthesis and Organic Green Chemistry, Department of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123 Perugia, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2020, 25(24), 5907; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245907
Submission received: 14 November 2020
/
Revised: 7 December 2020
/
Accepted: 9 December 2020
/
Published: 13 December 2020
(This article belongs to the Special Issue Organoselenium Reagents and Their Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
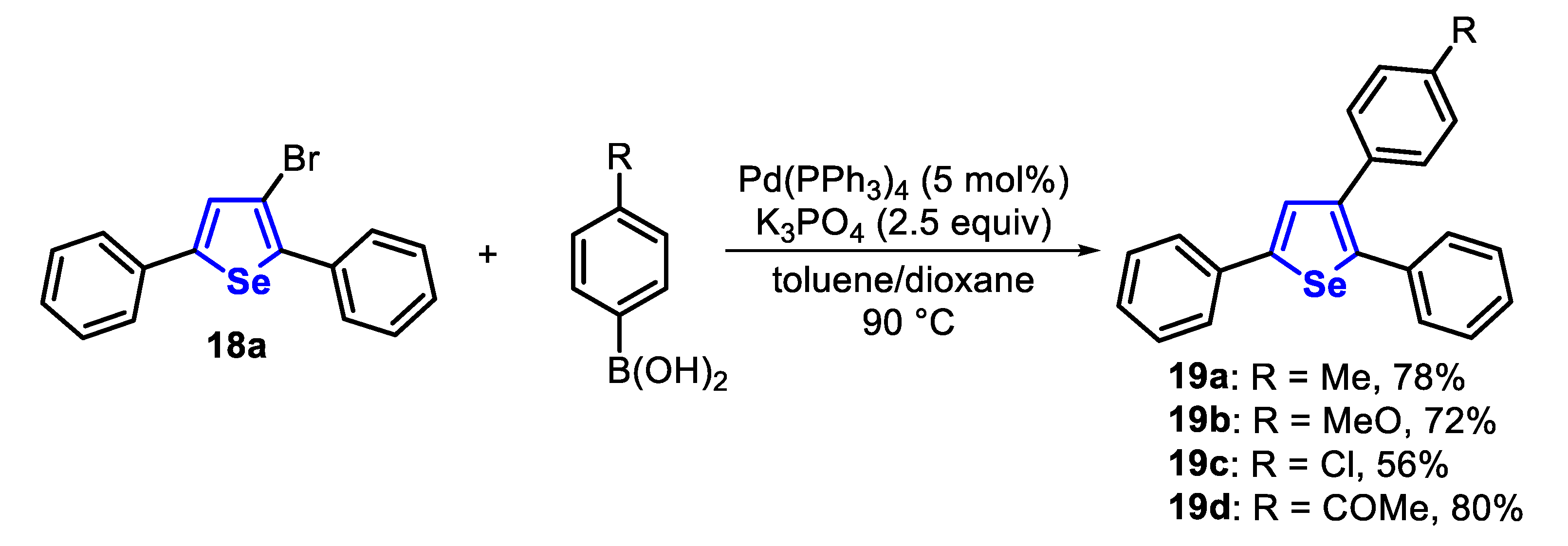{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
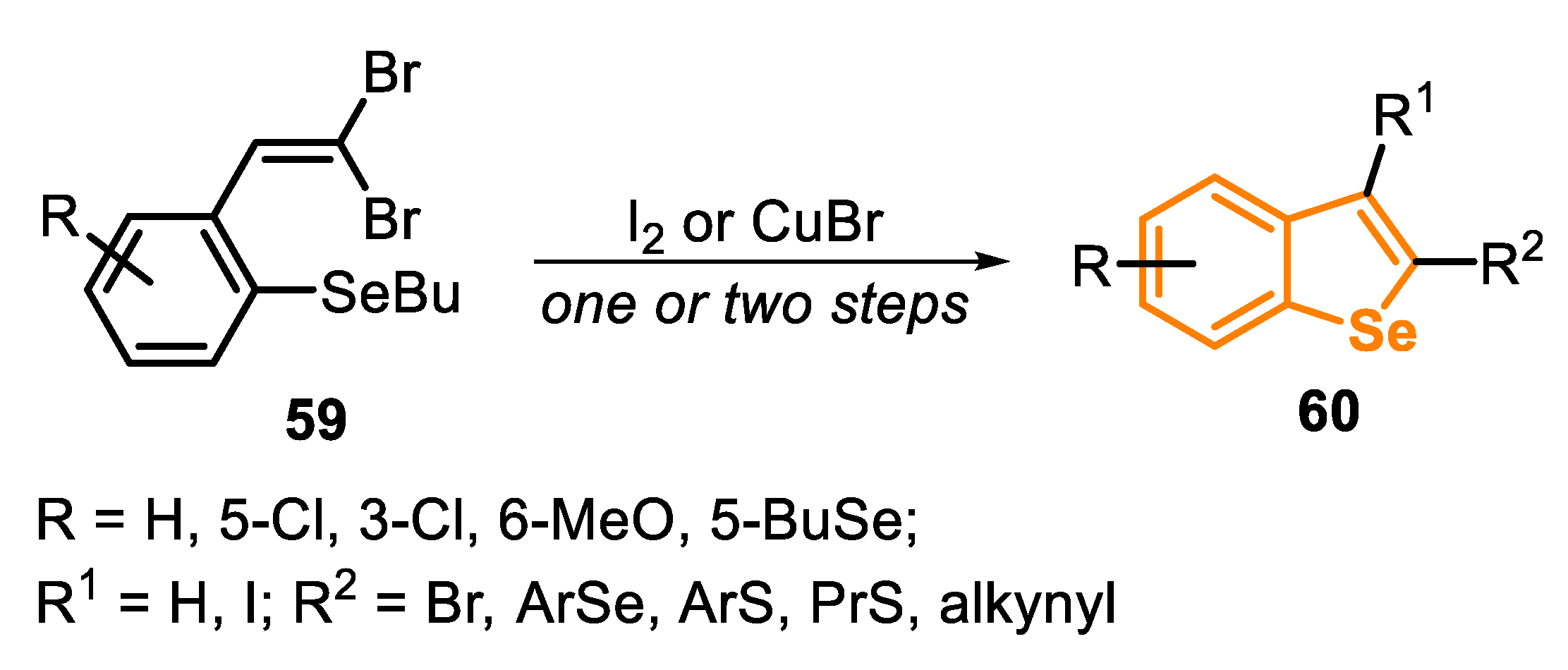{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The selenophene derivatives are an important class of selenium-based heterocyclics. These compounds play an important role in prospecting new drugs, as well as in the development of new light-emitting materials. During the last years, several methods have been emerging to access the selenophene scaffold, employing a diversity of cyclization-based synthetic strategies, involving specific reaction partners and particularities. This review presents a comprehensive discussion on the recent advances in the synthesis of selenophene-based compounds, starting from different precursors, highlighting the main differences, the advantages, and limitations among them.
1. Introduction
The importance of organoselenium compounds is linked to their synthetic applications as catalysts [1,2,3,4] and building blocks [5,6,7,8,9], allowing for several chemo-, regio- and stereoselective transformations [10,11,12,13,14,15]. Although organoselenium compounds have not yet reached the pharmacy shelves, studies in vitro and in vivo have demonstrated their effectiveness against several disorders [16,17,18,19]. Among the successful examples, those molecules presenting an organoselenium portion embedded in an aromatic or non-aromatic cycle—i.e., the selenium-containing heterocycles—have demonstrated powerful biological activities [20,21].
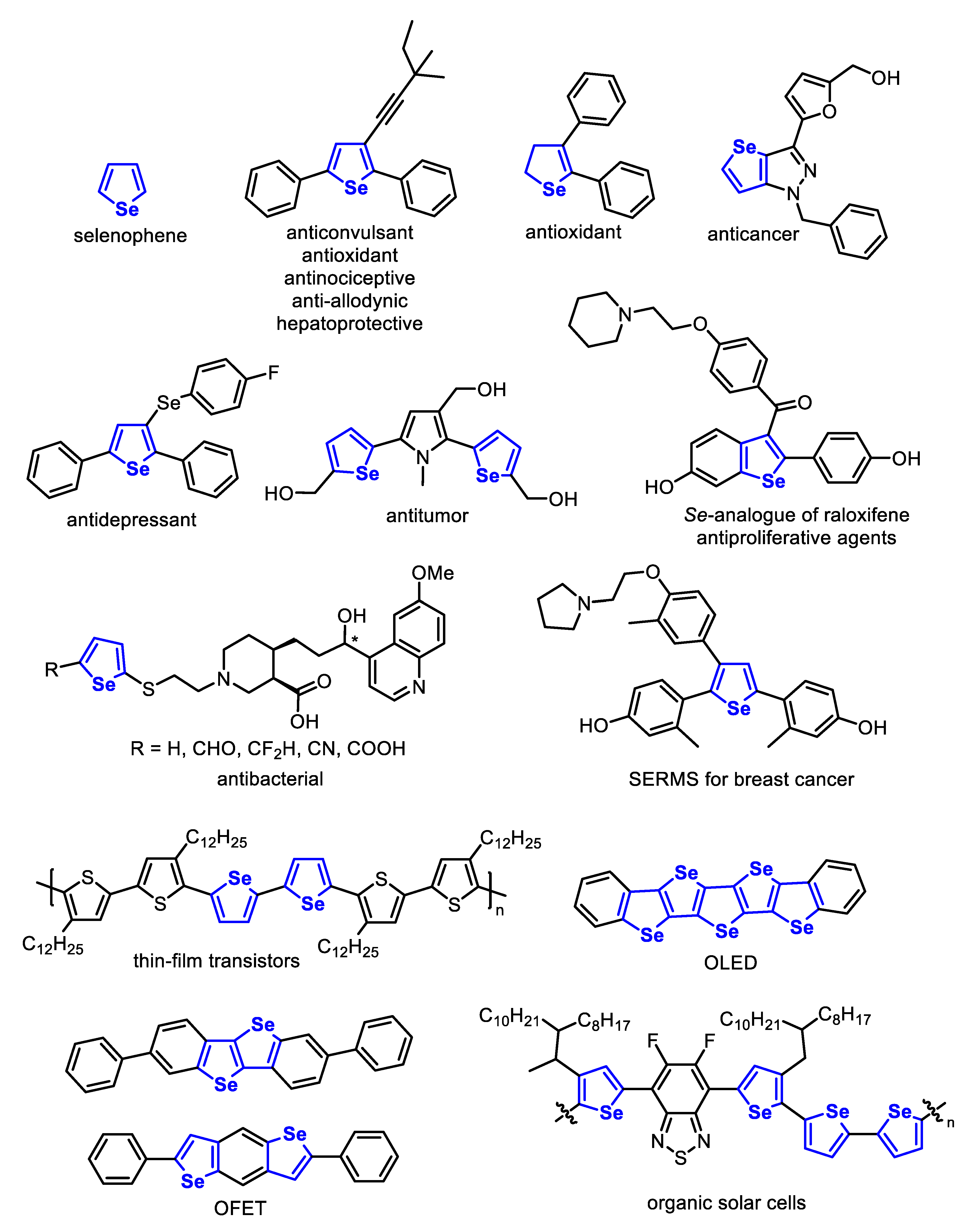
In this context, selenophenes and their derivatives have been widely studied due to their intrinsic biological activity [22], such as antidepressant [23,24,25,26,27,28], antioxidant [29,30,31,32,33,34,35], anticonvulsant [36,37], antibacterial [38], antitumor [39,40,41,42,43,44,45,46,47], antinociceptive [48], hepatoprotective [49,50] and antiapoptotic agents [51]. Besides, they have also played an important role in the materials science field, being used to build organic light emitting diodes (OLEDs) [52,53,54,55,56,57,58], organic field effect transistors (OFETs) [59,60,61,62,63,64,65,66,67,68,69], organic solar cells (OSC) [70,71,72,73,74,75,76,77,78,79,80,81,82] and thin-film transistors [83,84,85,86,87,88,89,90]. The structures of several biologically active and technologically interesting selenophenes are presented in Figure 1.
Selenophene derivatives have also been used as ligands in coordination chemistry [91,92,93,94,95,96,97]. After a pre-activation with a halide or an organometallic (B, Li, Mg, Sn or Zn), these compounds can be employed as reagents in the formation of new C-C [98,99,100,101,102,103,104,105,106,107], C-N [108,109,110] and C-S [111] bonds under Pd or Cu catalysis, through Heck, Stille, Negish, Kumada, Suzuki and Sonogashira coupling reactions. In recent years, unactivated selenophenes have been used as reagents in several synthetic transformations by palladium-catalyzed direct C-H bond activation [112,113,114,115,116].
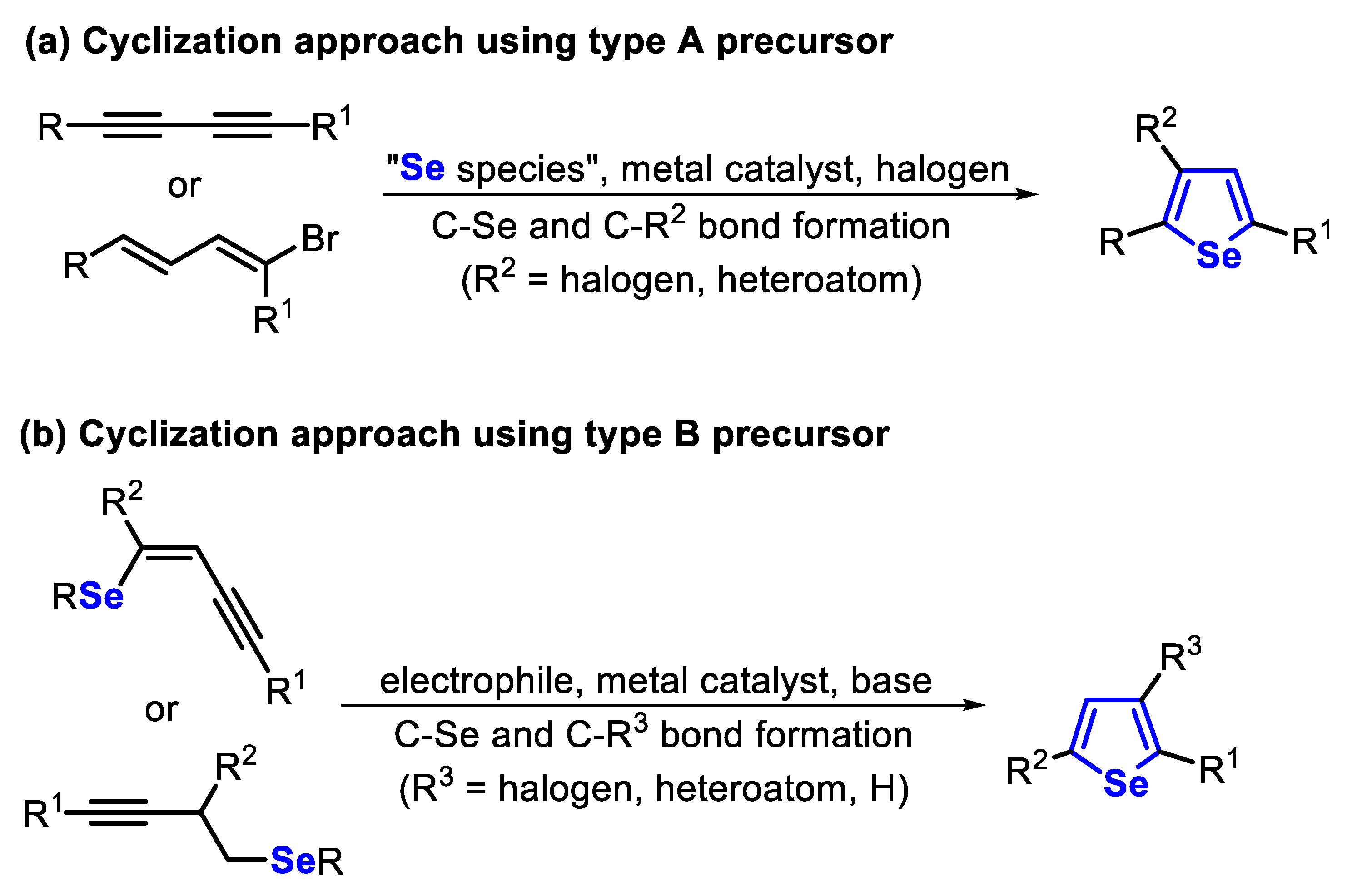
The traditional methods for the synthesis of selenophenes involve the addition of a selenium-based nucleophile or electrophile to an appropriate acyclic precursor containing a π-system, followed by an intramolecular cyclization (type A precursor). Alternatively, a previously prepared organoselenium precursor can easily undergo an intramolecular cyclization toward selenophenes (type B precursor). In general, these reactions are mediated by TM-based catalysts or electrophilic species (Scheme 1).
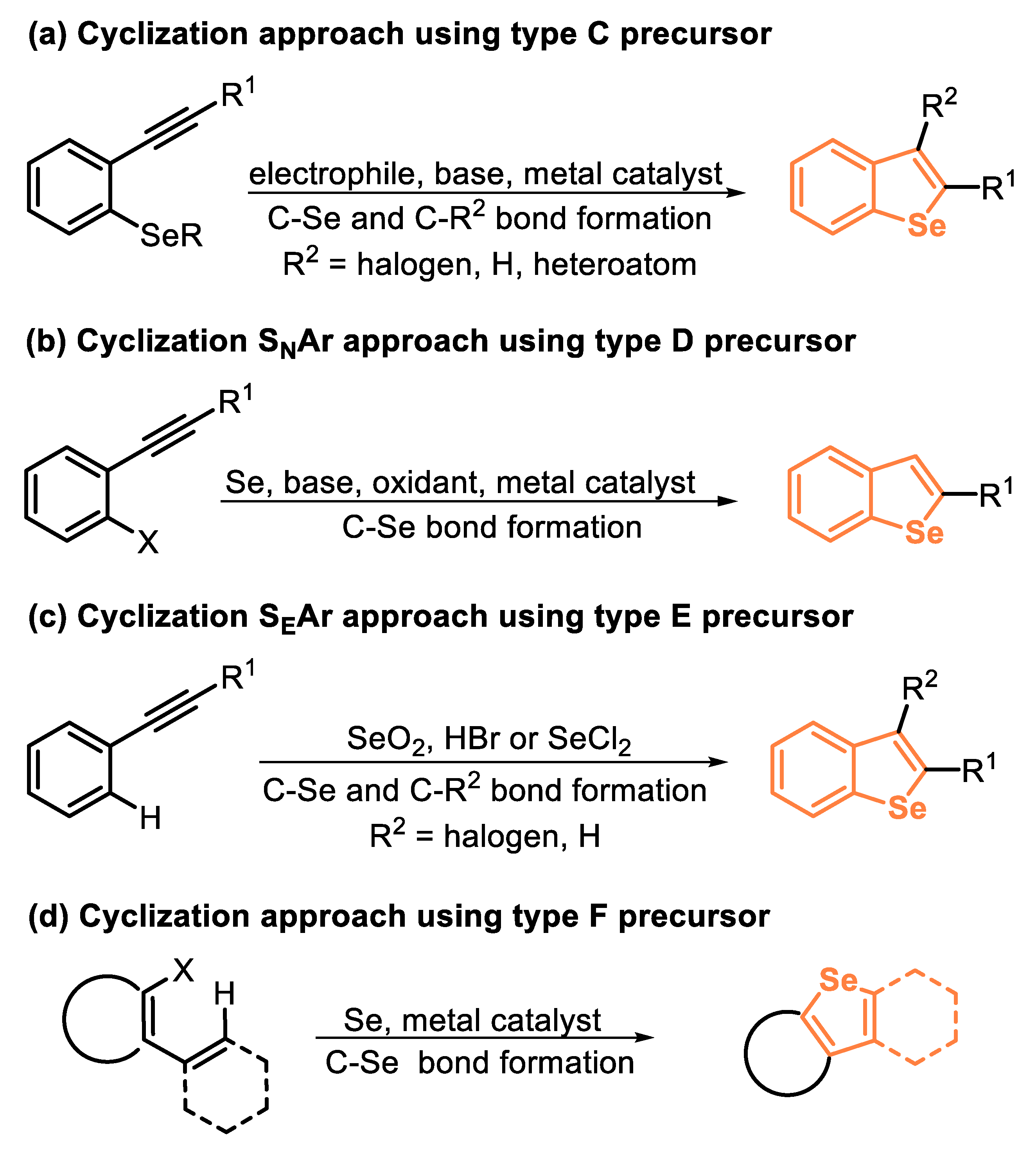
Another useful strategy to access benzoselenophene derivatives is through the intramolecular cyclization of selenium-functionalized arenes bearing ortho-alkynyl groups. These reactions can be mediated by TM-based catalysts and by electrophilic species (type C precursor). On the other hand, if alkynyl arenes bearing ortho-halogen groups are used in the presence of elemental selenium, under appropriate conditions, benzoselenophenes can be accessed through a SNAr reaction, followed by an intramolecular annulation (type D precursor). By replacing the ortho-halogen group by a hydrogen atom, a selane Friedel–Crafts cyclization affords the respective benzoselenophene (type E precursor). Finally, benzoselenophene derivatives can be satisfactorily accessed through a tandem cyclization of ortho-haloareneheterocycles, in the presence of a TM catalyst and elemental selenium (type F precursor) (Scheme 2).
Thus, considering the importance of the selenophene core, some reviews have been published during the last few decades that describe several strategies to access these compounds [117,118,119,120,121,122,123]. Among them, a short review was recently published [124] covering synthetic methods to access several selenophene derivatives, and the synthetic versatility of selenophenes in the formation of new C-C bonds.
Herein, we present a comprehensive review covering the recent synthetic methods to prepare selenophenes, benzoselenophenes and other heterocycles-fused derivates. For a better discussion and understanding, the range of synthetic methodologies is divided into three major groups: (i) selenophenes, (ii) benzoselenophenes and (iii) fused selenophenes and benzoselenophenes. In each section, methods employing the precursors described in the Scheme 1 and Scheme 2 are discussed first, followed by other important approaches.
2. Synthesis of Selenophenes
2.1. Starting from Type A Precursors
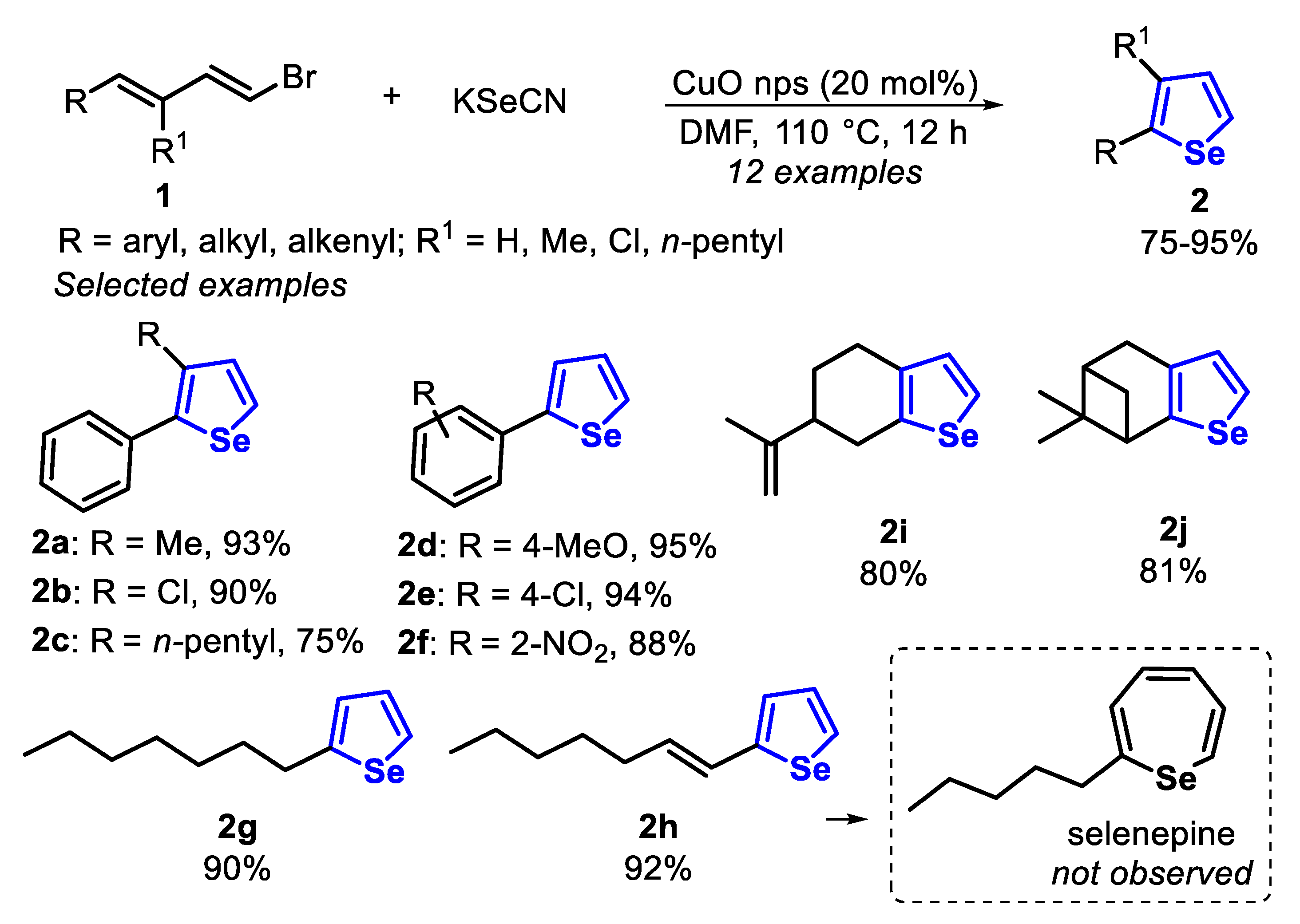
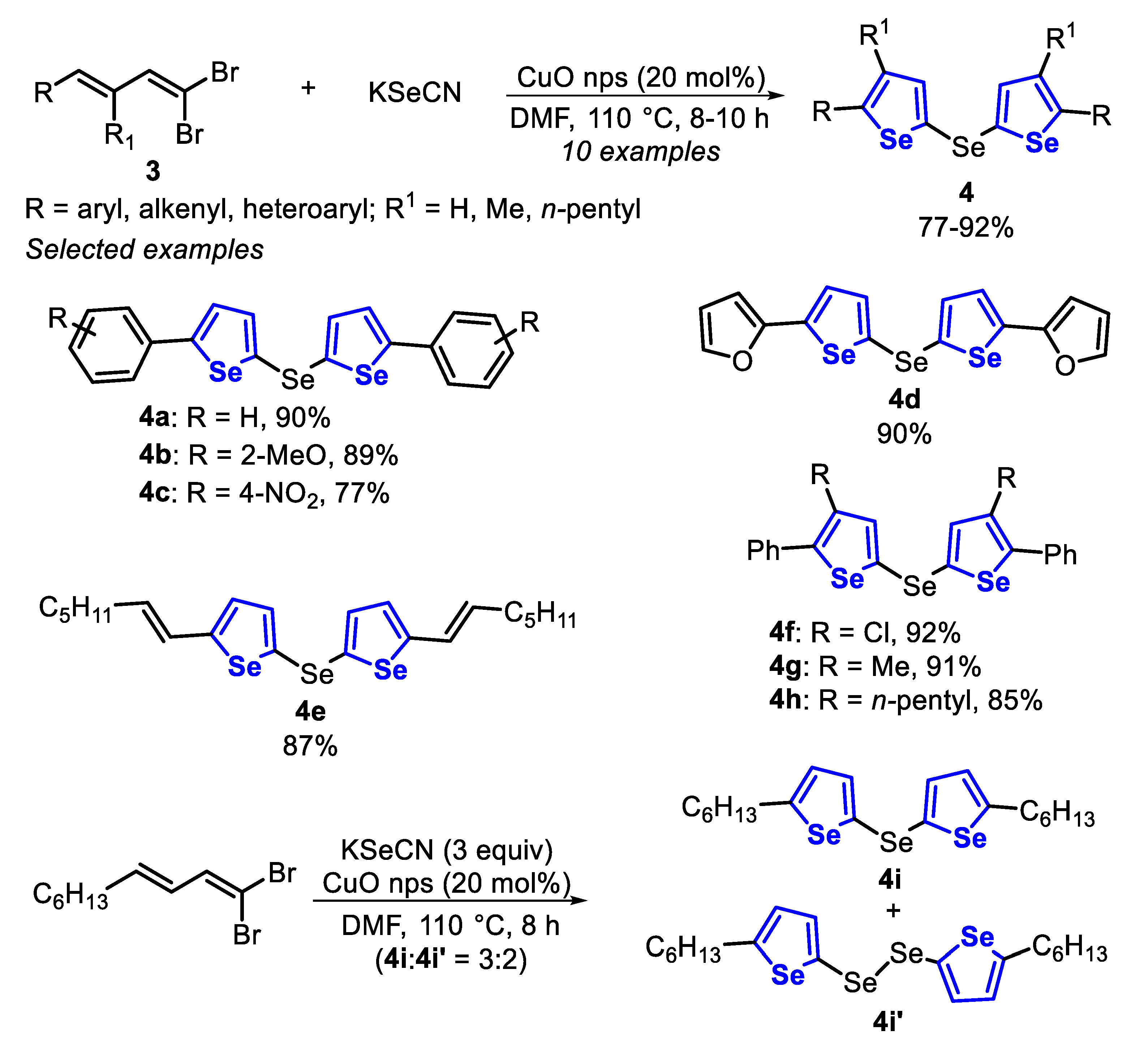
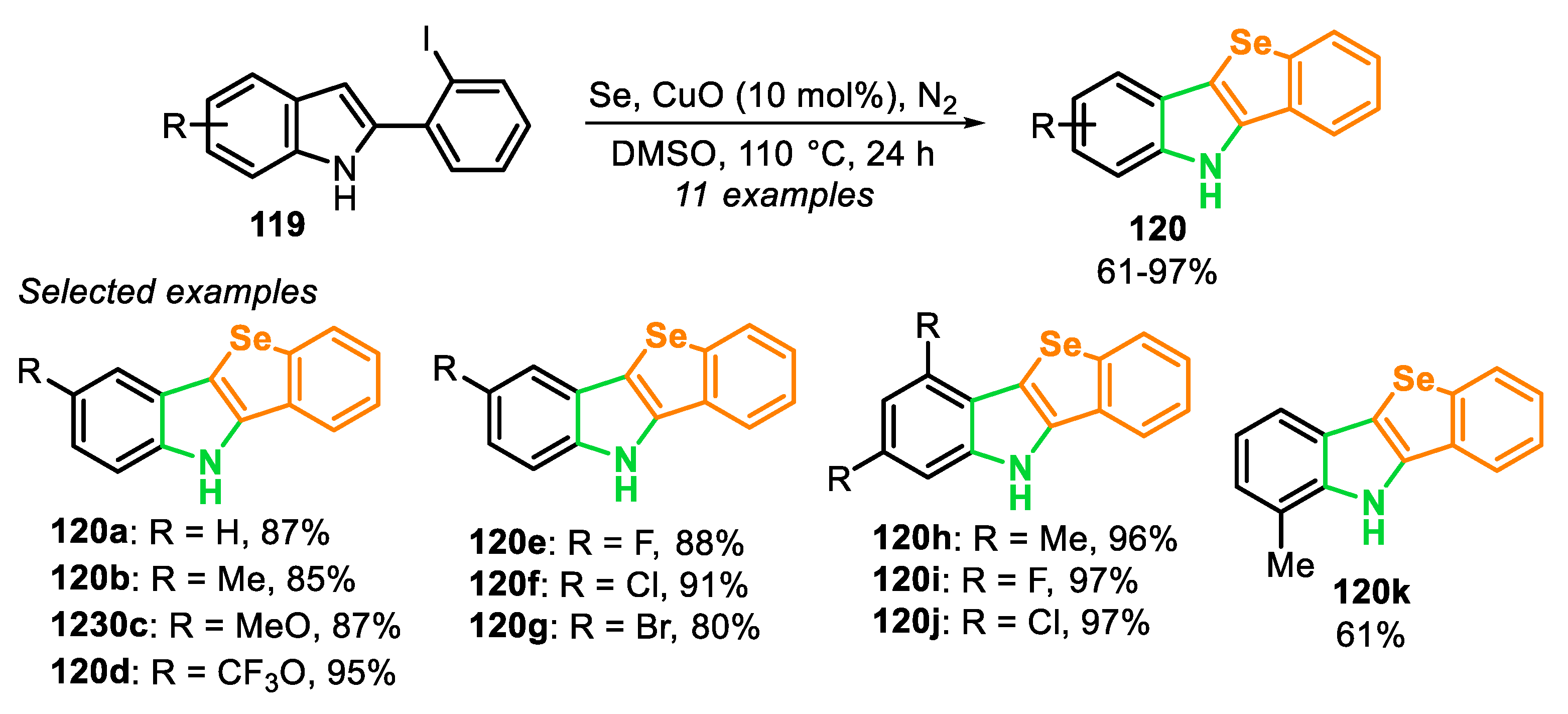
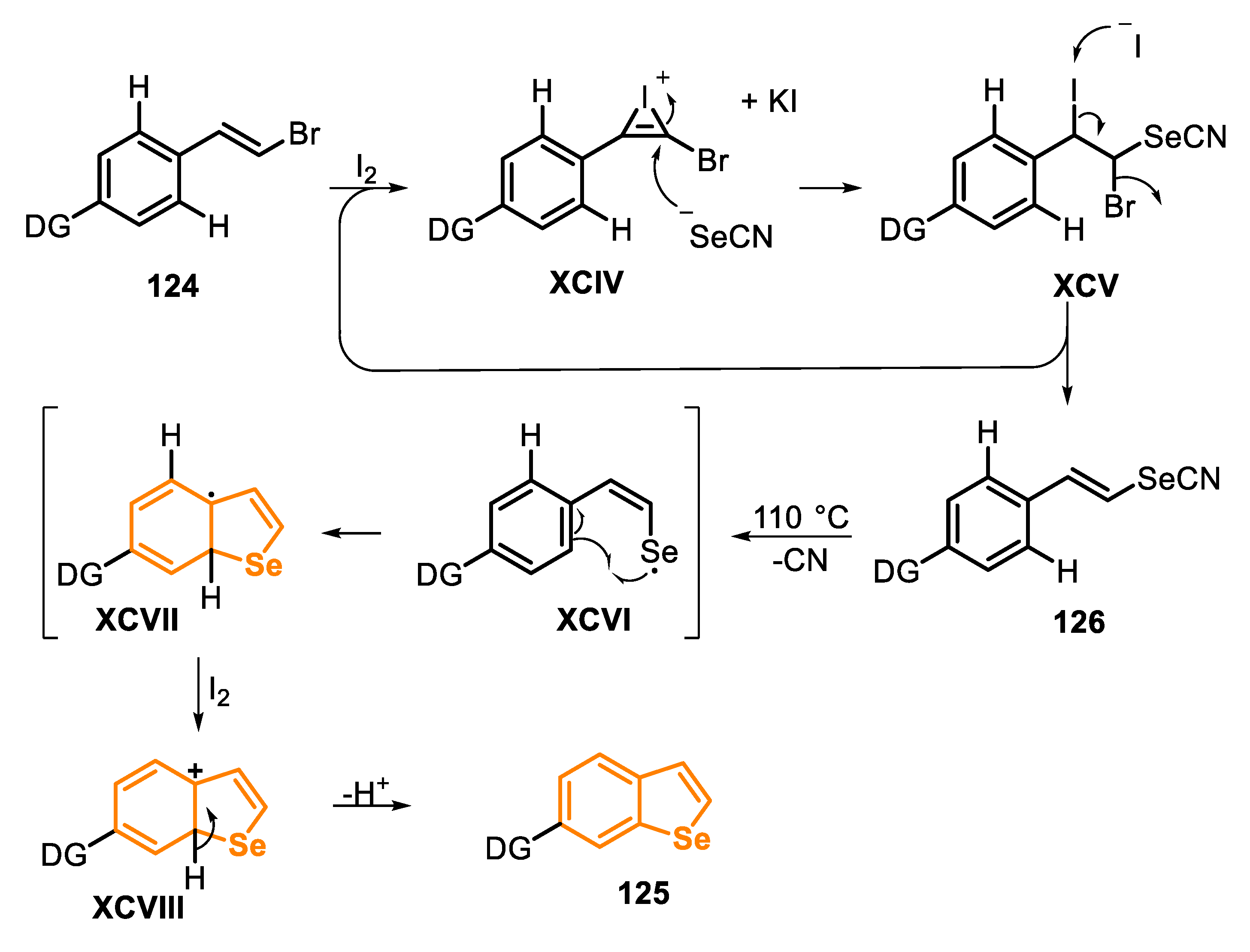
The first protocols for the synthesis of selenophenes used as precursors hexane-2,5-dione and phosphorous pentaselenide (Paal, 1885) [125], metallic selenium and acetylene (Briscoe and Peel, 1928) [126], acetylenes and a mixture of bauxite and aluminum selenide (McMahon and co-workers, 1933) [127], acetylene with dimethyl selenide or diselenide (Voronkov and co-workers, 1987) [128], 2,4-alkadienic esters by oxidation reaction with SeO2 (Takeda and co-workers, 1986) [129], among others [117], under high temperatures (180–600 °C). Since then, new protocols were developed for milder reaction conditions and that would afford selenophene derivatives in high yields. With this propose, in 2014, the one-pot synthesis of selenophenes 2 and selanyl selenophenes 4 by an intramolecular cyclization of 1,3-dienyl bromides 1 and 1,3-dienyl-gem-dibromides 3, in the presence of KSeCN and CuO nanoparticles (nps) was described [130]. The reactions were carried out in DMF, at 110 °C for 8–12 h (Scheme 3 and Scheme 4). Under this condition, aryl-1,3-dienyl bromides 1 bearing alkyl and electron-withdrawing substituents (R1 = Me, Cl and n-pentyl) reacted smoothly to produce the 2-phenyl-3-alkyl selenophenes 2a–c in good yields. The protocol was also able to employ 1,3-dienyl bromides 1, bearing activated and unactivated aryl groups (R = 4-MeO, 4-Cl and 2-NO2), affording the respective products 2d–f in excellent yields. Interestingly, 1,3,5-trienyl bromide 3 was able to furnish the 2-vinyl selenophene 2h in 92% yield, without the formation of selenepine, and with the C-C double bond remaining on the C2 position. Additionally, this protocol was also effective to access the tetrahydro-benzo[b]selenophenes 2i and 2j in good yields (Scheme 3).
In contrast, when aryl-substituted 1,3-dienyl-gem-dibromides 3 were submitted to the standard reaction condition, the corresponding selanyl selenophenes 4a–c were obtained in good yields. In addition, the 2-furyl-1,3-dienyl-gem-dibromide and 1,3,5-trienyl-gem-dibromide 3 furnished the selanyl selenophenes 4d and 4e in good yields. Aryl-1,3-dienyl-gem-bromides 3 bearing chloro- or alkyl groups (R1 = Cl, Me and n-pentyl), afforded the 2-phenyl-3-alkylselanylselenophenes 4f–h in very good to excellent yields. On the other hand, limitations were faced when 1,3-dienyl-gem-dibromides 3 (R = n-hexyl) were employed, producing a mixture of selanylselenophene 4i and diselanylselenophene 4i’ in different ratios (Scheme 4).
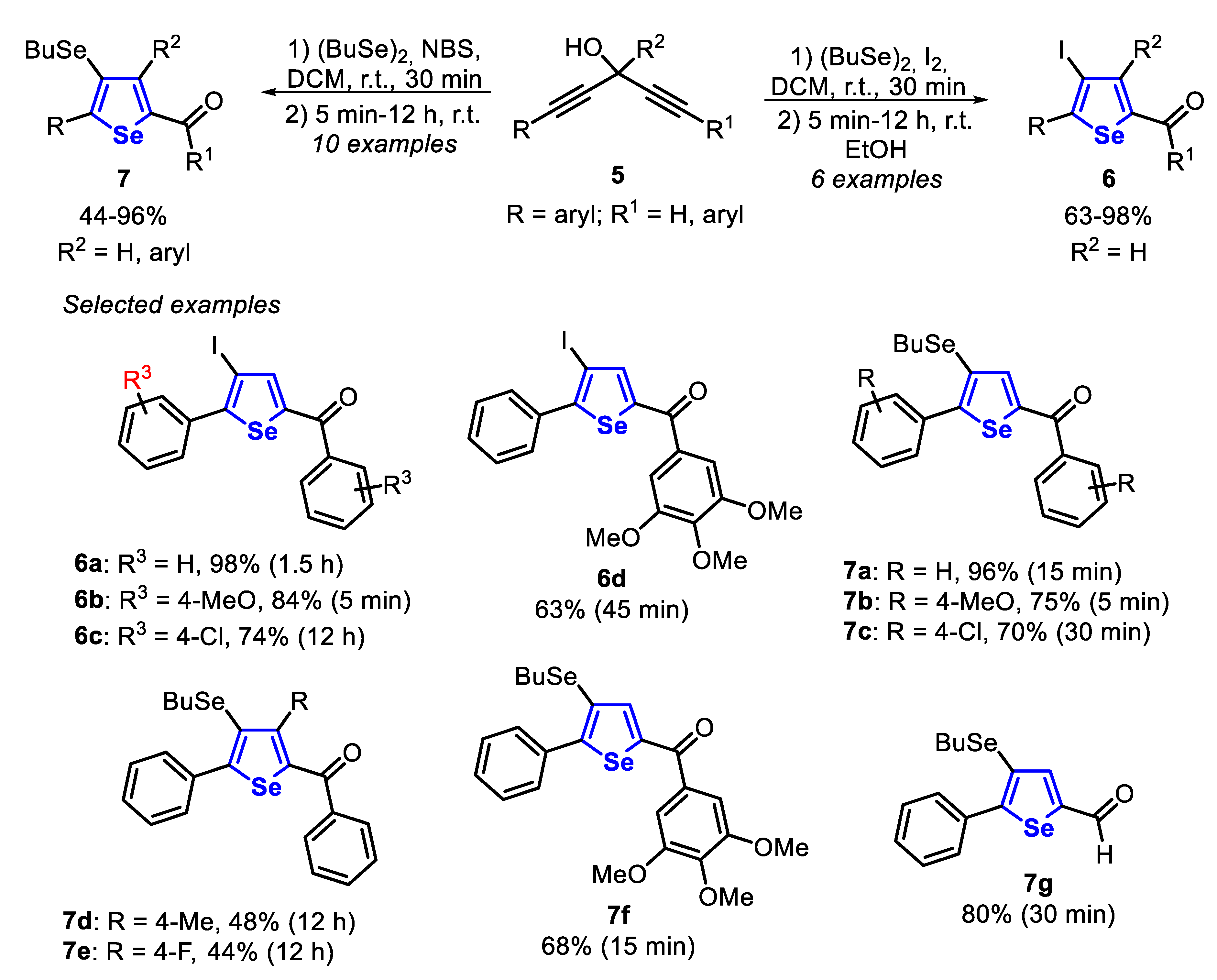
In 2015, was reported the synthesis of selenophenes through the cyclization of diynols 5, promoted by dibutyl diselenide and a halogen source [131]. The reaction optimization studies showed the best reaction condition as being the stirring of a solution of the diynol 5 in DCM, at room temperature, in the presence of dibutyl diselenide (1.5 equiv) and I2 or NBS (1.5 equiv), affording the desired products 6 and 7 in moderate to excellent yields. It is worth mentioning that the halogen source (I2 or NBS) directly affects the reaction regioselectivity. Thus, employing the symmetrical diynols 5 bearing neutral and electron-donating groups (R3 = H and 4-MeO) in the aromatic ring connected to C(sp), the products 6a and 6b were obtained in 98% and 84% yields, respectively, in short reaction times. However, the presence of an electron-withdrawing substituent (R3 = 4-Cl) reduced the reactivity, needing 12 h to access the 4-iodoselenophene 6c in 74% yield. A noteworthy result was obtained using an unsymmetrical diynol 5, which efficiently reacted to give the respective selenophene 6d in 63% yield after 45 min regioselectively. This result suggests that a sterically bulky group, bearing electron-donating groups, positively influences the regioselectivity, by stabilizing the seleniranium reaction intermediate. In contrast, 4-butylselanyl-selenophenes 7 were obtained when NBS was used instead of I2 in the reaction of diynol 5 with Bu2Se2 under the same conditions. In this case, symmetrical diynols 5 bearing neutral, activated and unactivated phenyl groups (R3 = H, MeO and Cl) reacted satisfactorily to give the respective selenophenes 7a–c in good to excellent yields, in short reaction times (5–30 min). Additionally, when tertiary diynols 5 were employed as substrates, the 4-butylselanyl-selenophenes 7d and 7e were obtained in 48% and 44% yields, respectively, after 12 h. Besides, the use of unsymmetrical diynols 5 led to the exclusive formation of the 4-butylselanylselenophenes 7f and 7g in 68% and 80% yields, respectively (Scheme 5).
The proposed mechanism of the transformation of the diynols 5 into 4-iodo-selenophenes 6 initiates by the in situ I2-promoted oxidation of dibutyl diselenide to give the electrophilic intermediate BuSeI, which is added to the C-C triple bond, affording the E-vinyl selenide I. In the sequence, another portion of BuSeI reacts with the second C-C triple bond, affording the seleniranium intermediate II, which undergoes an intramolecular 5-exo-dig cyclization, according to Baldwin’s rules [132], giving the cyclic selenonium intermediate III. In the sequence, the intermediate III is converted to the dihydroselenophene IV, regenerating the electrons to the selenium atom. Thus, the intermediate IV undergoes a 1,3-OH-migration, followed by the elimination of BuSeH, generating the desired selenophene 6 (Scheme 6).
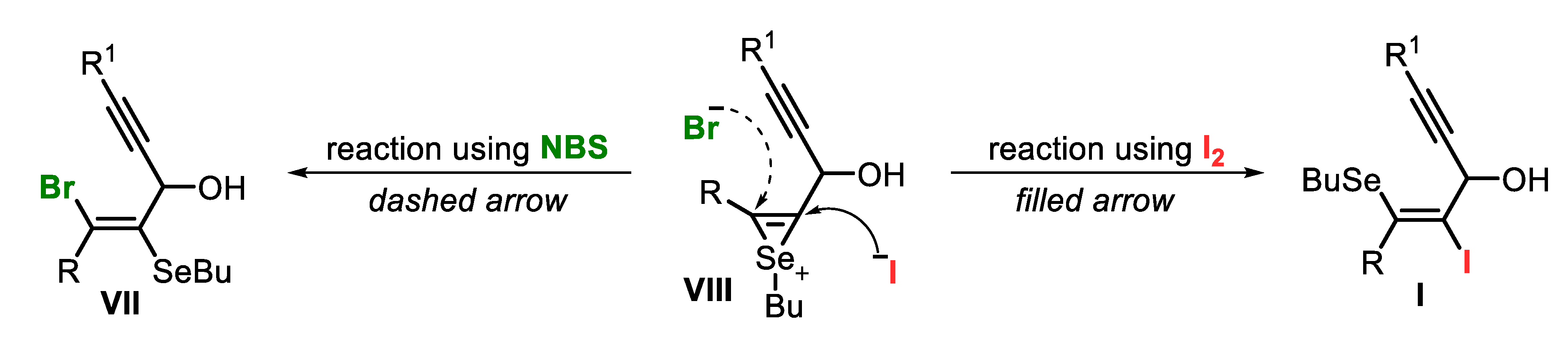
While the formation of the 4-iodo-selenophenes 6 may involve the formation of vinylic selenide I as the key reaction intermediate, instead, the synthesis of 4-butylselanyl-selenophenes 7 passes by the vinylic selenide VII (Scheme 7). After the initial formation of the seleniranium derivative VIII, the subsequent anti-addition of the halogen anion may follow two different ways: (i) the iodide, which is the larger anion, attacks the less hindered carbon, while (ii) bromide, the smaller one, is not affected by the bulky R group, and attacks the more reactive carbon (Scheme 7).
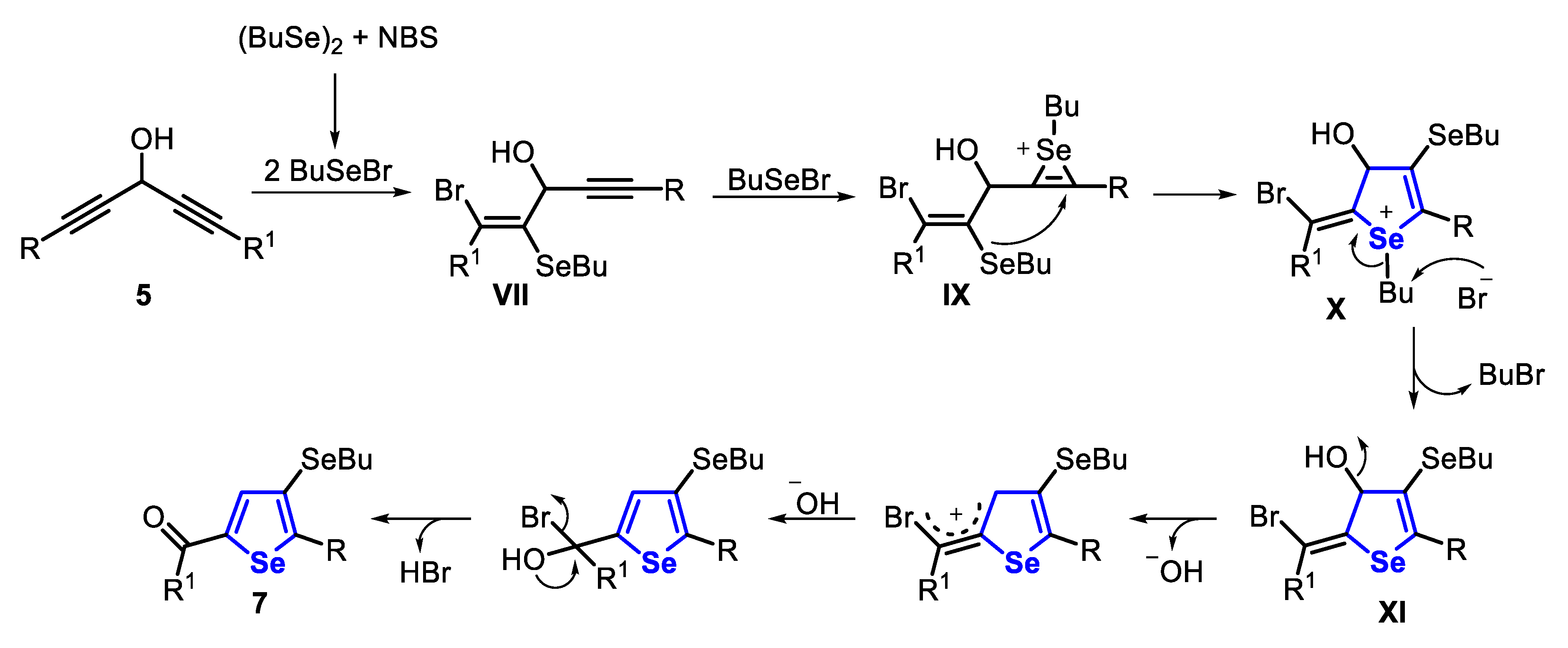
Once the intermediate VII is formed, it reacts with BuSeBr, generated in situ, to form the seleniranium IX, which, after an intramolecular annulation, affords the cyclic selenonium X. In the sequence, the intermediate XI is formed by the elimination of BuBr. Finally, a 1,3-migration of the OH group is followed by the elimination of HBr, giving the 4-butylselanyl-selenophene 7 (Scheme 8).
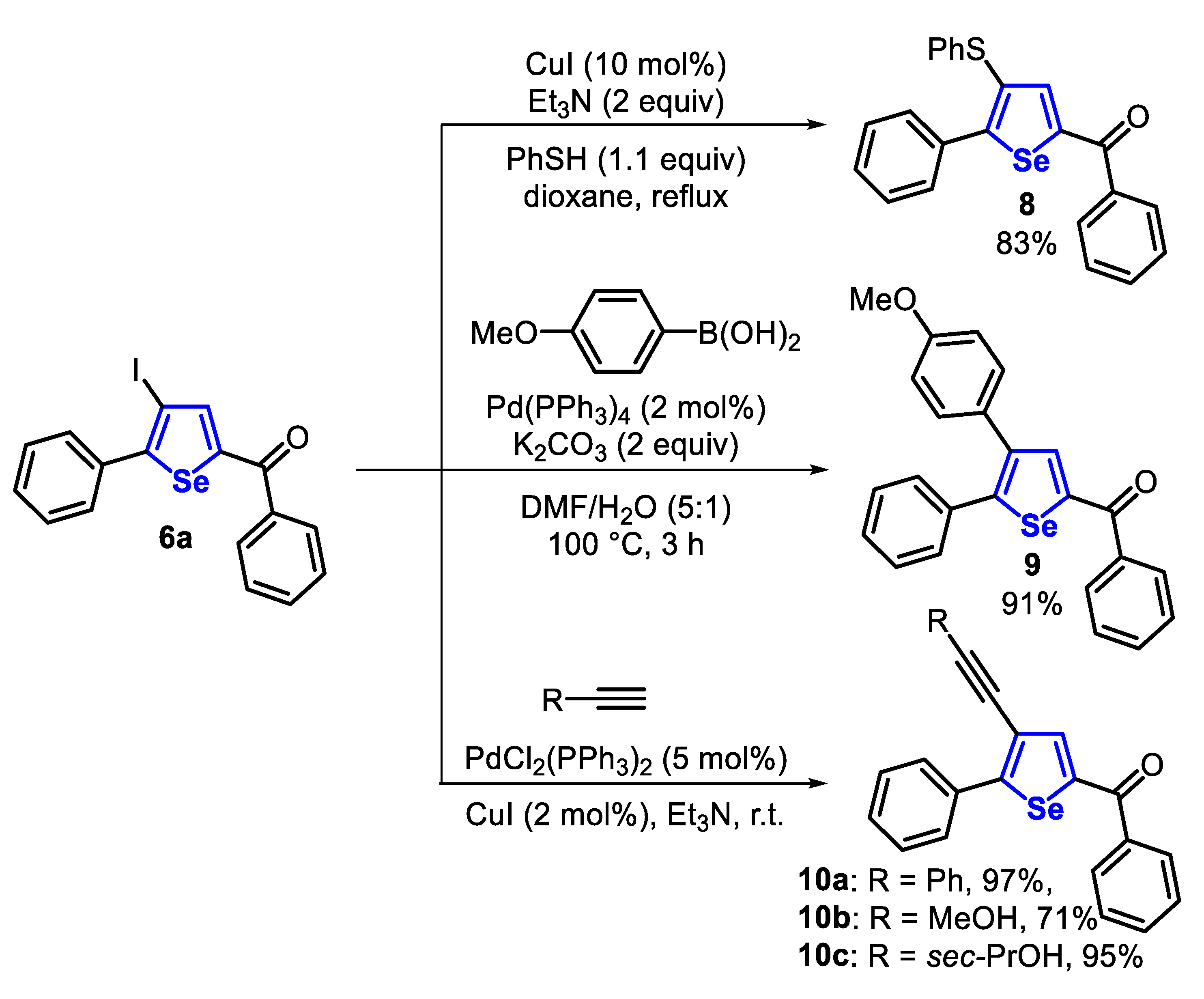
The reactivity of the carbon-halogen bond of 4-iodo-selenophene 6 was explored in Pd-catalyzed cross-coupling reactions (Scheme 9). In this sense, 4-iodo-selenophene 6a reacted with benzenethiol, under the Ullmann conditions, to access the 4-phenylthio-selenophene 8 in 83% yield. Additionally, 6a was a suitable substrate in the Suzuki cross-coupling with arylboronic acid, affording 4-methoxyphenyl-selenophene 9 in 91% yield, after 3 h. Finally, selenophene 6a was subjected to the Sonogashira cross-coupling with terminal acetylenes, giving the respective alkynyl selenophenes 10a-c in good to excellent yields (Scheme 9).
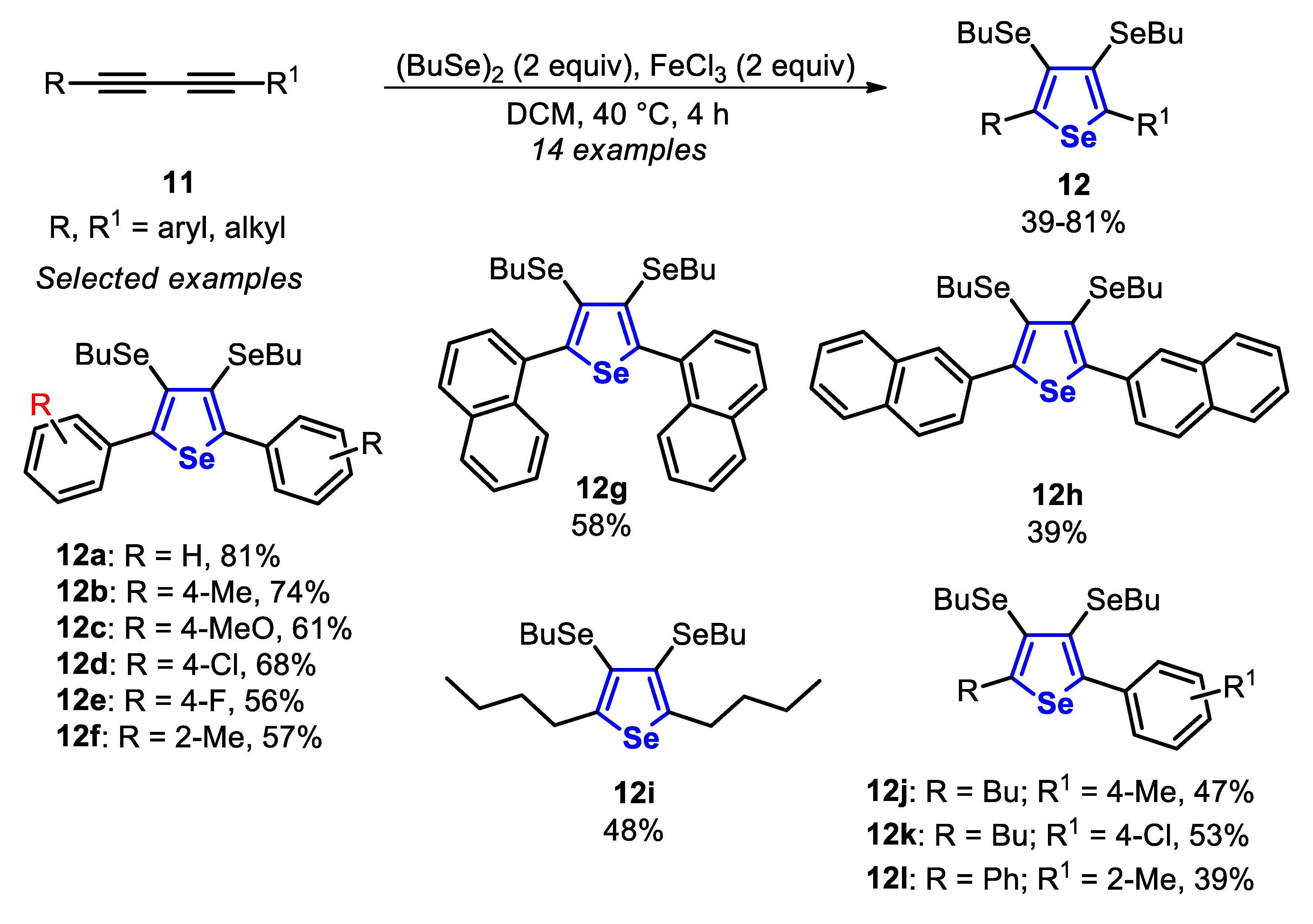
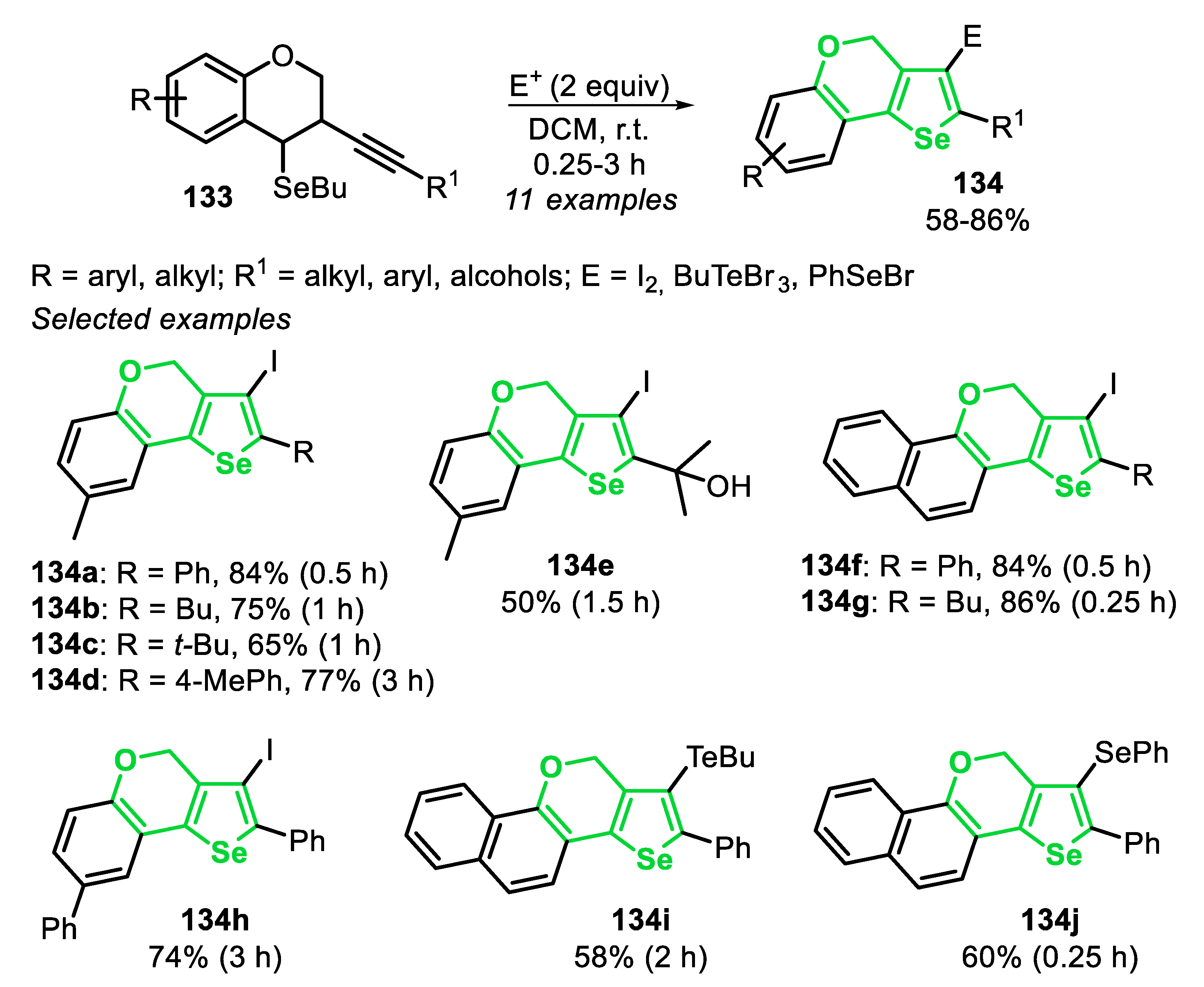
In 2015, the synthesis of 3,4-bis(butylselanyl)selenophenes 12 through the cyclization reaction between dibutyl diselenide and 1,3-diynes 11, in the presence of equimolar amounts of FeCl3 in DCM, at 40 °C for 4 h, under Ar atmosphere was described (Scheme 10) [133]. In order to achieve better results, a 1:2 ratio of 1,3-diynes 11 and dibutyl diselenide was required, once 2 equivalents of dibutyl diselenide give 4 equivalents of the reactive species BuSe−. Among them, 3 equivalents are incorporated in the final product, while 1 equivalent acts as a nucleophile in an SN2 step, to remove the butyl group directly bonded to the selenium atom. The optimal conditions were applied to a wide range of electron-rich and electron-poor symmetrical 1,3-diynes 11, affording the 3,4-bis(butylselanyl)selenophenes 12a–e in moderate to good yields. The symmetrical 1,3-diynes 11, substituted with ortho-methylaryl and naphthyl groups, reacted satisfactorily to afford the selenophenes 12f–h in moderate yields. When the reaction was carried out using dodeca-5,7-diyne, the alkyl-substituted selenophene 12i was obtained in 48% yield. In addition, the reactivity of unsymmetrical 1,3-diynes 11 was also evaluated, and the respective selenophenes 12j–l were obtained in 39–53% yields (Scheme 10).
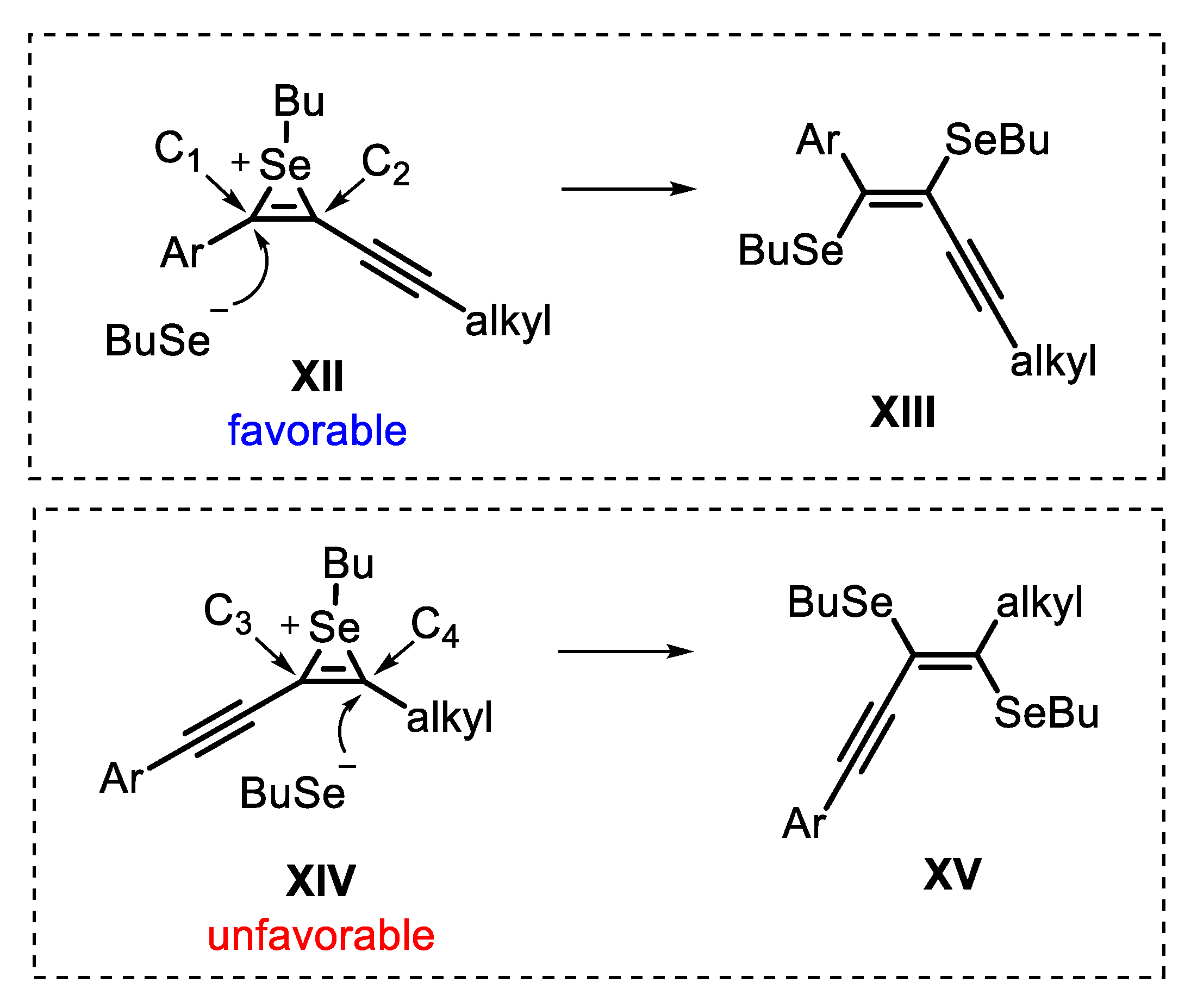
Regarding the regioselectivity of the reaction, the cyclization of unsymmetrical 1,3-diynes 11 can be led to two possible regioisomers. Thus, a detailed analysis of the key intermediates could be helpful to determine the exact position of each selenium atom, as well as which one performs the nucleophilic attack in the annulation step (Scheme 11). The first possibility is the nucleophilic anti-attack of BuSe- at C1 in the seleniranium ion XII, giving the selenoenyne XIII. On the other hand, an anti-attack at C4 in the seleniranium ion XIV, affords selenoenyne XV. Once the aromatic π-system is richer in electrons than the C-C triple bond, the aromatic ring stabilizes the seleniranium ring, favoring the formation of the intermediate XII over XIV. Due to this effect, the reaction regioselectivity is achieved, passing by the selenoenyne XIII, which is converted into the respective selenophene 12 (Scheme 11).
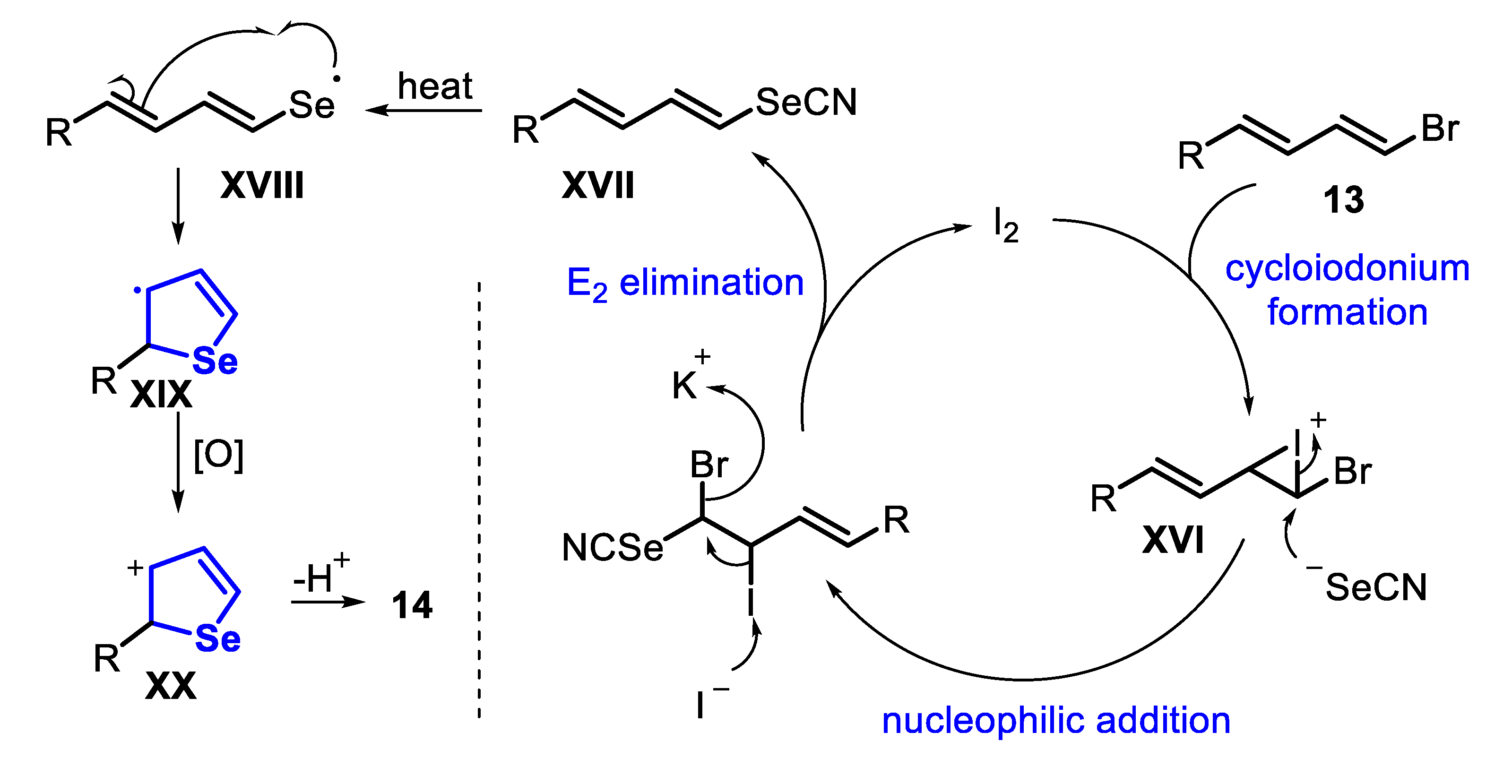
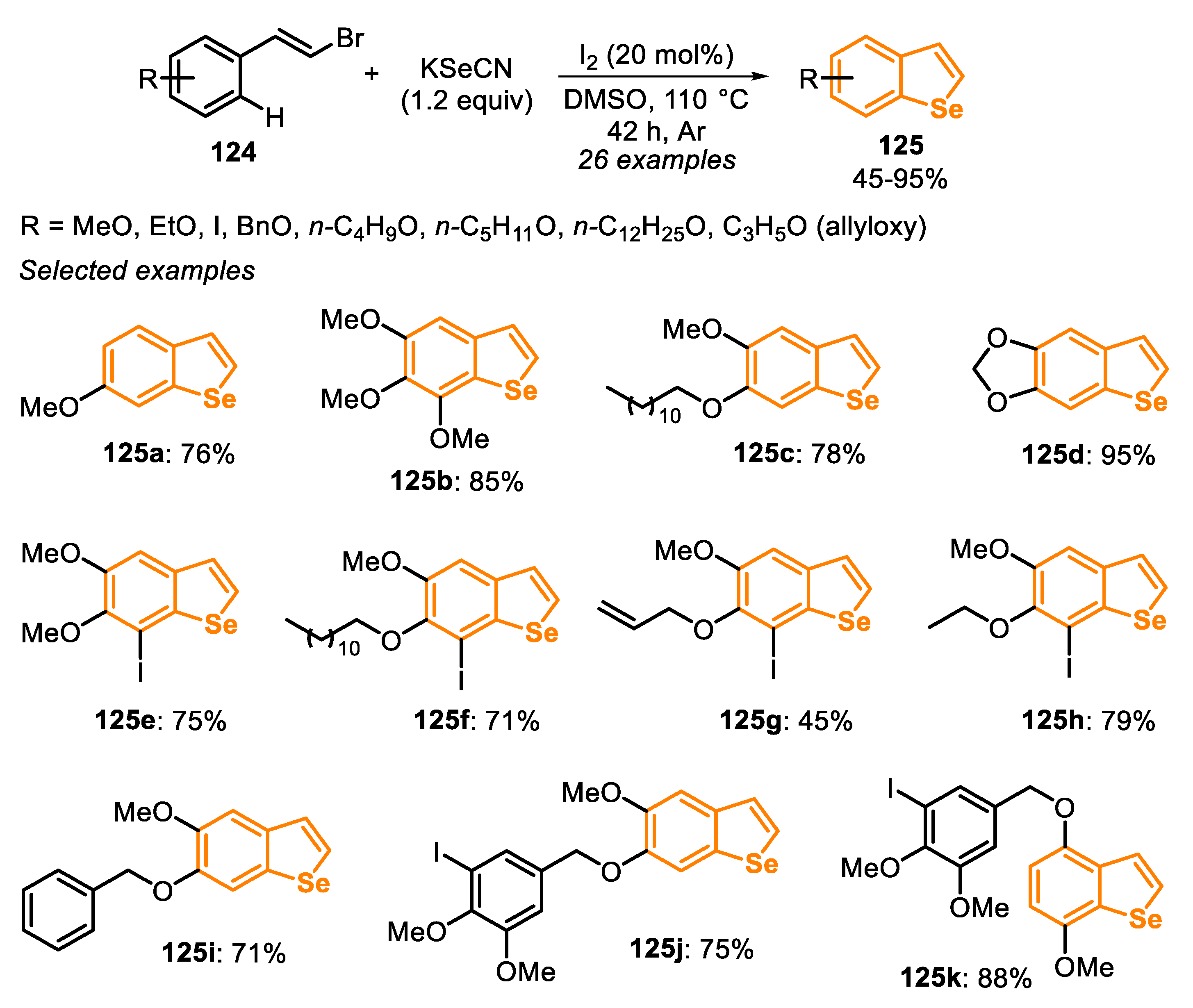
In 2017, the synthesis of selenophenes 14 by the reaction between 1,3-dienyl bromides 13 and potassium selenocyanate in the presence of I2 (20 mol%) as a benign, cheap and easily available catalyst was reported (Scheme 12) [134]. The reactions were carried out in DMSO, for 10–12 h under heating (90–100 °C) and argon atmosphere. The optimal conditions were extended to 1,3-dienyl bromides 13 bearing electron-donating (R2 = 4-MeO, 2-MeO) and electron-withdrawing groups (R2 = 4-F, 4-Cl, 4-Br, 4-NO2, 4-NH2, 2-NO2) in the aromatic ring, reaching the desired 2-arylselenophenes 14a–h in 74% to 95% yield. Reactions between KSeCN and aryl-1,3-dienyl bromides 13 bearing different groups pendant to the vinyl carbon (R1 = Cl, Ph, n-pentyl and n-hexyl) reacted smoothly to afford the products 14i–l in moderate to good yields. Additionally, anthracenyl- and furyl-1,3-dienyl bromide 13 derivatives were suitable substrates for the reaction, and the products 14m and 14o were obtained in 75% and 77% yields, respectively. The 4-[(1E,3E)-4-Bromobuta-1,3-dien-1-yl]-2-methoxyphenyl acetate 13 efficiently reacted with KSeCN to produce the selenophene 14n in 89% yield. Finally, it is worth mentioning that the 1,3,5-trienyl bromide 13 furnished the 2-vinylselenophene 14p at 55% yield, without the formation of selenepine (Scheme 12).
Control experiments suggested that the reaction mechanism involves the formation of the 1,3-dienylselenocyanate XVII as the key intermediate. Initially, 1,3-dienyl bromide 13 reacts with I2 to give a cycloiodonium XVI, which undergoes a nucleophilic addition of NCSe-, followed by an elimination step to give the 1,3-dienylselenocyanate XVII and regenerating I2. Thus, the intermediate XVII undergoes a homolytic cleavage of the Se-CN bond to give the Se-centered radical species XVIII. In the sequence, an intramolecular cyclization occurs by the reaction of the styryl C-C double bond, to give the five-membered cyclic intermediate XIX. The radical intermediate XIX is then oxidized to the carbocation XX, which is finally converted into the desired selenophene 14 through a proton elimination step (Scheme 13).
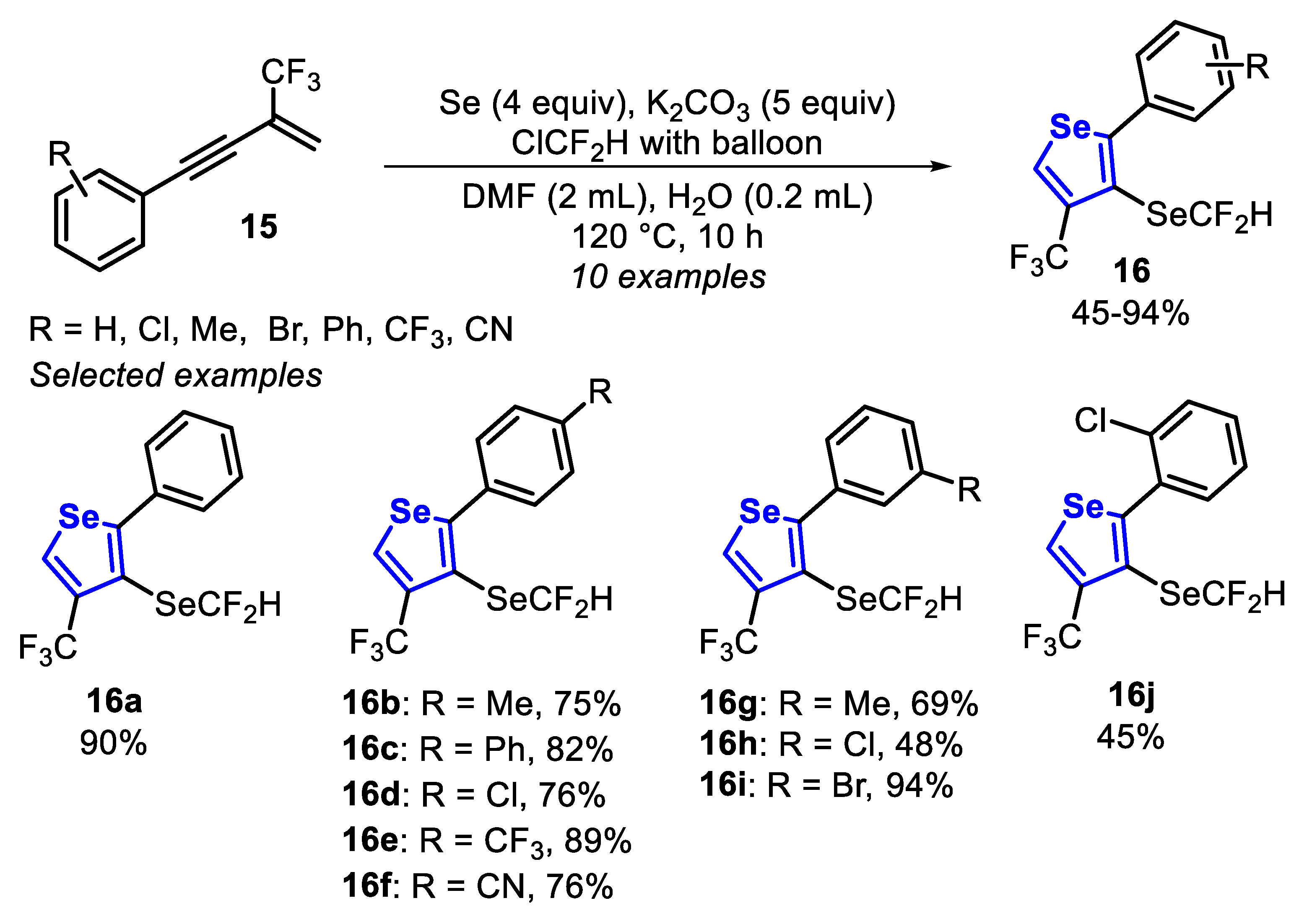
Recently, a divergent strategy was developed for the construction of 3-SeCF2H-4-CF3-selenophenes 16 from readily available CF3-containing 1,3-enynes 15 and elemental Se through a tandem selenophene construction/selective C3-selenation/difluoromethylselenation, under a ClCF2H atmosphere (in a balloon) in the presence of K2CO3 and DMF at 120 °C for 10 h (Scheme 14) [135]. In general, the reaction to access the products 16a–i was efficient and robust once the substituents of the enyne 15 did not influence the reactivity. It is worth mentioning that steric effects were faced when the ortho-chlorophenyl pendant to the triple bond was employed, affording the expected product 16j in 45% yield (Scheme 14).
2.2. Starting from Type B Precursors
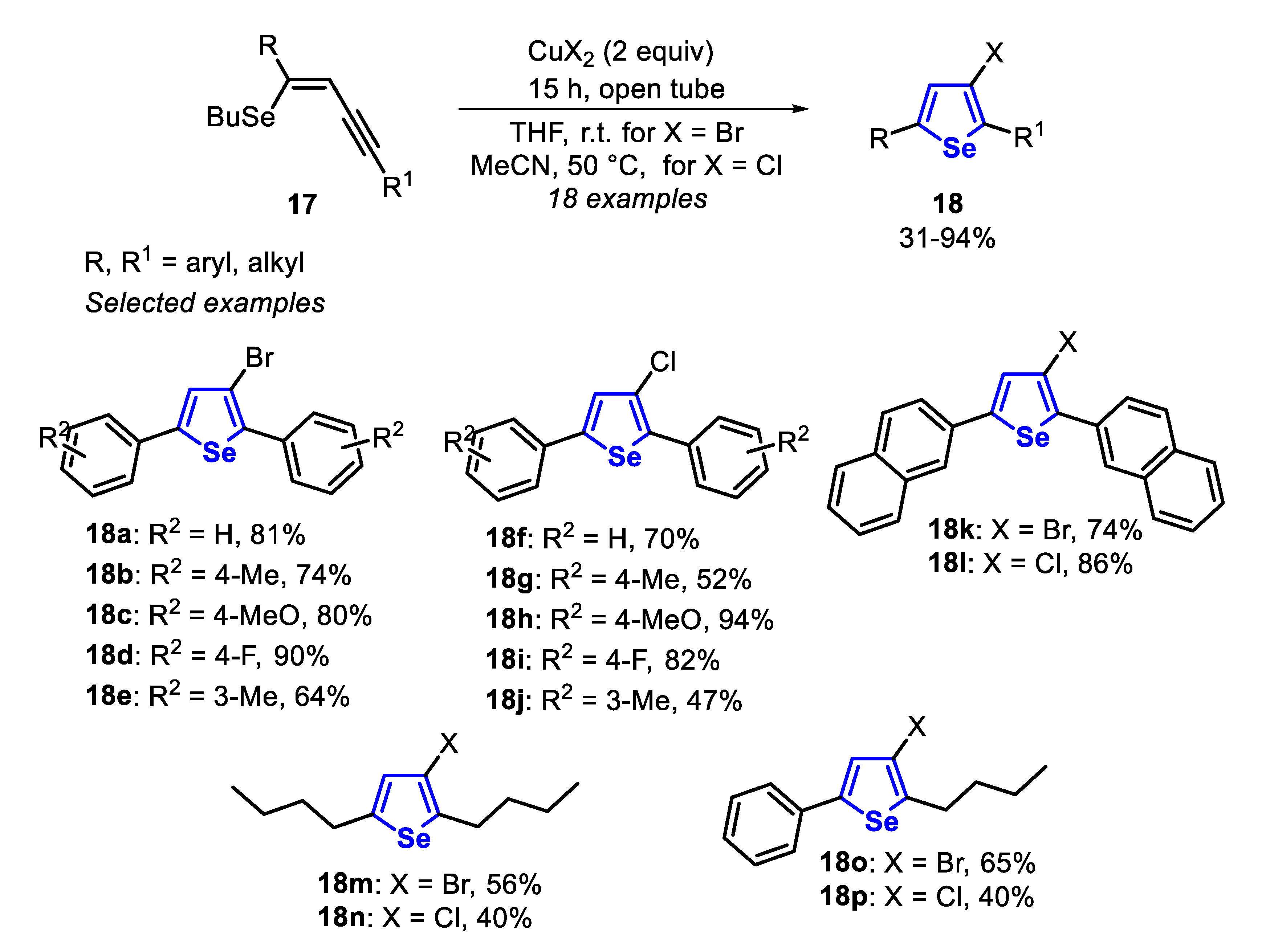
In 2011, the intramolecular 5-endo-dig cyclization of (Z)-selenoenynes 17 mediated by copper(II) salts (CuBr2 at room temperature in THF, or CuCl2 at 50 °C in MeCN) to afford the corresponding 3-halochalcogenophenes 18 was reported (Scheme 15) [136]. Under these conditions, (Z)-selenoenynes 17, substituted with neutral phenyl, electron-rich (R2 = 4-Me, 4-MeO, 3-Me) and electron-deficient (R2 = 4-F) aryl groups afforded the respective 3-haloselenophenes 18a–j in moderate to good yields after 15 h. When bulky (Z)-selenoenyne was employed, the products 18k and 18l were obtained in 74% and 86% yields, respectively. The reaction was also tolerant to alkyl groups directly bonded to the alkyne, producing the desired 4-haloselenophenes 18m–p in moderate yields (Scheme 15).
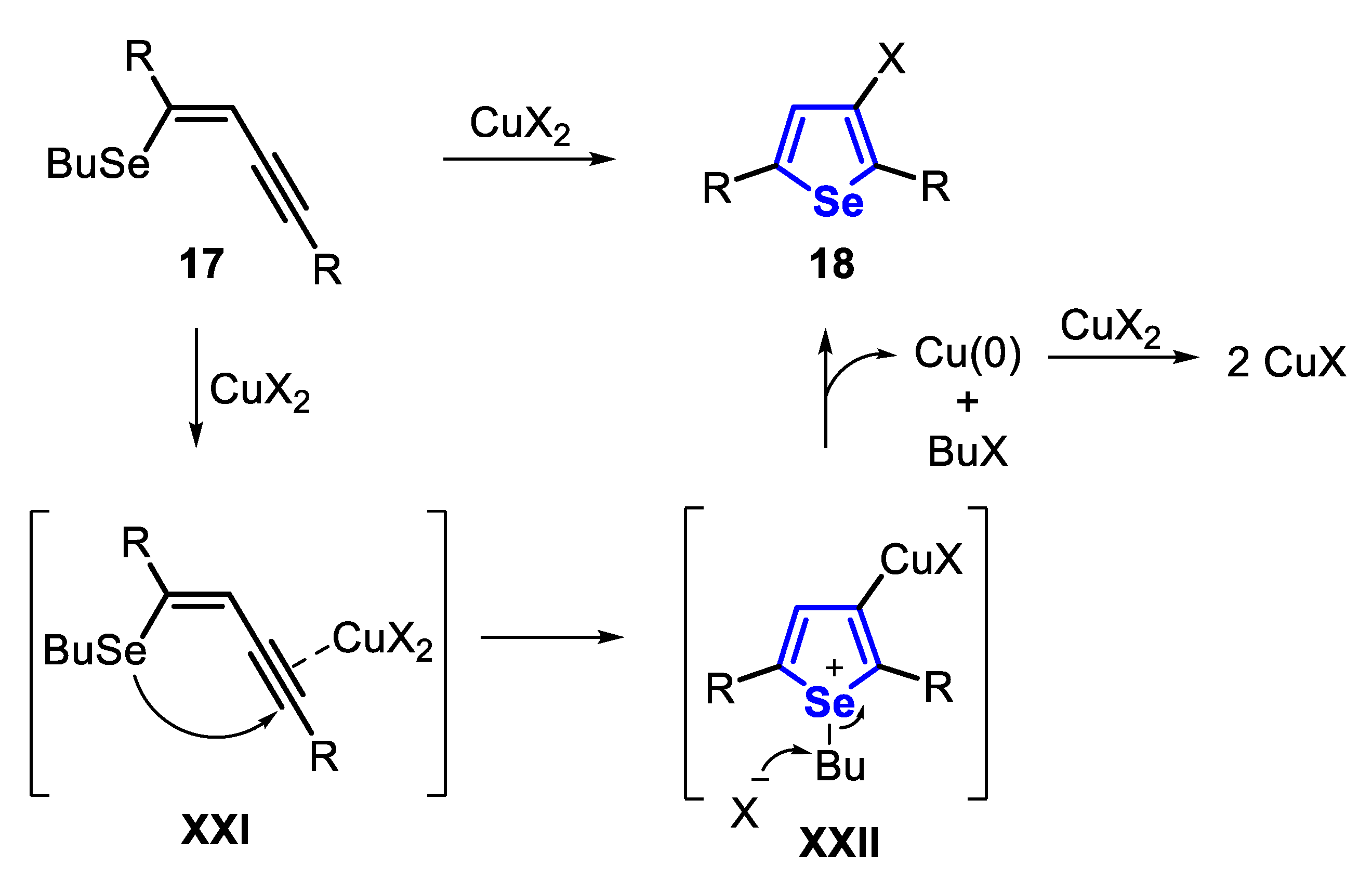
The proposed mechanism starts with the coordination of the C-C triple bond to the copper salt, generating the intermediate XXI. In the sequence, an intramolecular anti-attack of the selenium atom to the activated triple bond affords the selenonium salt XXII. After this, an SN2 displacement of the butyl group delivers the 3-haloselenophene derivative 18, Cu(0) and 1-halobutane as a co-product. The formed Cu(0) can be oxidized by CuX2 to CuX, which explains the need for two equivalents of CuX2 in this reaction (Scheme 16).
In order to demonstrate the applicability of the synthesized compounds, 3-bromo-selenophene 18a was subjected to the Pd-catalyzed Suzuki cross-coupling reaction with arylboronic acids. The reaction between the selenophene 18a and electron-rich (R = Me, MeO) and deficient (R = Cl, COMe) arylboronic acids afforded the corresponding products 19a–d in 56–80% yields (Scheme 17).
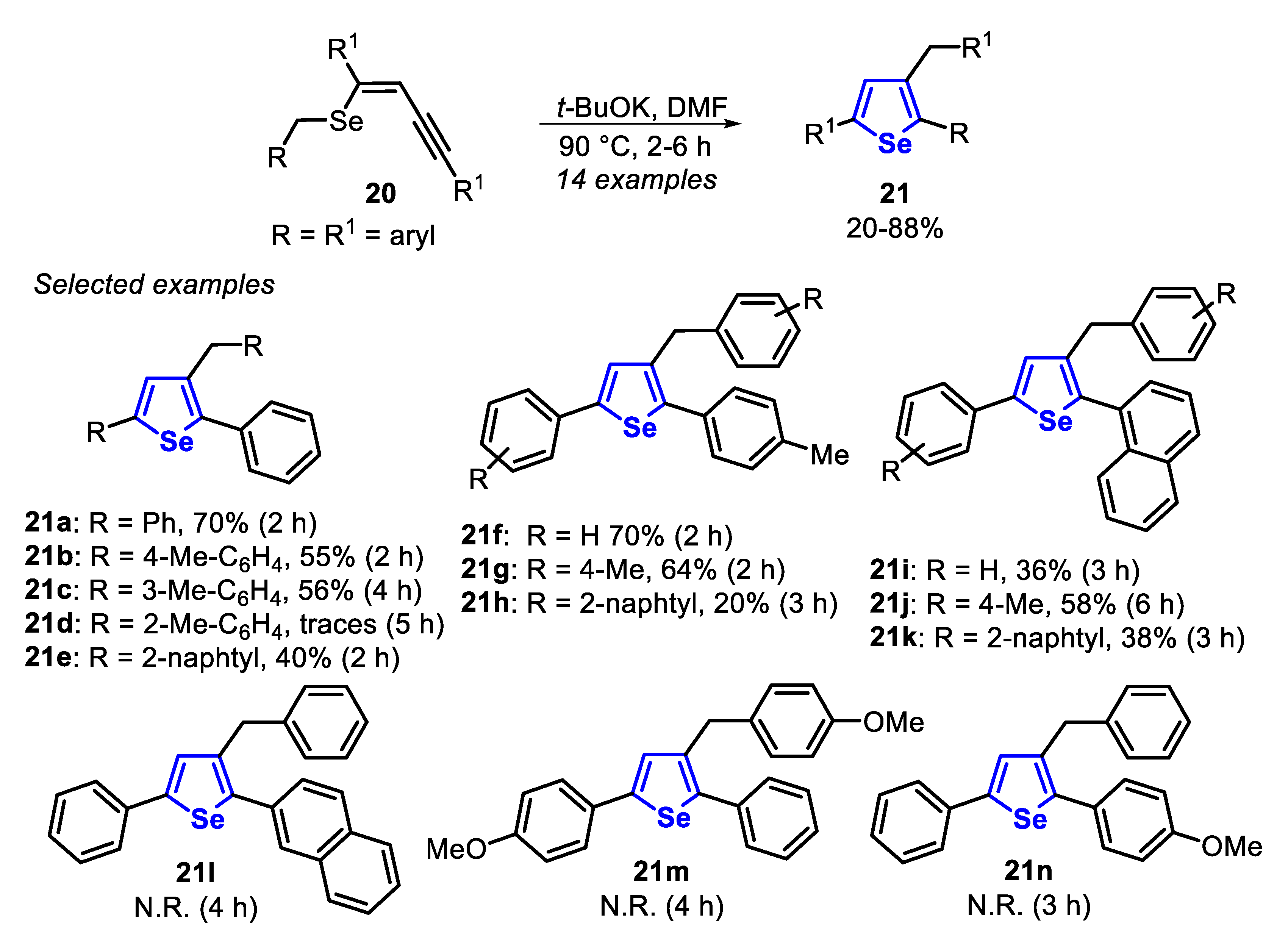
In the same year, the carbocyclization of (Z)-benzylselenoenynes 20, using t-BuOK in DMF, at 90 °C for 2 to 6 h, to prepare the 3-benzyl-2,5-diarylselenophenes 21 (Scheme 18) was described [137]. Firstly, in order to evaluate the influence of the substituent at the pendant aromatic ring of the C-C triple bond (R1), the authors fixed the vinyl selenobenzyl group. The efficiency of the cyclization was significantly influenced by steric effects of the aromatic ring, affording the ortho-substituted selenophenes 21d and 21e in lower yields than the respective neutral, para- and meta-substituted ones, 21a, 21b and 21c. In addition, when the (Z)-benzylselenoenynes 20, bearing different substituents at the BnSe-vinyl groups, were used, the corresponding selenophenes 21f-l were obtained in low to good yields. Limitations were found when the electron-donor MeO group was attached at the para-position of the pendant aromatic ring of the C-C triple bond (R1) or of the BnSe-vinyl group. In these cases, the desired selenophenes 21m and 21n were not formed (Scheme 18).
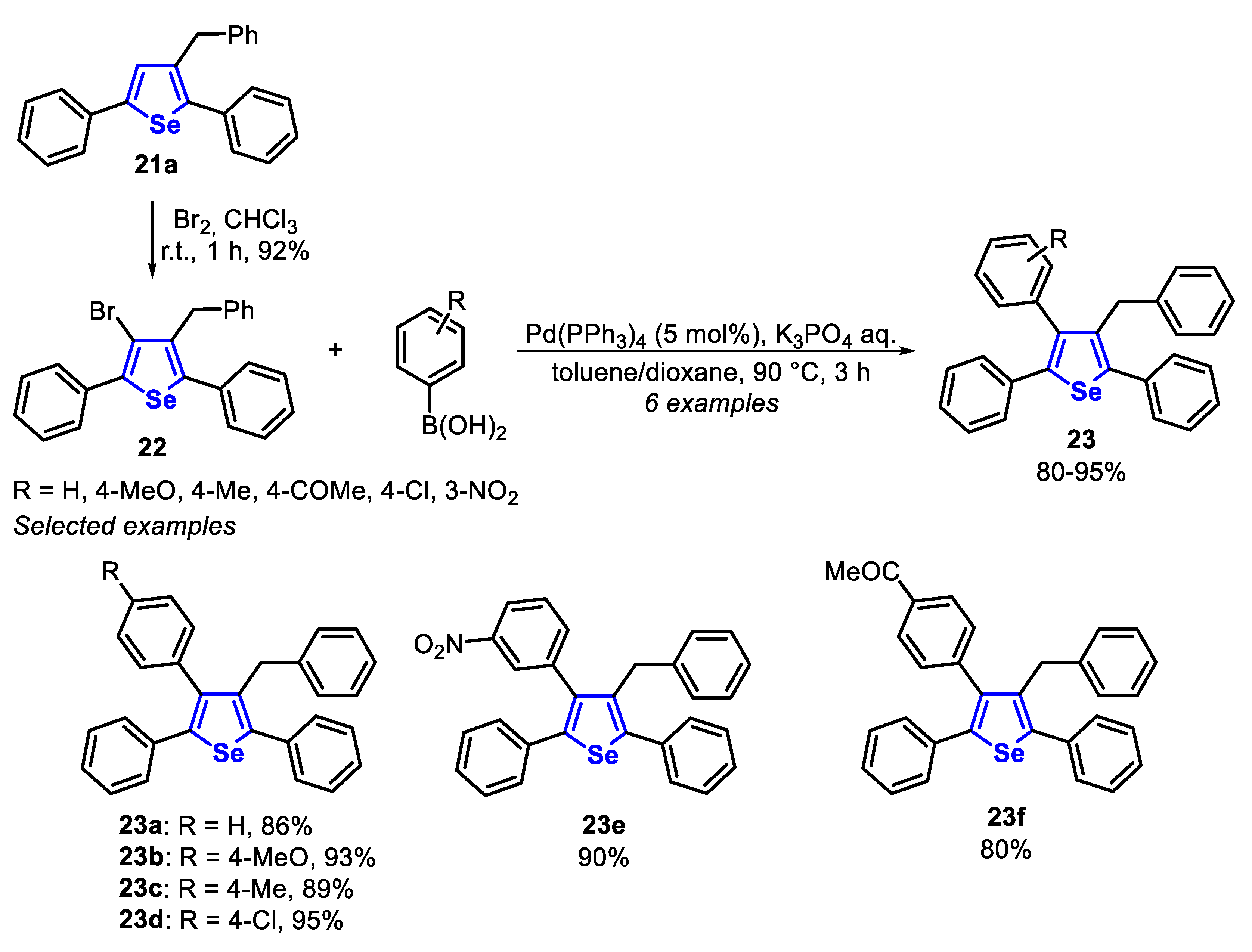
The synthetic versatility of the synthesized compounds was demonstrated in the Pd-catalyzed Suzuki reaction using different arylboronic acids. Initially, the selenophene 21a was functionalized by the reaction with Br2 (2.5 equiv) in CHCl3, at room temperature, to afford the 4-Br substituted selenophene 22 in 92% yield. The functionalized selenophene 22 was then submitted to the reaction with different electron-rich and electron-deficient arylboronic acids in the presence of Pd(PPh3)4 (5 mol%) and an aqueous solution of K3PO4 (1 mmol), using a 1:1 mixture of dioxane/toluene as solvent, at 90 °C for 3 h. The reaction was not sensitive to electronic effect, and electron-rich and electron-poor arylboronic acids gave equally good yields of the respective selenophenes 23a–f (Scheme 19).
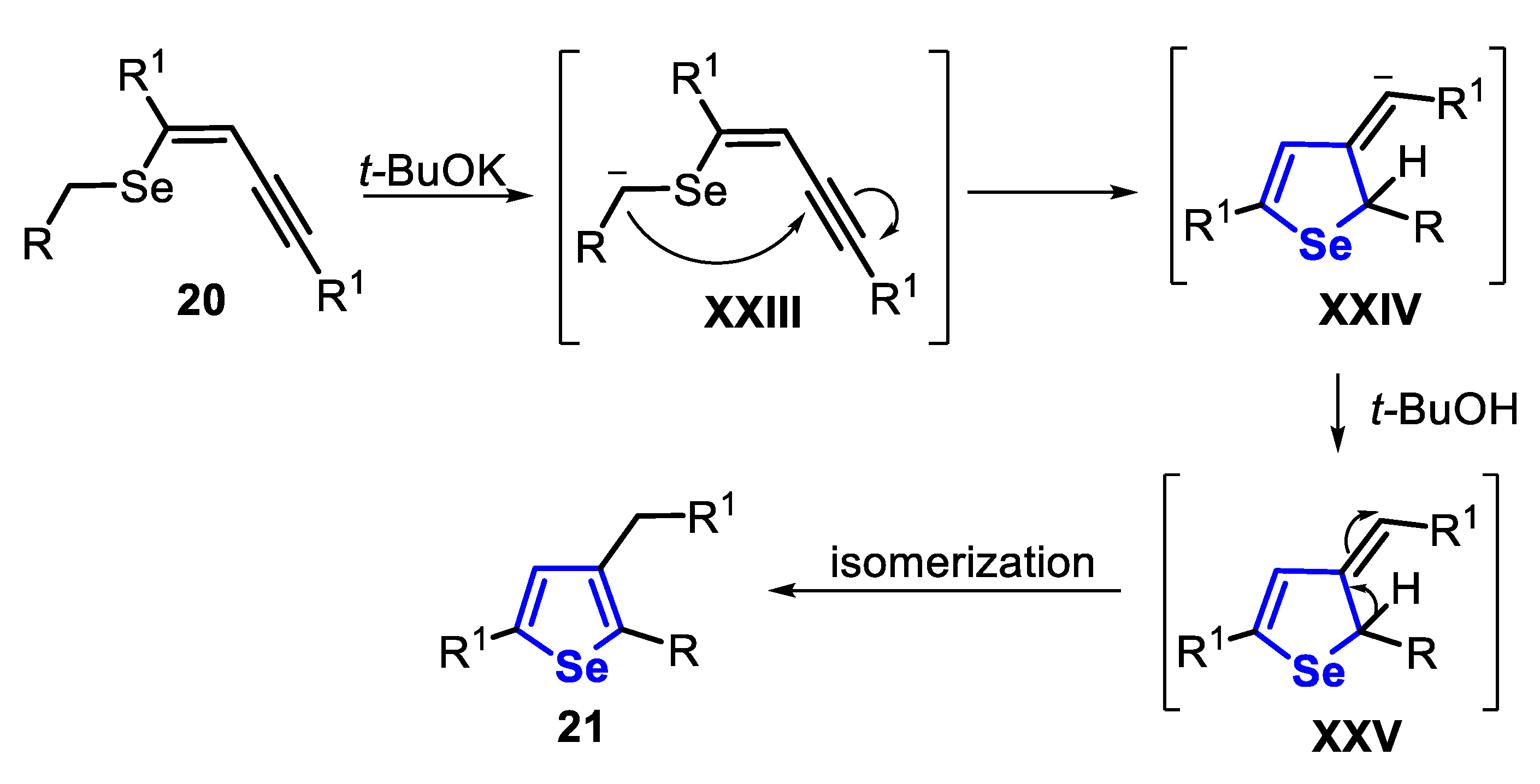
The proposed mechanism for the synthesis of the product 21 initiates with the deprotonation of the (Z)-benzylselenoenyne 20 by t-BuOK to generate the α-Se-stabilized carbanion XXIII, which undergoes an intramolecular cyclization to produce the vinyl carbanion intermediate XXIV. Thus, the intermediate XXIV is protonated, being converted to the intermediate XXV, which finally undergoes an isomerization reaction to afford the desired selenophene 21 (Scheme 20).
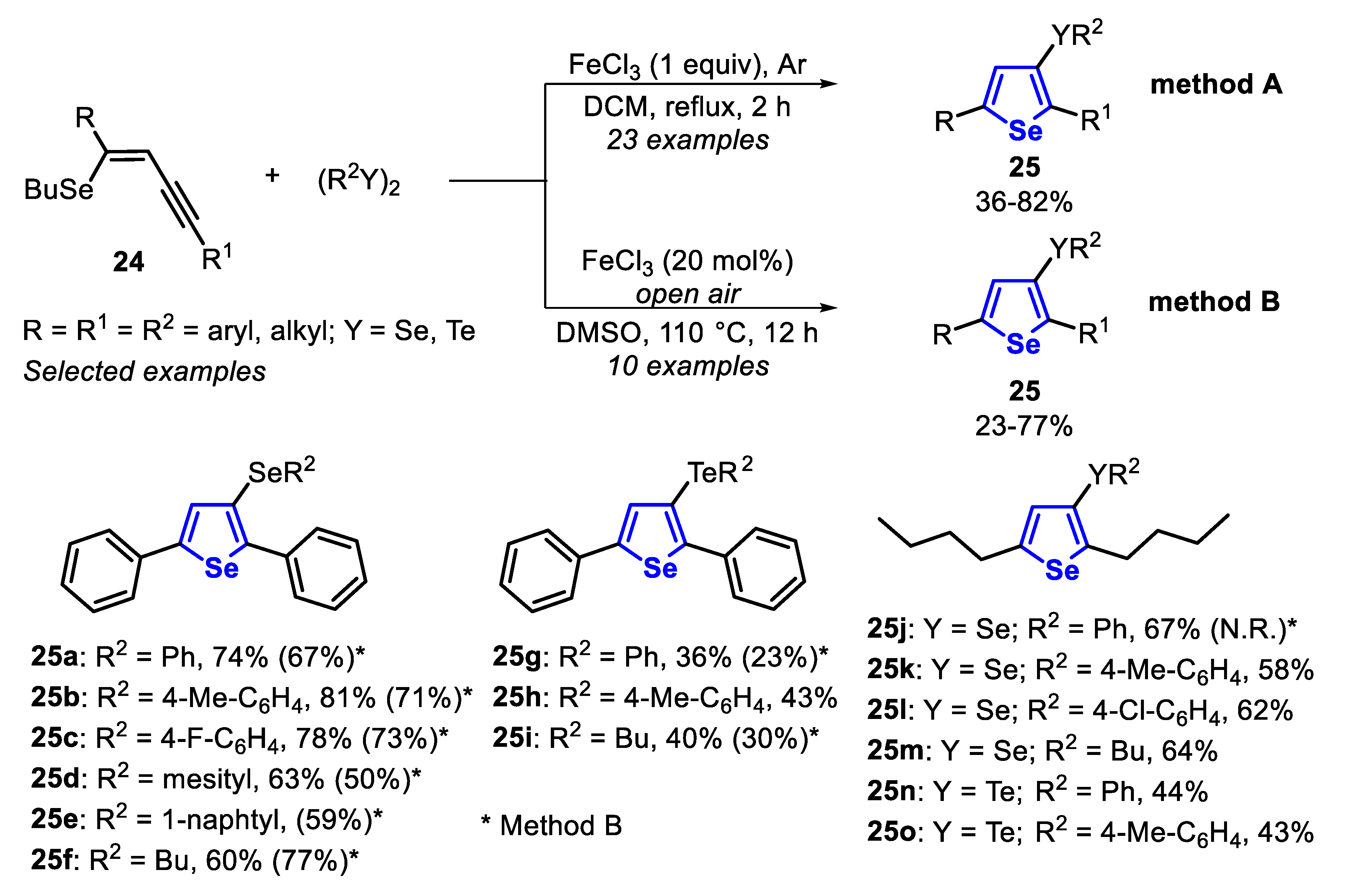
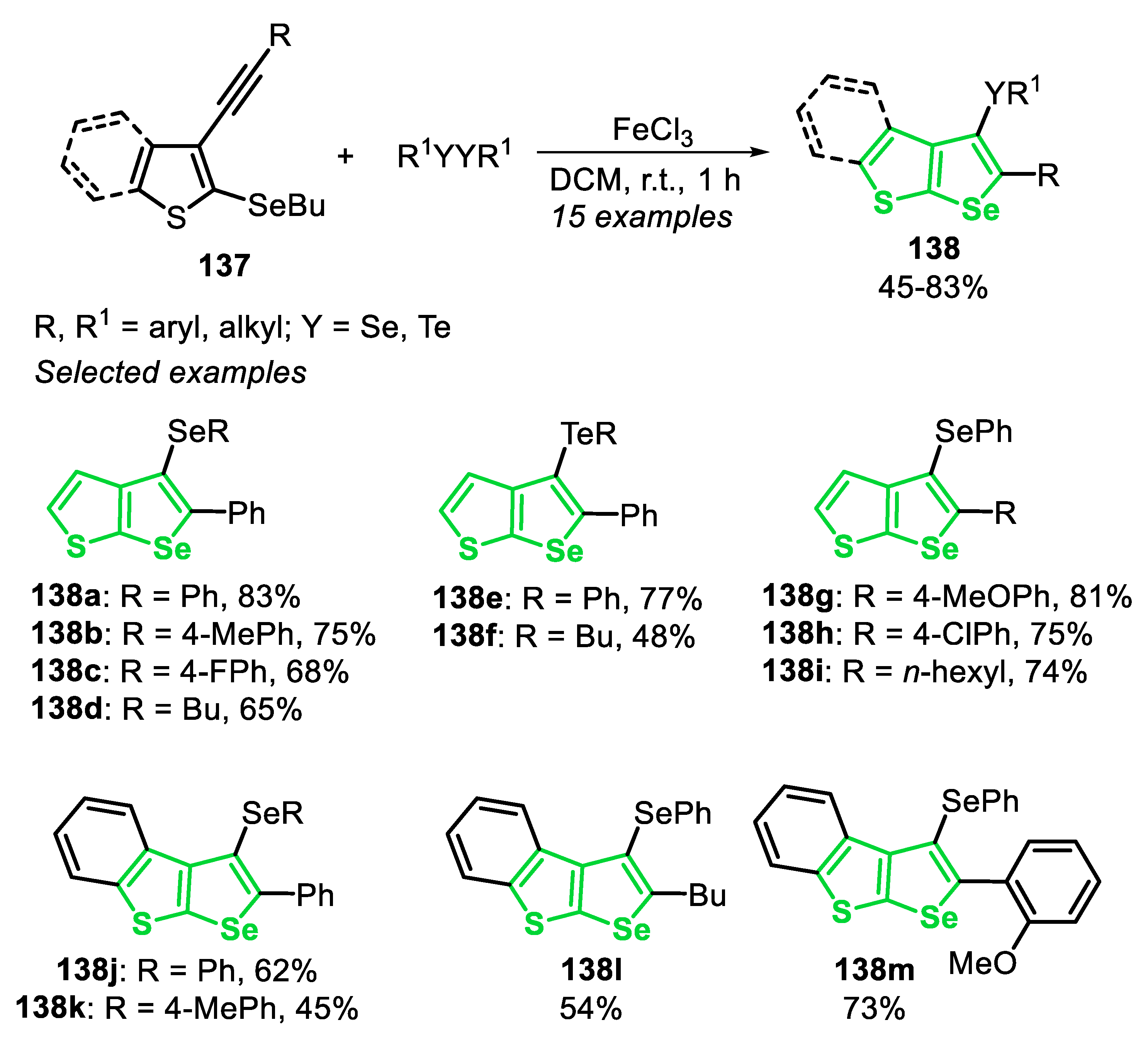
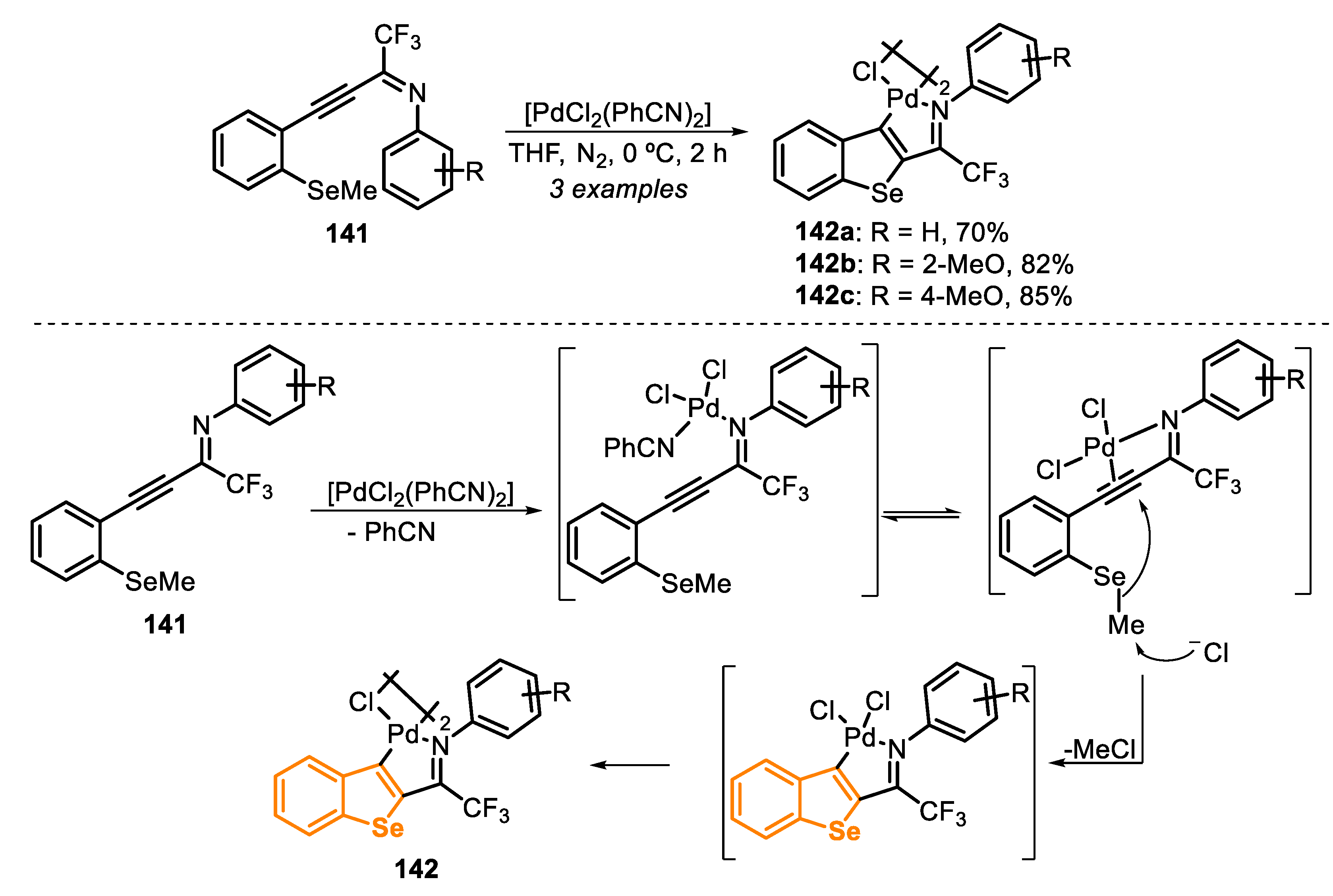
In 2012, a FeCl3/dichalcogenide-mediated intramolecular cyclization of (Z)-selenoenynes 24 for the synthesis of 2,5-disubstituted-3-(organoseleno)-selenophenes 25 [24] was reported through two different methods: using one equivalent or a catalytic amount (20 mol%) of FeCl3. In method A, using a stoichiometric amount of FeCl3, the (Z)-selenoenynes 24 reacted with diorganyl dichalcogenide (0.55 equiv), in DCM at 40 °C, under argon atmosphere, while in method B (20 mol% of FeCl3) the reaction was conducted in DMSO at 110 °C in an open flask condition (Scheme 21). The protocol has proved to be robust and general, affording 33 different derivatives of the 2,5-disubstituted-3-(organoseleno)-selenophenes 25. No significant differences were found between electron-rich and electron-deficient diorganyl dichalcogenides, and the selenophenes 25a–c and 25g–h were accessed in moderate to good yields. However, the reaction efficiency was hampered when diorganyl diselenides bearing sterically hindered groups (R2 = mesityl or 1-naphthyl) were employed, giving the selenophenes 25d and 25e in moderate yields, using both methods. Regarding the dichalcogenide reaction partner, the results suggest that electronic effect did not influence the reaction directly; however, the transformation is sensitive to steric effects. Additionally, dialkyl diselenides and ditellurides were satisfactorily employed as a substrate, giving the products 25f and 25i in acceptable yields. It is worth mentioning that, in general, the results reached with diorganyl diselenides are consistently better than those obtained with ditellurides. This trend is intricately linked to the fact that tellurides are more susceptible to undergoing a telluroxide elimination reaction than the corresponding selenides. The protocols were successfully applied to alkyl selenoenynes 24, in the presence of a wide range of functionalized diorganyl dichalcogenides, giving the respective selenophenes 25j–o in moderate to good yields (Scheme 21).
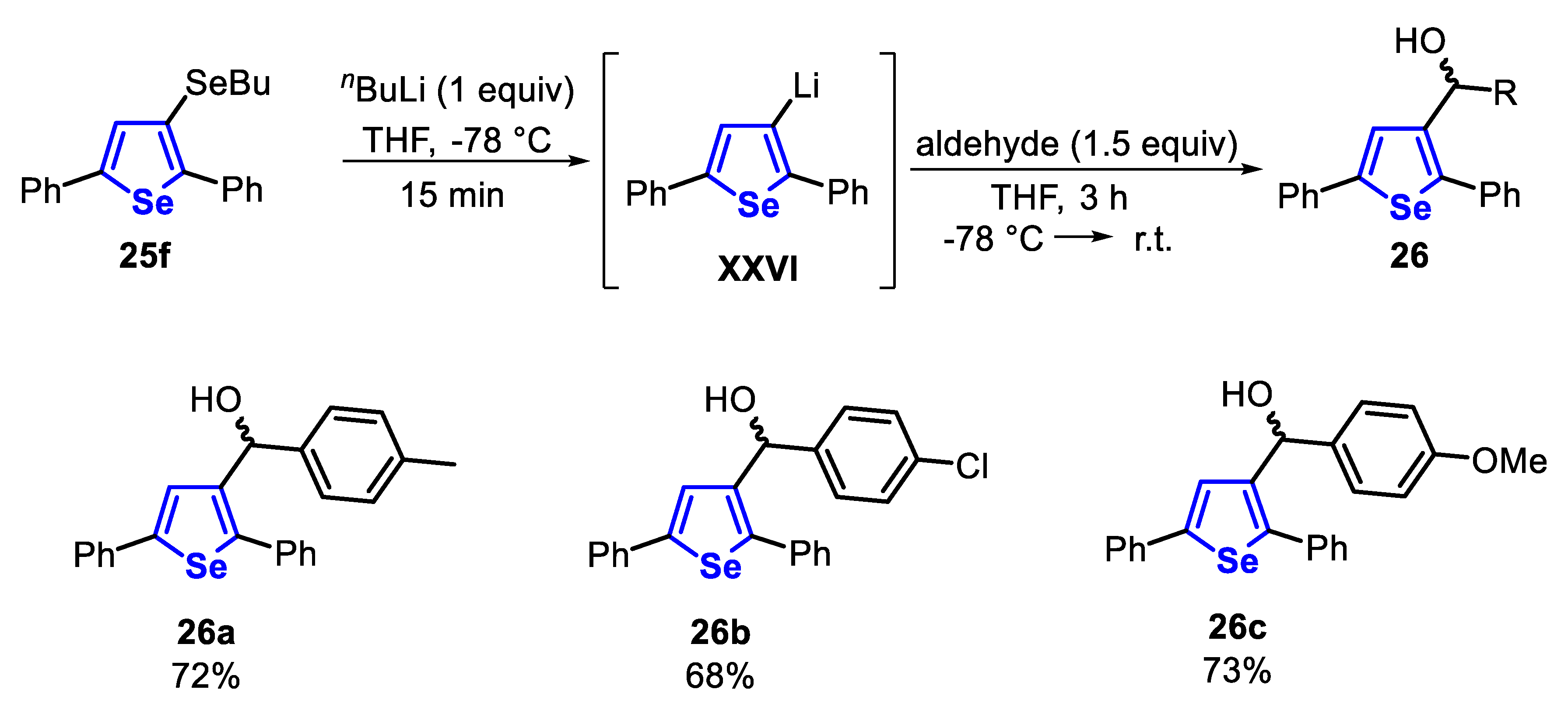
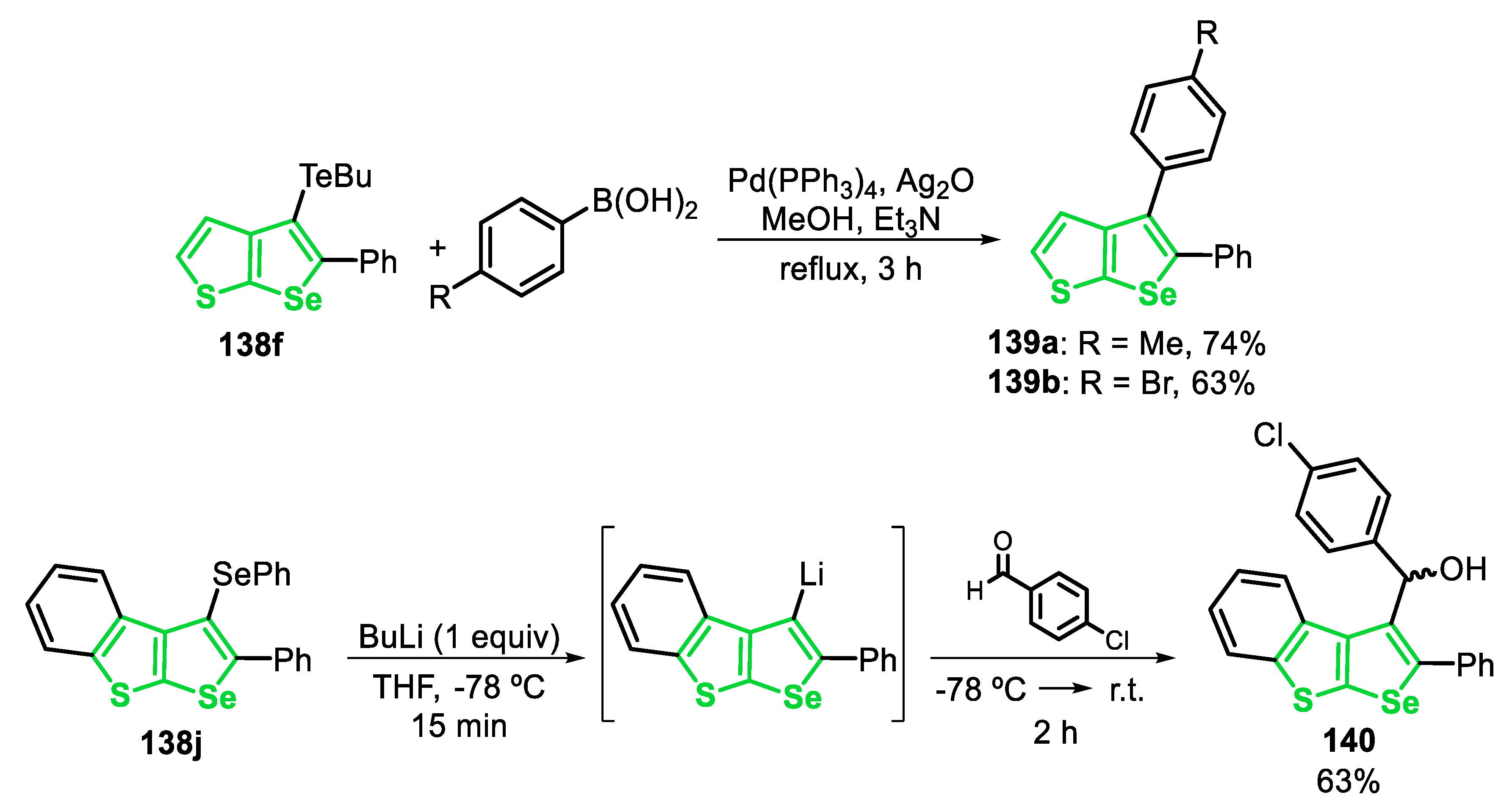
The reactivity of the 2,5-disubstituted-3-(organoseleno)-selenophenes 25 was explored in the transmetalation Li/Se. Thus, the selenophene 25j reacted with butyllithium (1 equiv) in THF at −78 °C, affording the Li-containing species XXVI. After this, the species XXVI was trapped with aldehydes (1.5 equiv) at −78 °C and the system was allowed to reach the room temperature. The secondary alcohols 26a-c were satisfactorily obtained in 68–73% yields after 3 h (Scheme 22).
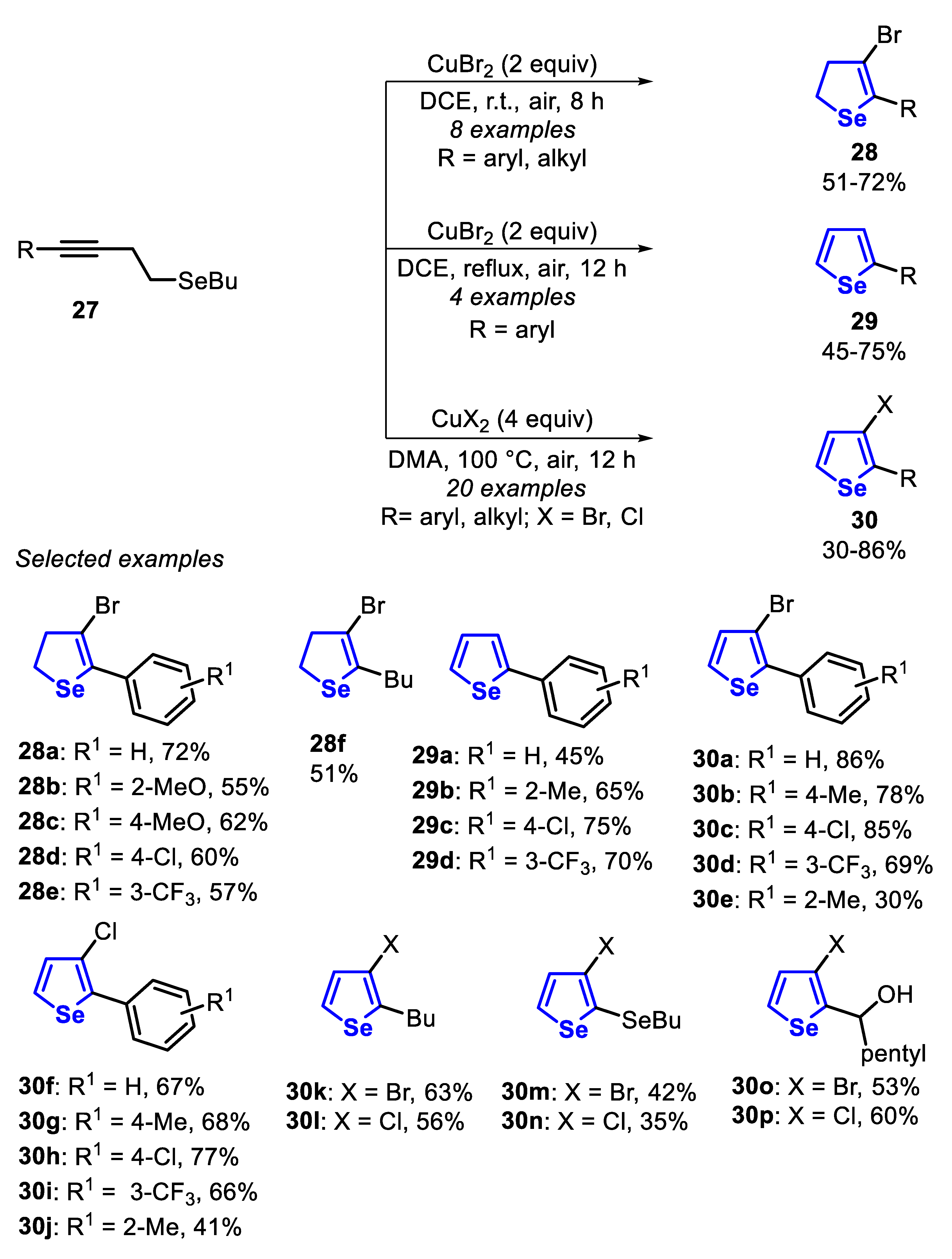
In 2013, the Cu(II)/halide-mediated cyclization of homopropargyl selenides 27, to give selectively 2,3-dihydroselenophenes 28, 2-arylselenophenes 29 and 3-haloselenophenes 30 was reported (Scheme 23) [138]. The selectivity of the reaction was achieved by controlling the solvent and the reaction temperature. Thus, using CuBr2 (2 equiv) and 1,2-dichloroethane, at room temperature under air atmosphere, the 4-bromodihydroselenophenes 28 were obtained after 8 h. Electron-rich and electron-deficient homopropargyl selenides 27 reacted smoothly to produce the 3-bromodihydroselenophenes 28a–e in moderate yields. Intriguingly, when the ortho-alkynylanisole selenide 27 was used, the dihydroselenophene 28b was exclusively obtained, and the benzo[b]furan derivative, a possible by-product, was not detected. This remarkable selectivity may be attributed to the electronic effect (relative nucleophilicity of the Se atom and the cationic nature of the intermediate), as well as to the resistance of the methoxyl group to undergo a demethylation, followed by a ring closure. The dihydroselenophene 28f, derived from the homopropargyl selenide bearing alkyl group (R = Bu), was obtained at a lower yield of 51%. When the reaction was conducted at a reflux instead of at room temperature, 2-arylselenophenes 29 were obtained. Neutral- and electron-rich homopropargyl selenides 27 (R1 = H and 2-Me) afforded the products 29a and 29b in 45% and 65% yield, respectively, while electron-deficient substrates (R1 = 4-Cl and 3-CF3) were more efficient, delivering the products 29c and 29d in 75% and 70% yield, respectively (Scheme 23).
When the reaction was performed using dimethylacetamide (DMA) as a solvent at 100 °C, the reaction selectivity was changed and 3-halo-selenophenes 30 were exclusively obtained. It is worth mentioning that this protocol is not influenced by the electronic effect and the selenophenes 30a–d, f–i were obtained in moderate to very good yields. However, the steric hindrance affected the reactivity, and a remarkable decrease in the efficiency was observed when the ortho-Me substituent was present, and the products 30e and 30j were obtained in 30% and 41% yields, respectively. This optimized condition was suitable for differently substituted homopropargyl selenides 27 (R = Bu, BuSe and propargyl alcohol), affording the respective 3-bromo- and 4-chloroselenophenes 30k-p in poor to moderate yields (Scheme 23).
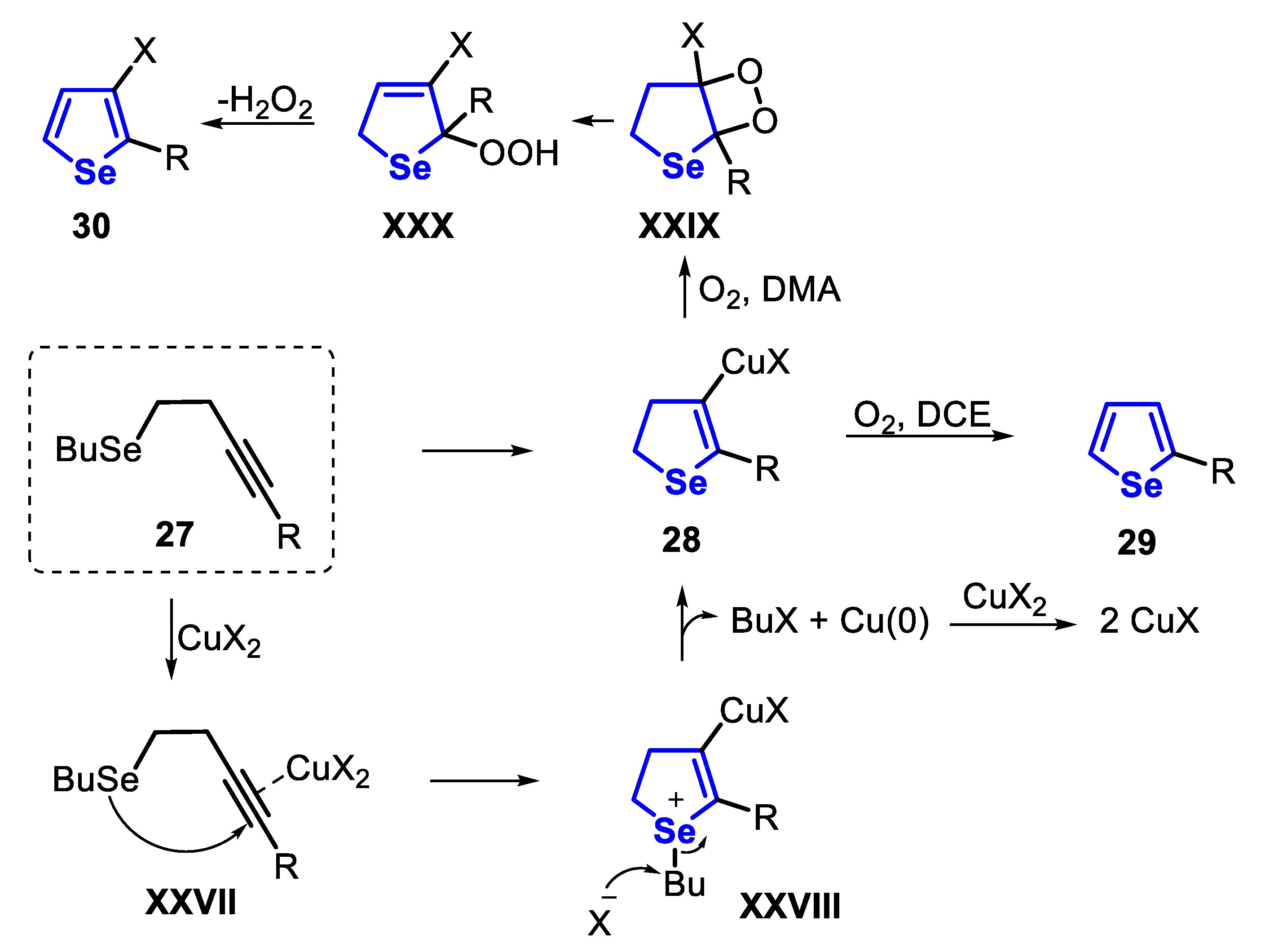
The proposed mechanism of the cyclization reaction of the homopropargyl selenides 27 starts with the coordination between the Cu catalyst and the C-C triple bond generates the intermediate XXVII, which undergoes an anti-attack of the Se to the activated triple bond, to produce the selenonium intermediate XXVIII. Finally, a reductive elimination yields the 3-halodihydroselenophene derivative 28, which, in the presence of atmospheric oxygen, can be oxidized to the selenophene 29. The Cu(0) species reacts with CuX2 spontaneously to produce two equivalents of CuX. On other hand, the synthesis of the 3-haloselenophenes 30 is performed in the presence of DMA at 100 °C under air atmosphere via an oxygen-promoted oxidation reaction of the dihydroselenophene 28, through hydroperoxide intermediates XXIX and XXX (Scheme 24).
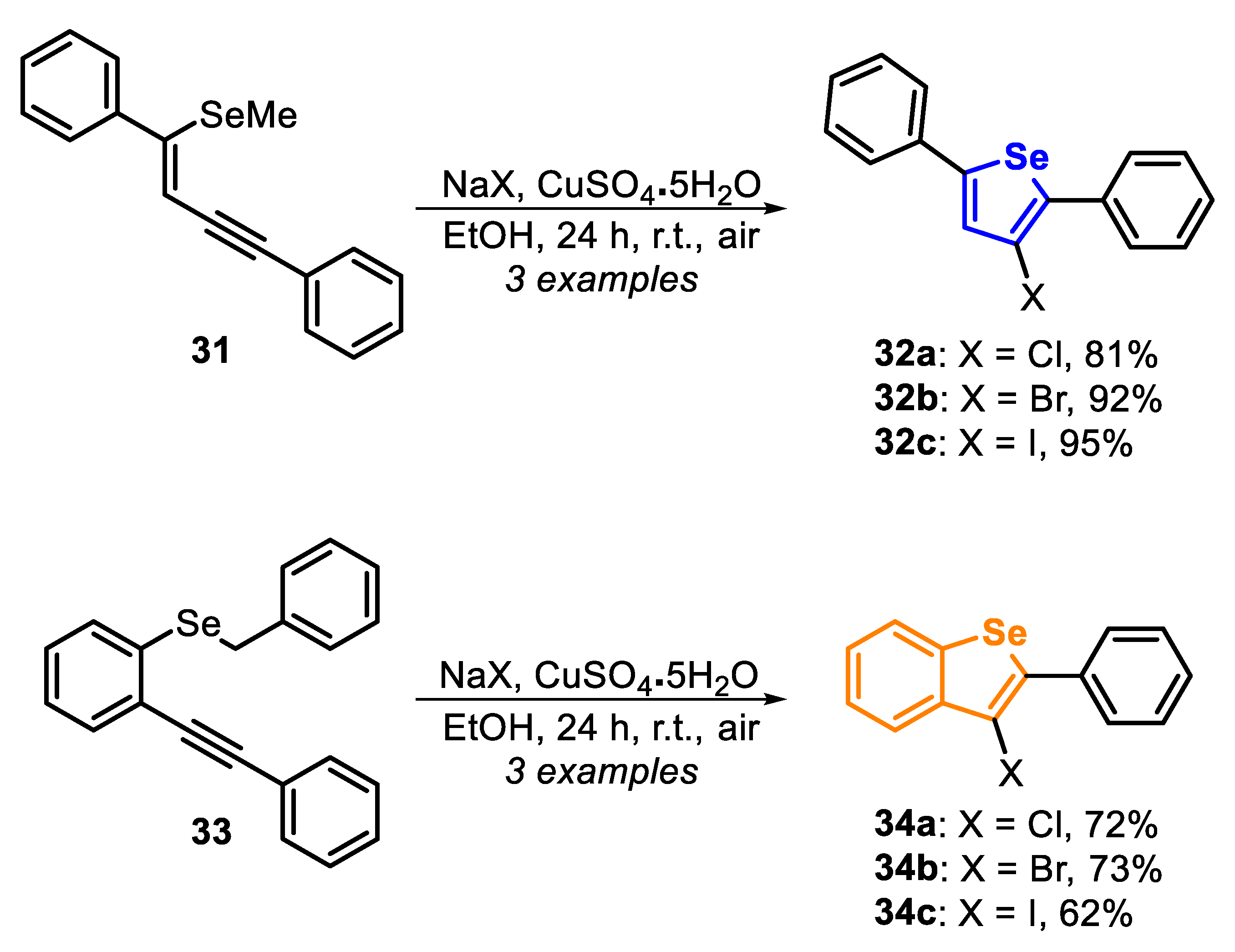
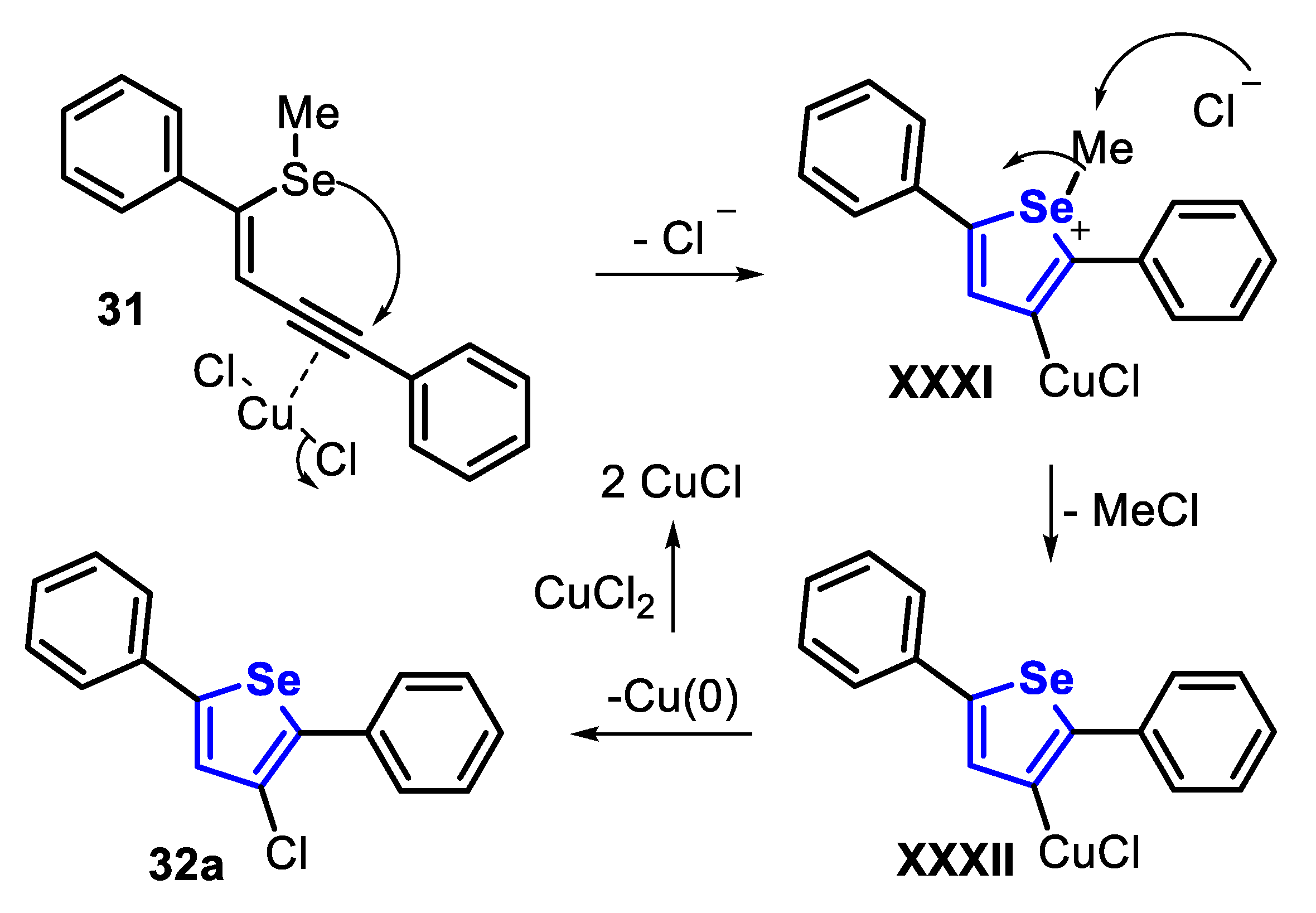
In 2017, the green synthesis of halogenated selenophenes 32 and benzo[b]selenophenes 34 through the electrophilic cyclization of selenoenyne 31 and selenoalkyne 33, using sodium halides as electrophilic reaction partner, in EtOH, at room temperature and under air atmosphere was described [139]. When the enyne 31 was submitted to the optimized condition, the desired chloro-, bromo-, and iodoselenophenes 32a–c were satisfactorily accessed in 81%, 92% and 95% yields, respectively. The authors claimed that this was the first Cl-promoted cyclization of the 2-alkynylmethylselenobenzene 33, affording the respective 3-chlorobenzo[b]selenophene 34a in 72% yield. In addition, this protocol was extended to the synthesis of the 3-bromobenzo[b]selenophene 34b and 3-iodobenzo[b]selenophene 34c in 73% and 62% yield, by using NaBr and NaI, respectively (Scheme 25).
The first step in the synthesis of chloroselenophene 32a is the in situ generation of CuCl2, by the reaction between Cu2SO4·5H2O and NaCl. Then, the resulting CuCl2 weakly coordinates to the C-C triple bond of selenoenyne 31, which undergoes an intramolecular anti-attack from the nearby Se nucleophile, to afford the selenonium intermediate XXXI. The methyl group can be subsequently removed by an SN2 displacement, promoted by the chloride anion, to form the intermediate XXXII. Finally, a reductive elimination leads to the desired product 32a. The resulting Cu(0) is easily oxidized by CuCl2 to produce CuCl (Scheme 26). Similarly, Cu2SO4·5H2O in the presence of NaI or NaBr results in the formation of CuI2 or CuBr2, respectively, which are converted in situ to I2 and Br2. The mechanism of the cyclization involving I2 and Br2 electrophiles is well established [140].
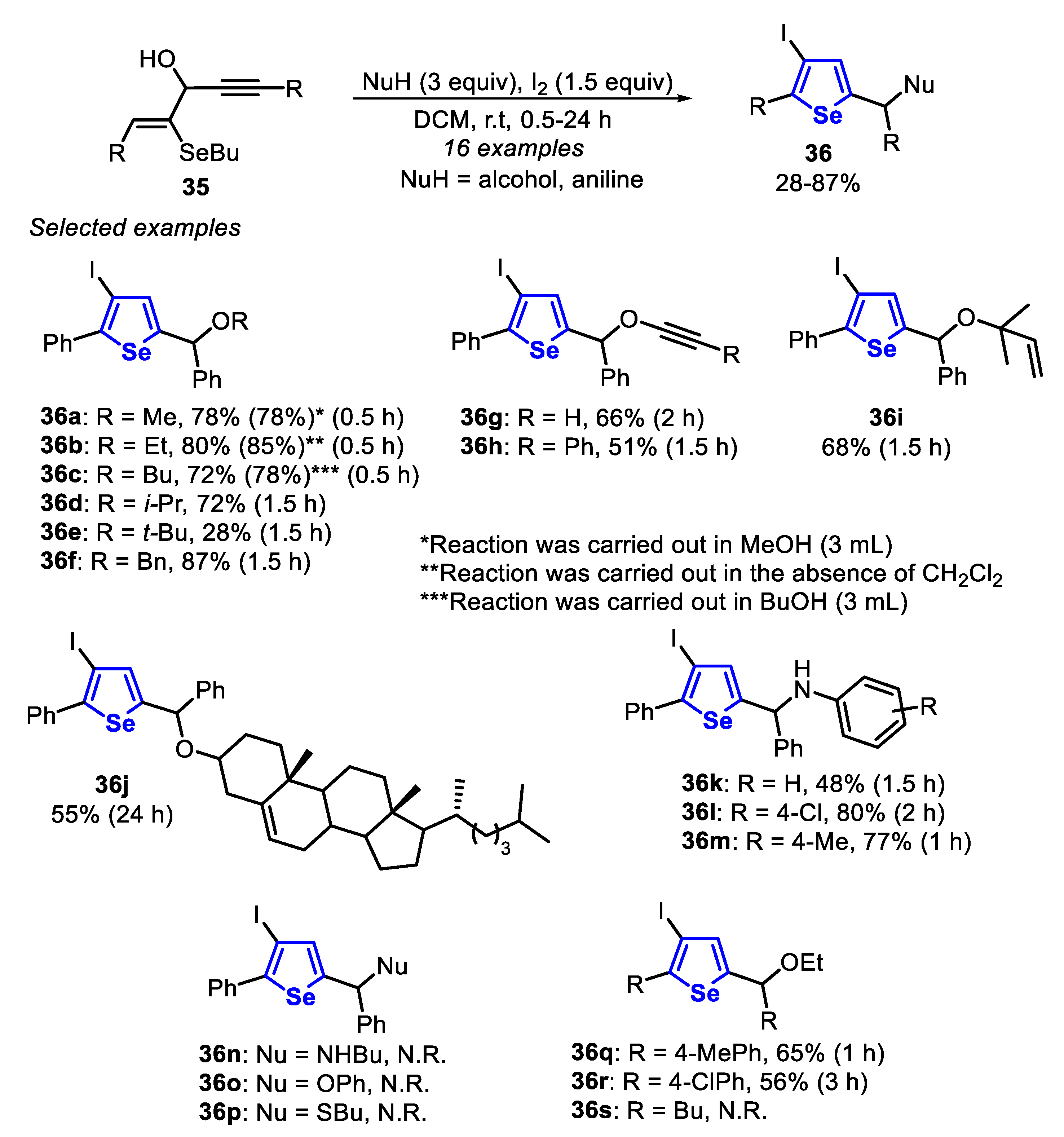
In 2017, the I2-promoted electrophilic cyclization of selenoenynes 35, in the presence of an appropriate nucleophile, to give the 3-iodo-selenophenes 36 and the 3-organoselanyl-selenophenes 37 was described (Scheme 27 and Scheme 28) [141]. The optimized condition to prepare the 3-iodo-selenophenes 36 involves the addition of I2 (1.5 equiv) and a nucleophile (3 equiv) to a solution of selenoenynes 35 in DCM. The resulting mixture was stirred at room temperature and in an open flask for 0.5 to 24 h. The behavior of different nucleophiles was evaluated, causing the selenoenyne 35 to react with a range of alcohols and amines. Thus, when primary alcohols (NuH = MeOH, EtOH and BuOH) were employed, the respective selenophenes 36a-c were obtained in 78–85% yields, after just a half hour. In contrast, the use of i-PrOH and t-BuOH resulted in the formation of products 36d and 36e in 72% and 28% yield, respectively, after 1.5 h. Satisfactorily, benzyl alcohol, alkynols and allyl alcohols were successful employed as nucleophiles, giving the corresponding selenophenes 36f-i in 51–87% yields, after 1.5 to 2 h. Cholesterol was also a suitable nucleophile, giving the respective selenophene 36j in 55% yield after 24 h. Aniline derivatives were suitable substrates in the reaction with selenoenyne 35, with p-chloro and p-toluidine giving the respective selenophenes 36l and 36m in 80% and 77% yields, respectively. In contrast, unsubstituted aniline afforded the respective selenophene 36k in just 48% yield. Additionally, the effect of the presence of electron-donor and electron-withdrawing groups in the pendant phenyl of the selenoenynes 35 was evaluated in the reaction with EtOH, and the selenophenes 36q and 36r were obtained in 65% and 56% yields, respectively. No reaction was observed using alkyl amine, phenol and thiols as nucleophiles, or alkyl-substituted selenoenyne (R = Bu) as substrates. The authors claimed that, in these cases, the selenoenynes were totally consumed, producing a complex mixture of products (Scheme 27).
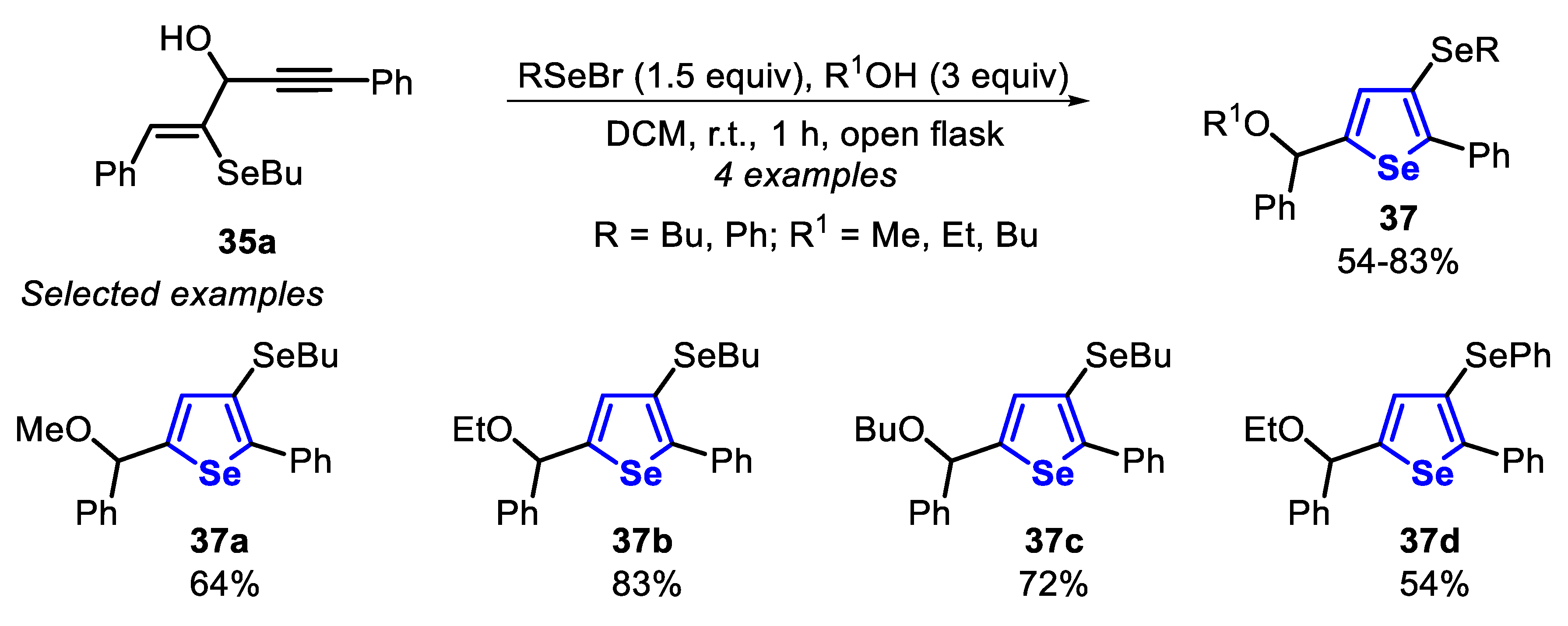
In the same work, 3-organoselanyl-selenophenes 37 were accessed through the reaction between selenoenyne 35a, alcohols (3 equiv) and organoselenyl bromide (1.5 equiv), generated in situ. For instance, the product 37a was obtained in 64% yield by the addition of BuSeBr, freshly prepared in situ by the reaction of N-bromosuccinimide (1.5 equiv) with BuSeSeBu (1.5 equiv), to a solution of selenoenyne 35a in MeOH. EtOH and BuOH were also satisfactorily employed as nucleophiles, giving the selenophenes 37b and 37c in 83% and 72% yields, respectively. The less reactive electrophile PhSeBr was a suitable substrate, promoting the cyclization of the selenoenyne 35a to the selenophene 37d in 54% yield (Scheme 28).
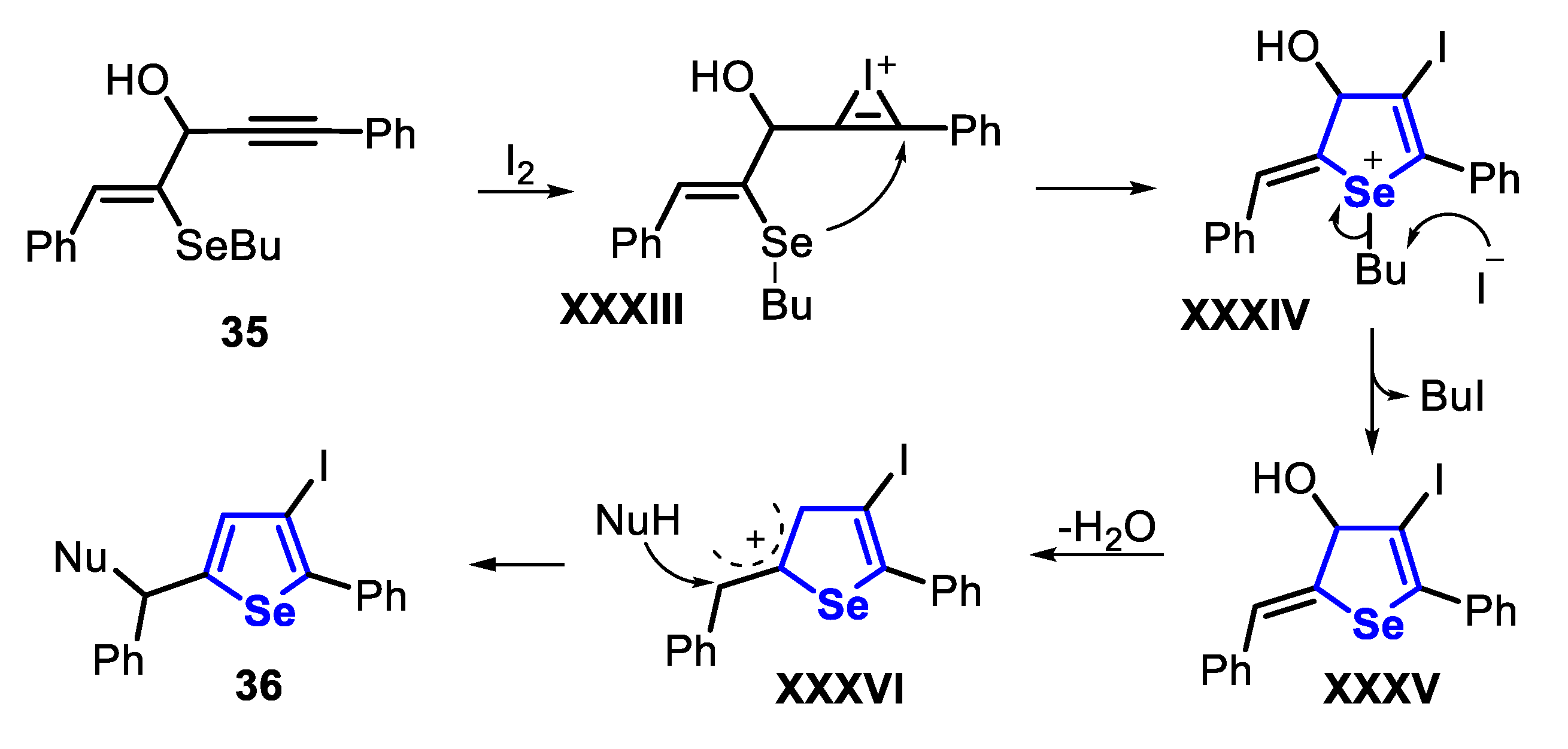
The proposed mechanism for the synthesis of 3-iodoselenophenes 36 involves the initial formation of an iodonium intermediate XXXIII, followed by a regioselective 5-endo-dig intramolecular nucleophilic attack of the selenium atom to the C-C triple bond, giving the selenonium intermediate XXXIV. The removal of the butyl group via an SN2 displacement, promoted by the iodide anion, affords the dihydroselenophene XXXV and BuI as a byproduct. The aromatization of dihydroselenophene XXXV leads to allylic cation XXXVI, which is trapped by a nitrogen- or/and oxygen-based nucleophile, producing the desired selenophene 36 (Scheme 29).
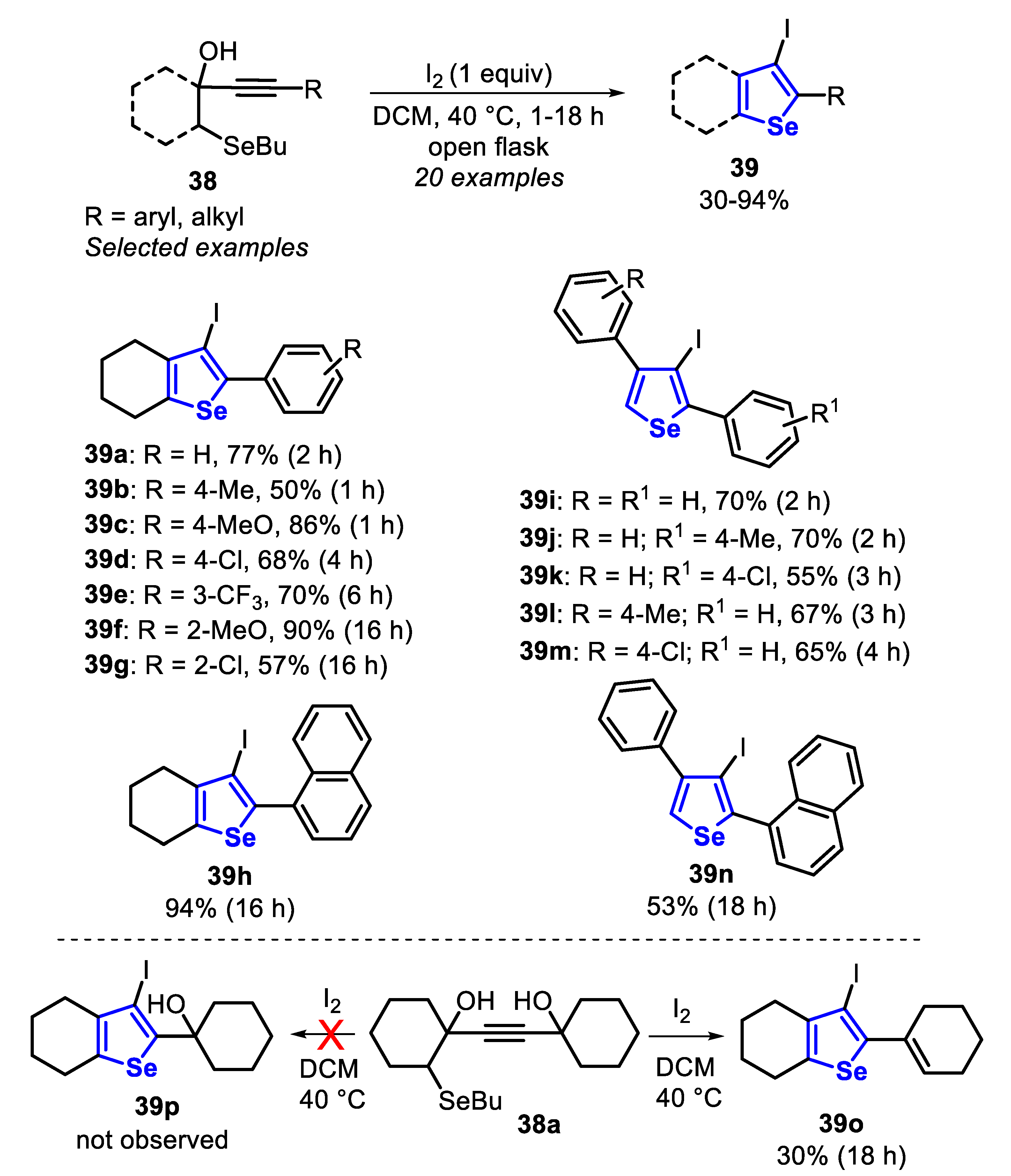
In 2018, the I2-promoted electrophilic cyclization of butylselanyl propargyl alcohols 38, for the synthesis of 3-substituted selenophenes 39–41 was described (Scheme 30, Scheme 31 and Scheme 32) [142]. The 3-iodoselenophenes 39 were obtained by the reaction between butylselanyl propargyl alcohols 38 and I2 (1 equiv) in DCM under air atmosphere at 40 °C for 1 to 18 h (Scheme 30). The effect of different substituents in the phenyl group directly attached to the C-C triple bond of the butylselanyl propargyl alcohols 38 was examined. The presence of electron-donor groups at the para-position positively influenced the reaction, and selenophene 39b (R = 4-MePh) and 39c (R = 4-MeOPh) were obtained in 50% and 86% yield after 1 h. The electron-withdrawing groups, however, caused a decrease in the reactivity, and the compounds 39d (R = 4-ClPh) and 39e (R = 3-CF3Ph) were obtained in 68% and 70% yield after 4 and 6 h of reaction, respectively. Sterically hindered butylselanyl propargyl alcohols 38 (R = 2-MeOPh, 2-ClPh and 1-naphthyl) were able to give the desired selenophenes 39f-h in moderate to good yields after longer reaction times (16 h), suggesting that the steric effects are more prominent than the electronic effect. Substrates bearing electron-rich and electron-deficient aryl substituents at the propargyl position provided the corresponding selenophenes 39i-m in 55% to 70% yields, after 2 to 4 h. The sterically hindered selenophene 39n was obtained in 53% yield after 18 h of reaction. The reaction of the alkynyl diol 38a under the optimal conditions afforded the elimination product cyclohexenylselenophene 39o instead the expected alcohol 39p. This event is promoted by iodine, which, besides acting to promote the selenophene core aromatization, also promotes the C-C double bond formation in cyclohexane, via an eliminative dehydration process (Scheme 30).
Regarding the formation of the 3-bromo-selenophenes 40, Br2 and NBS were not efficient in promoting the cyclization of the substrate 38 in an efficient way. However, CuBr2 (2 equiv) smoothly promoted the formation of the selenophenes 40a-f in 50% to 94% yields, after 2 to 18 h, in the presence THF at room temperature and under air atmosphere. Similar to what was previously observed in the reaction with iodine (Scheme 30), electron-rich and electron-deficient butylselanyl propargyl alcohols 38 afforded the 3-bromo-selenophenes 40a-c in moderate to very good yields, after 2–5 h of reaction. Additionally, sterically hindered butylselanyl propargyl alcohols 38 (R = 2-Cl and 1-naphthyl) reacted satisfactorily, affording the products 40d and 40e in 57% and 94% yields, after 18 and 16 h (Scheme 31).
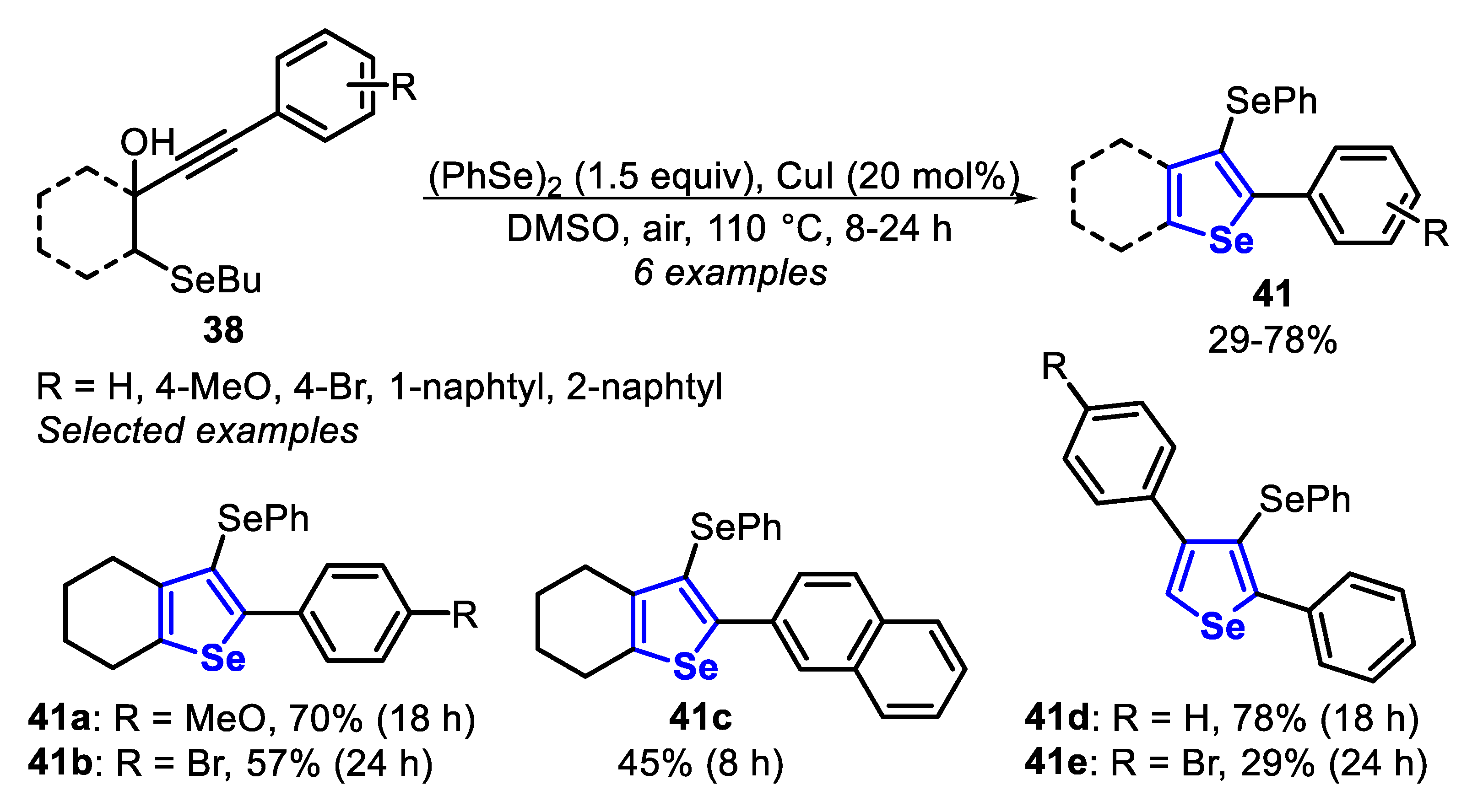
Finally, the 3-(phenylselanyl)selenophenes 41 were obtained through the CuI-catalyzed reaction between butylselanyl propargyl alcohols 38 and diphenyl diselenide (1.5 equiv), in DMSO as solvent at 110 °C and under an air atmosphere (Scheme 32). Under these conditions, six 3-(phenylselanyl)selenophenes were prepared in poor to acceptable yields, after 8–24 h of reaction. When the aryl group attached to the C-C triple bond was substituted with a methoxyl group (R = 4-MeO) a better yield was obtained in comparison to that substituted with a bromine group (R = 4-Br) (41a vs. 41b). This difference in the reactivity is due to the electron density increasing, caused by the mesomeric effect (R = 4-MeO), favoring the formation of a Cu-π-complex intermediate. In contrast, when the reaction was carried out with butylselanyl propargyl alcohol 38, bearing a naphthyl group (R = 2-naphthyl), the respective selenophene 41c was obtained in only 45% yield after 8 h, due to the steric hinderance around the C-C triple bond. When a neutral phenyl group was employed, the corresponding selenophene 41d was obtained in good yield, while the presence of the para-substituted aryl group at the propargyl position negatively affected the reaction (Scheme 32).
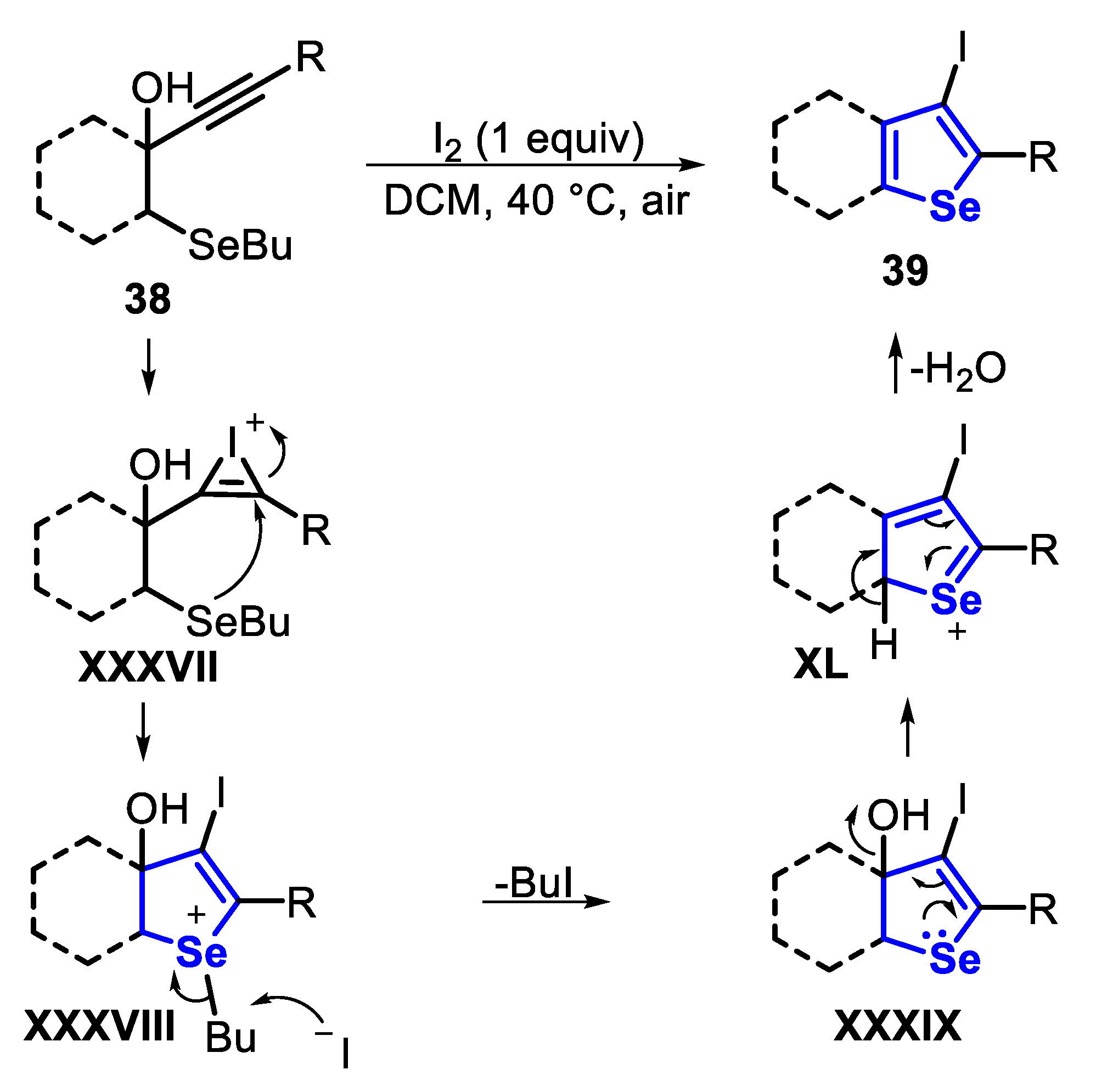
In order to collect data on the identity of the intermediates and then proposing a mechanism, the reaction for the synthesis of 3-iodo-selenophene 39 was monitored by 1H NMR and mass spectrometry. Based on these data, it was suggested that the cyclization and removing of the butyl group from the selenonium species occur before the aromatization step. Thus, initially, I2 adds to the C-C triple bond to give the iodonium intermediate XXXVII. The intramolecular nucleophilic anti-attack of Se to C1 produces the 2,3-dihydroselenophene selenonium salt XXXVIII. Thus, the iodide anion promotes butyl group removal, through an SN2 displacement, giving the 2,3-dihydroselenophene XXXIX, which undergoes an aromatization step to afford the 3-iodo-selenophene 39 (Scheme 33).
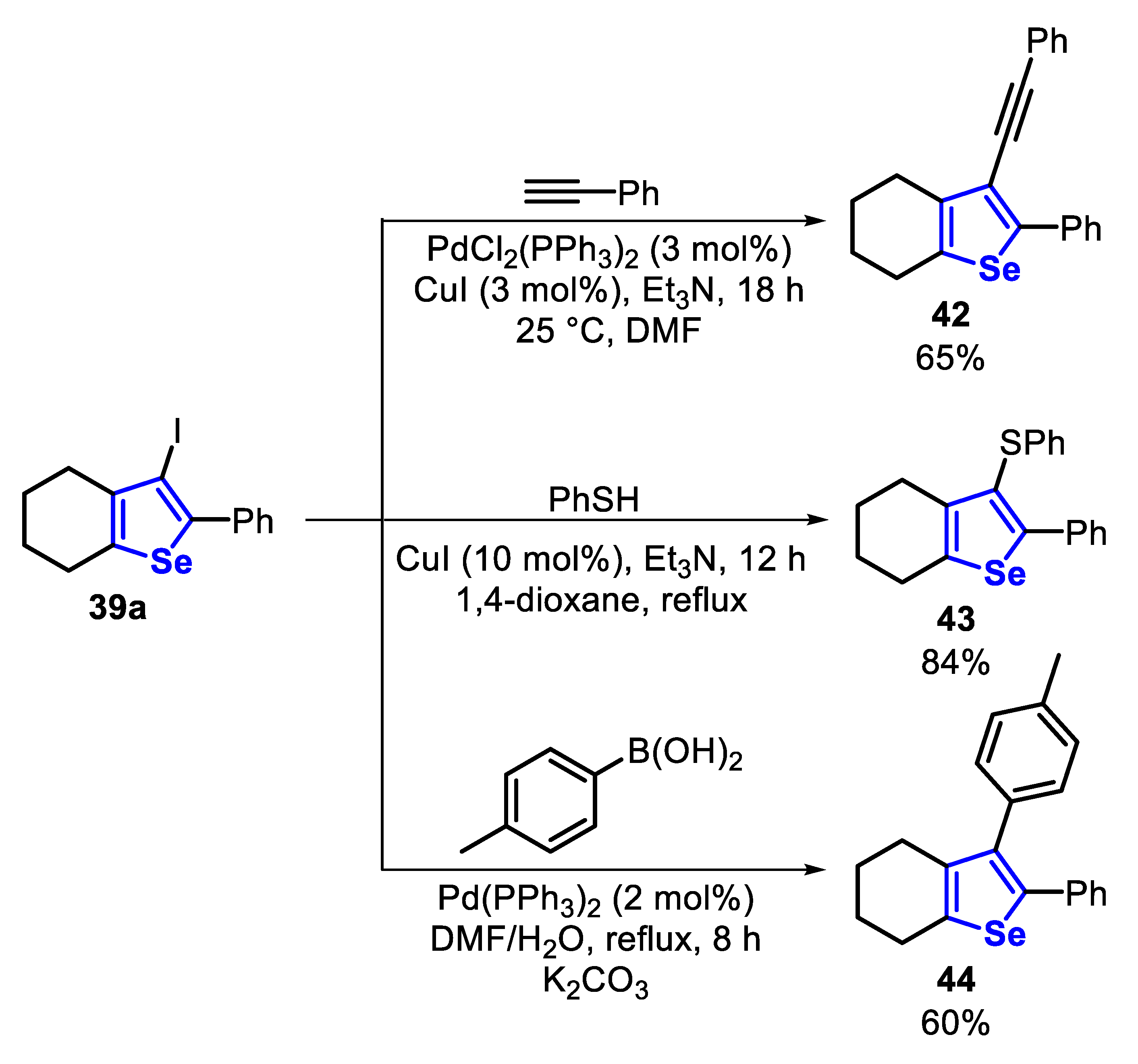
The prepared 3-iodoselenophenes 39 were used as starting materials for the synthesis of multifunctional selenophenes, through Cu- and Pd-catalyzed Sonogashira, Ullmann and Suzuki cross-coupling reactions (Scheme 34). Thus, under the Sonogashira condition, 3-iodoselenophe 39a reacted with phenylacetylene, at room temperature, in the presence of PdCl2(PPh3)2 (3 mol%), CuI (3 mol%), Et3N and DMF to give the 3-alkynylselenophene 42 in 65% yield after 12 h. Additionally, under the Ullmann protocol, 39a reacted with benzenethiol in the presence of CuI (10 mol%), Et3N and 1,4-dioxane under reflux to afford the 3-thiophenylselenophene 43 in 84% yield, after 18 h. Finally, under Suzuki conditions, 2-arylselenophene 44 was obtained in 60% yield, through the reaction between 39a and 4-tolylboronic acid, in the presence of Pd(PPh)3 (2 mol%) as the catalyst, K2CO3 as the base and a mixture of DMF/H2O as the solvent under reflux (Scheme 34).
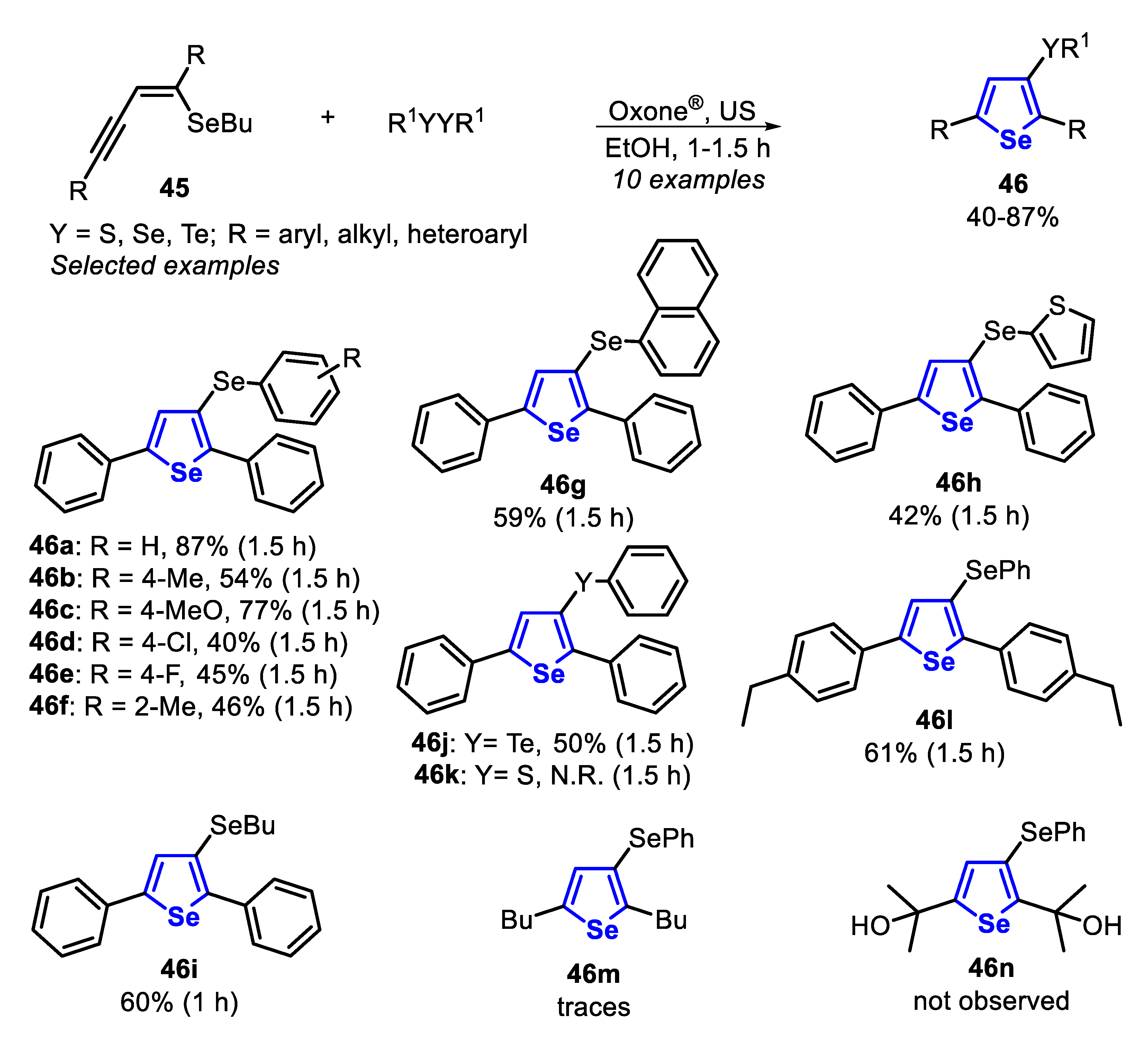
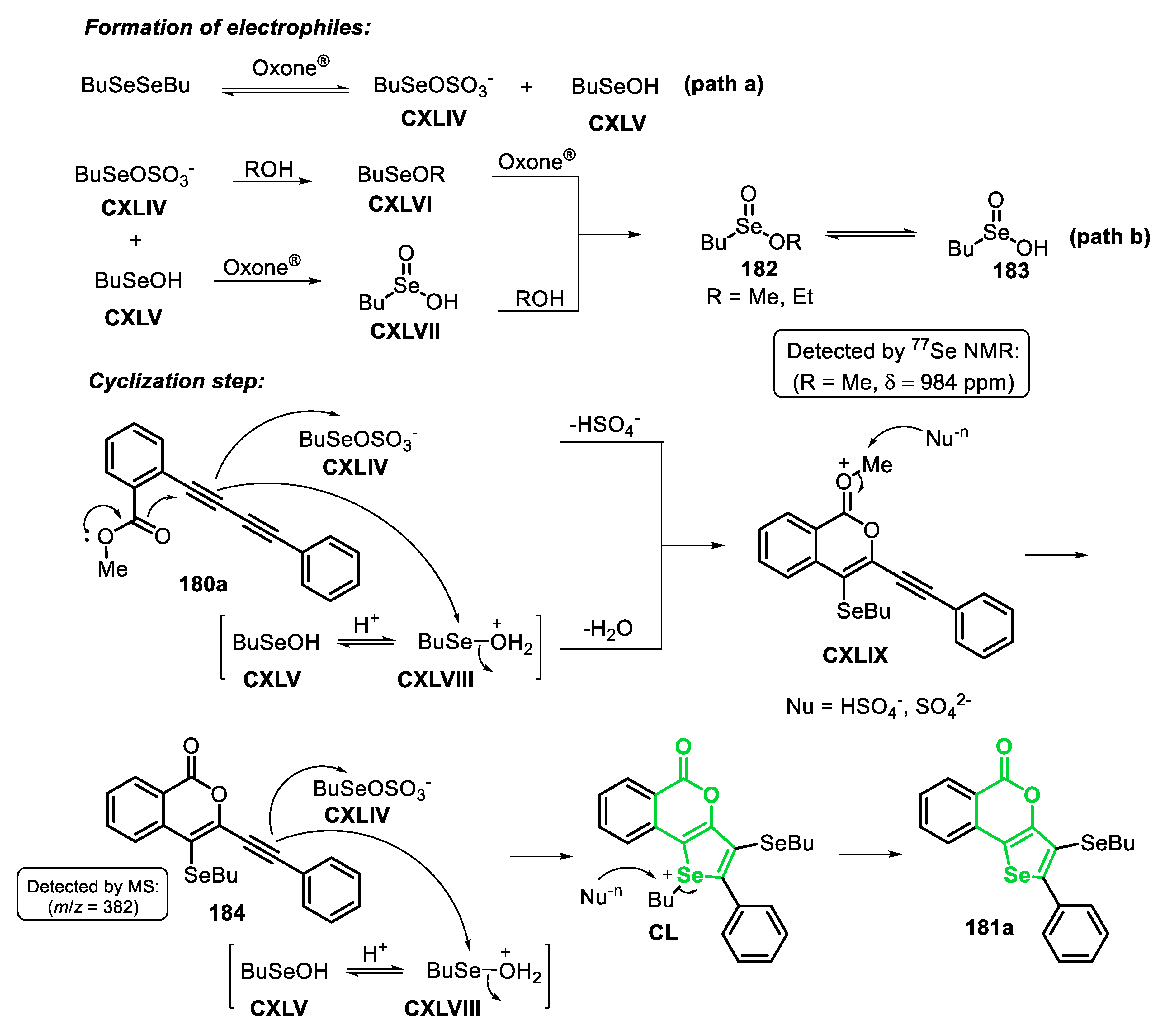
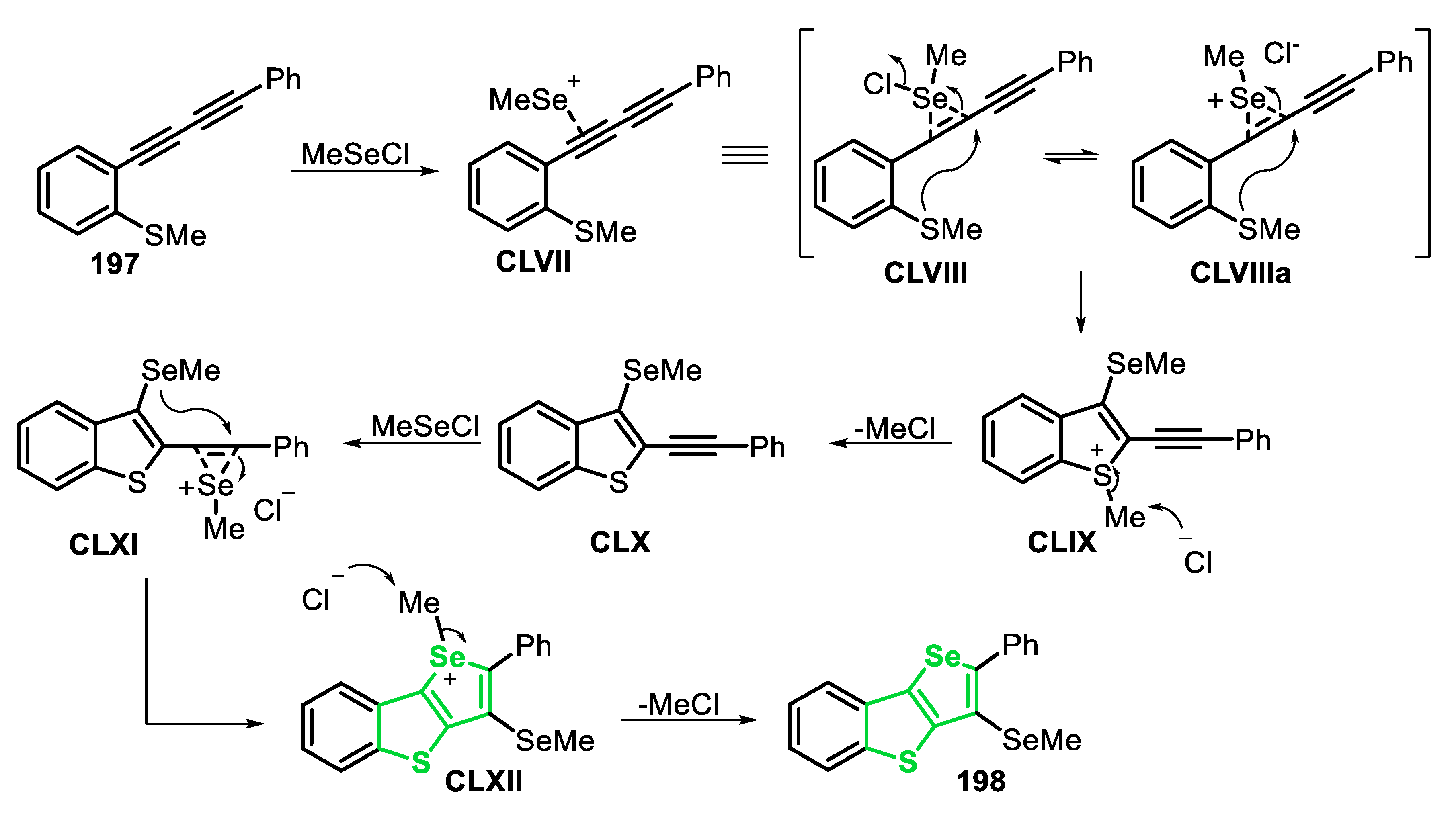
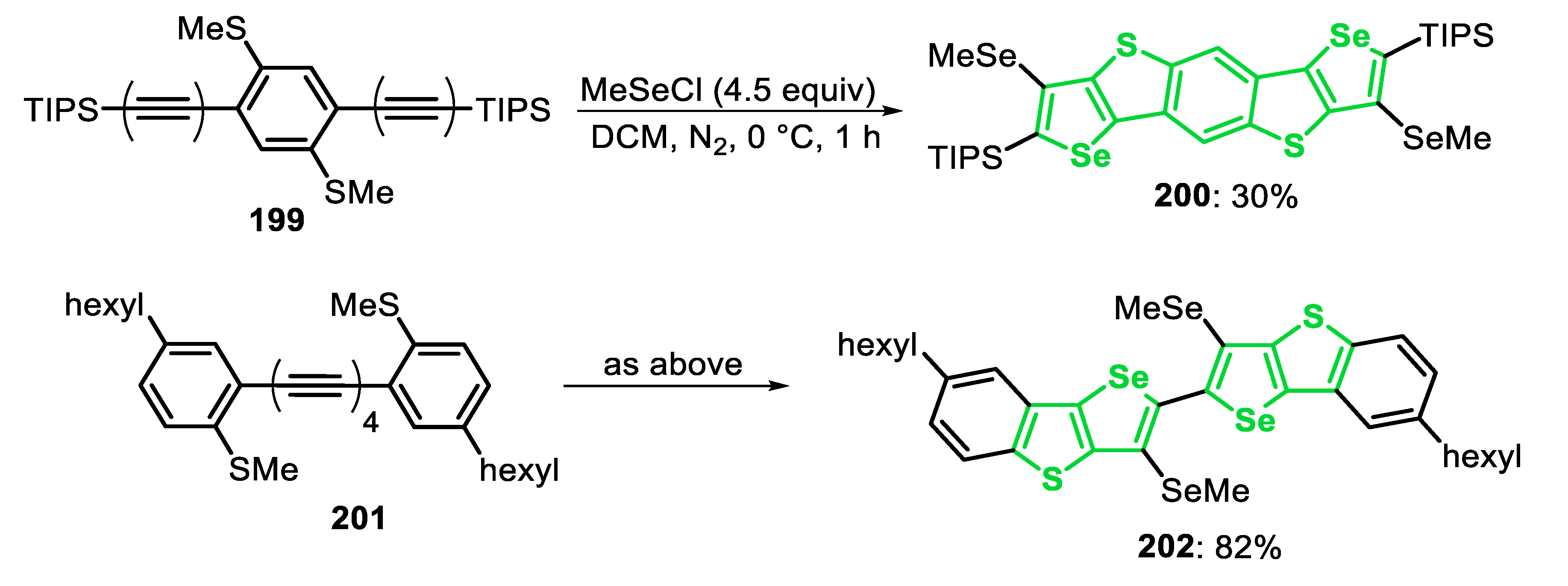
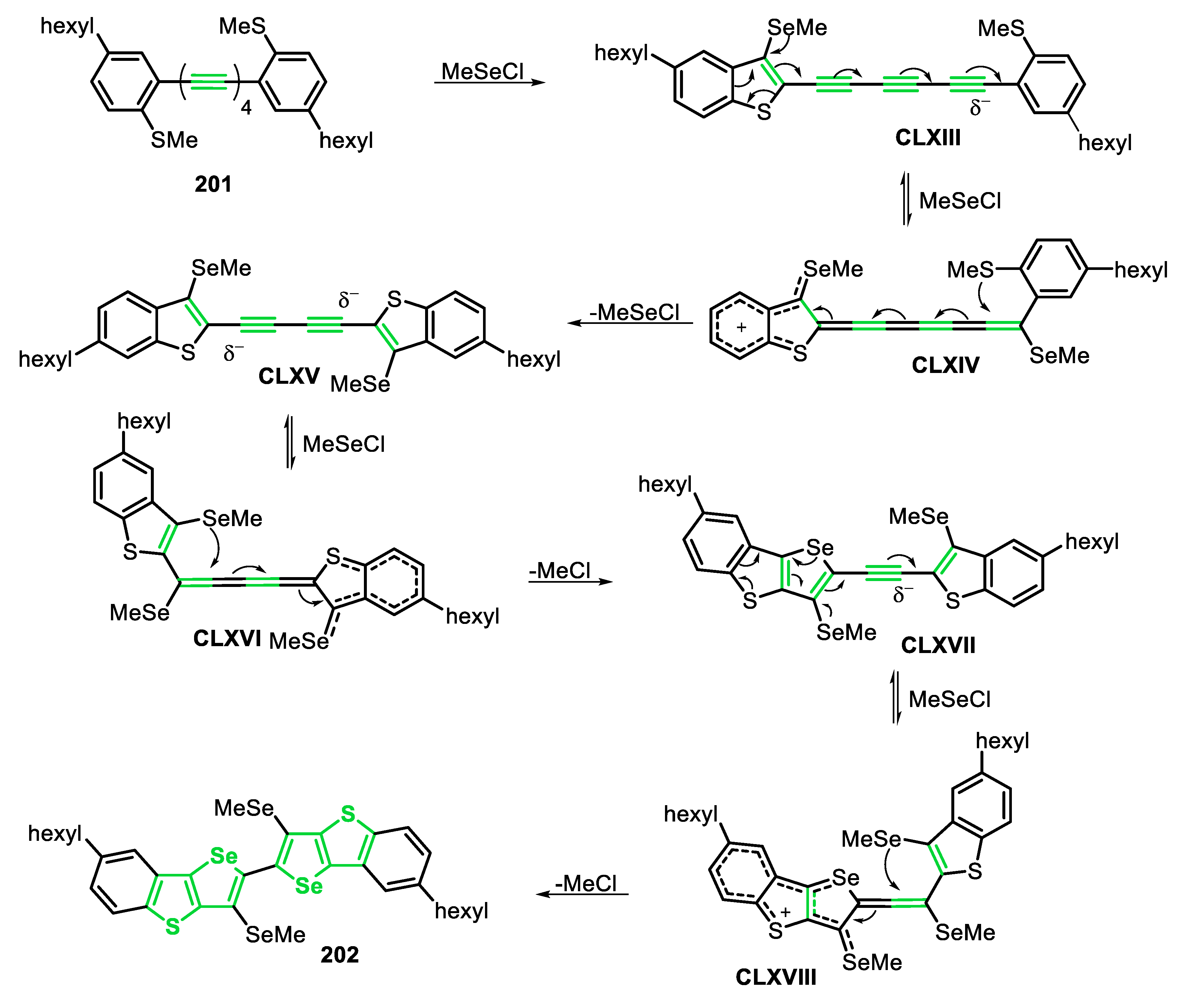
Very recently, some of us [143] reported the electrophilic cyclization of (Z)-selenoenynes 45 under ultrasound irradiation (US) conditions, promoted by the system R1YYR1/Oxone®/ethanol (Y = Se, Te), to access 3-selanyl/telanylselenophenes 46 (Scheme 35). This green alternative was carried out using Oxone® as an inexpensive and non-toxic oxidant agent to promote the oxidative cleavage of Se-Se and Te-Te bonds. In order to evaluate the generality and limitations of this protocol, the (Z)-butyl(1,4-diphenylbut-1-en-3-yn-1-yl)selane 45a (R = Ph) was reacted with several diorganyl diselenides. Electron-rich and electron-deficient diaryl diselenides were suitable reagents, affording the corresponding selenophenes 46a-f in moderate to good yields, after 1.5 h. In these cases, the presence of substituents at the para-position of the aryl ring reduced the reactivity of the diselenide (46a vs. 46b-e). This decreasing in the reactivity was less pronounced in the case of electron-rich diselenides, and the selenophenes 46b (R = 4-Me) and 46c (R = 4-MeO) were obtained in 54% and 77% yields, respectively. The electron-deficient (R = 4-Cl and 4-F) and ortho-tolyl (R = 2-Me) diselenides were less reactive, giving the respective selenophenes 46d-f in 40%, 45% and 46% yields, respectively. This protocol was also compatible with aromatic bulky, heteroaromatic and aliphatic diselenides, affording the selenophenes 46g-i in 42–60% yields. In addition, when diphenyl ditelluride was submitted to the optimal conditions, the corresponding Te-functionalized selenophene 46j was obtained in 50% yield. Regarding the enyne counterpart, good result was obtained in the cyclization of the aryl substituted (Z)-enyne 45b (R = 4-EtPh) using PhSeSePh, which was converted to the respective selenophene 46l, in 61% yield. The reaction failed when diphenyl disulfide was the chalcogen source (46k), as well as starting from the (Z)-selenoenynes bearing diol (46n) and aliphatic groups (46m) as substrates (Scheme 35).
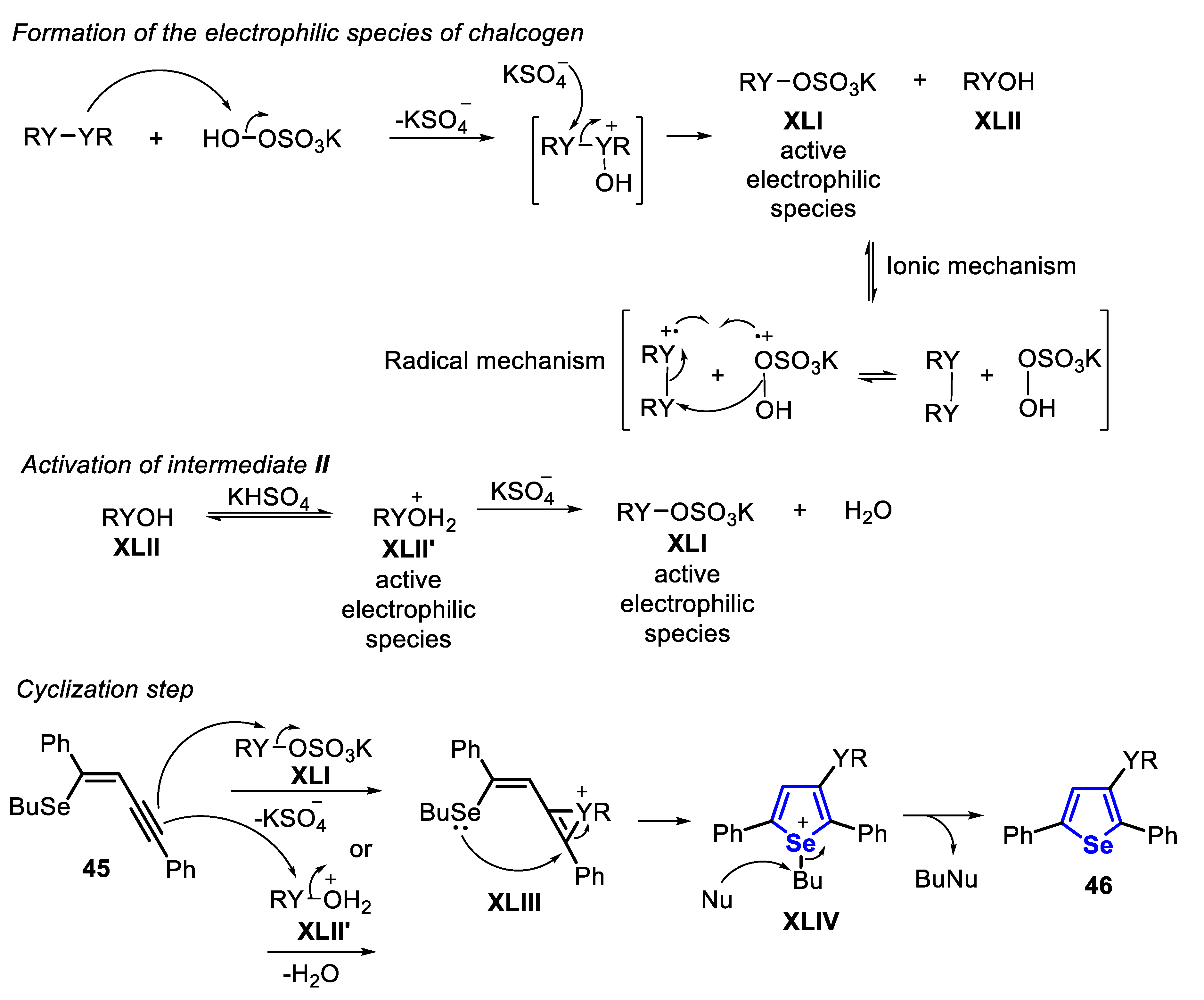
The proposed mechanism of the reaction starts with the US-promoted oxidative cleavage of the Y-Y bond of the diorganyl dichalcogenide in the presence peroxymonosulfate (KHSO5), the active component of Oxone®, to give the reactive intermediates XLI and XLII. Once formed, intermediate XLII is activated by the acidic medium to form the strongest electrophile XLII’. Then, the selenoenyne 45 reacts with XLI or XLII’ to form the seleniranium intermediate XLIII, that after an intramolecular attack by the electron pair of Se, gives the selenonium intermediate XLIV. Finally, XLIV undergoes a nucleophilic attack by one of the nucleophilic species present in the medium (SO42−, HSO4−) to form the desired selenophene 46 (Scheme 36).
3. Synthesis of Benzoselenophenes
3.1. Starting from Type C Precursors
There are several protocols to obtain benzoselenophenes through intra- or intermolecular reactions between functionalized arenes and alkynes. Among them, electrophilic and radical cyclizations of type C precursors are the most common approaches, with modifications in the electrophilic source and the reaction conditions.
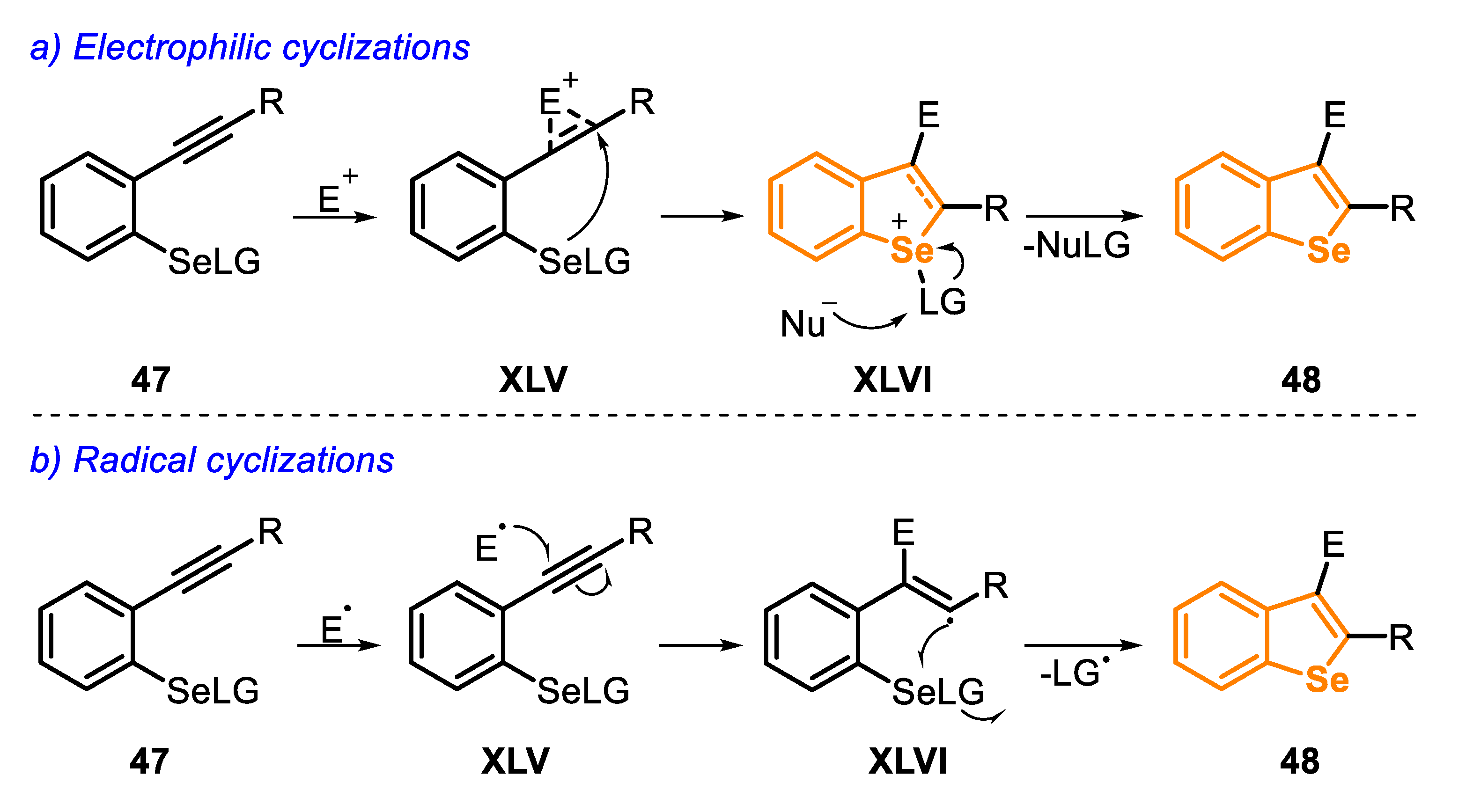
In general, these reactions involve the addition of an electrophile to a C(sp) bond of Se-functionalized arenes, bearing an ortho-alkynyl group, such as compound 47 (type C precursor). In the electrophilic cyclization, there is initially a coordination of the electrophile (E+) to the C-C triple bond, generating the three-membered cyclic intermediate XLV (activation step). The selenium anti-attack takes place on the intermediate XLV (via a 5-endo-dig cyclization) to produce the intermediate XLVI. Finally, the leaving group attached to the Se atom is removed by a nucleophile (Nu−), present in the reaction mixture, via an SN2 path, affording the cyclized heterocyclic product 48 (Scheme 37a). On the other hand, in the radical cyclization reactions, the first step is the generation of the radical species, followed by an attack on the C(sp) bond in the type C precursor. Finally, the vinyl radical XLVI undergoes an intramolecular cyclization (a 5-exo-trig cyclization), followed by the elimination of the LG radical, to generate the desired benzoselenophene 48 (Scheme 37b).
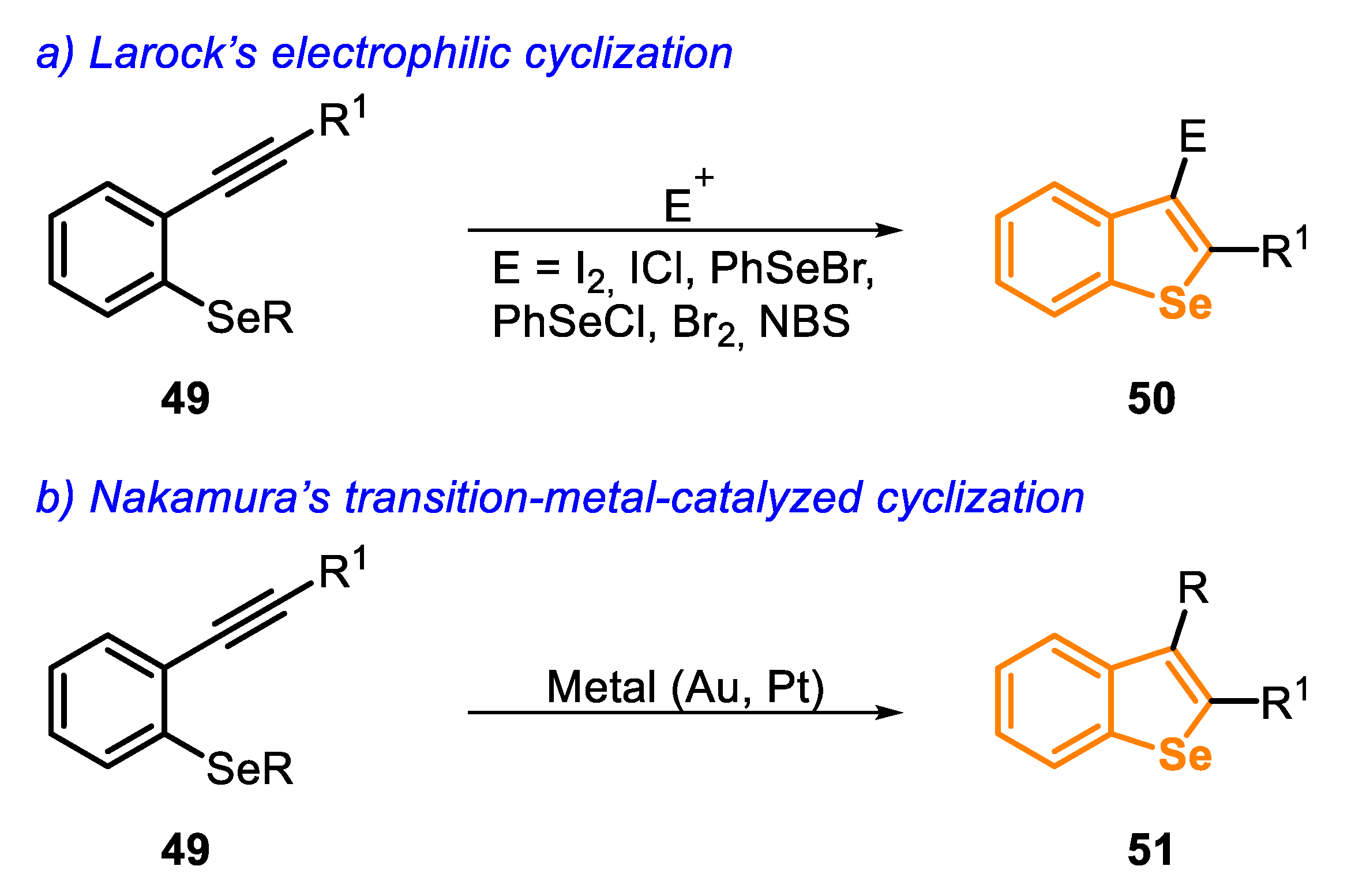
In the early 2000s, Larock’s and Nakamura’s groups [144,145,146] described valuable protocols for the synthesis of benzo[b]selenophenes, starting from type C precursors, using different electrophilic sources or transition-metal-catalyzed strategies (Scheme 38).
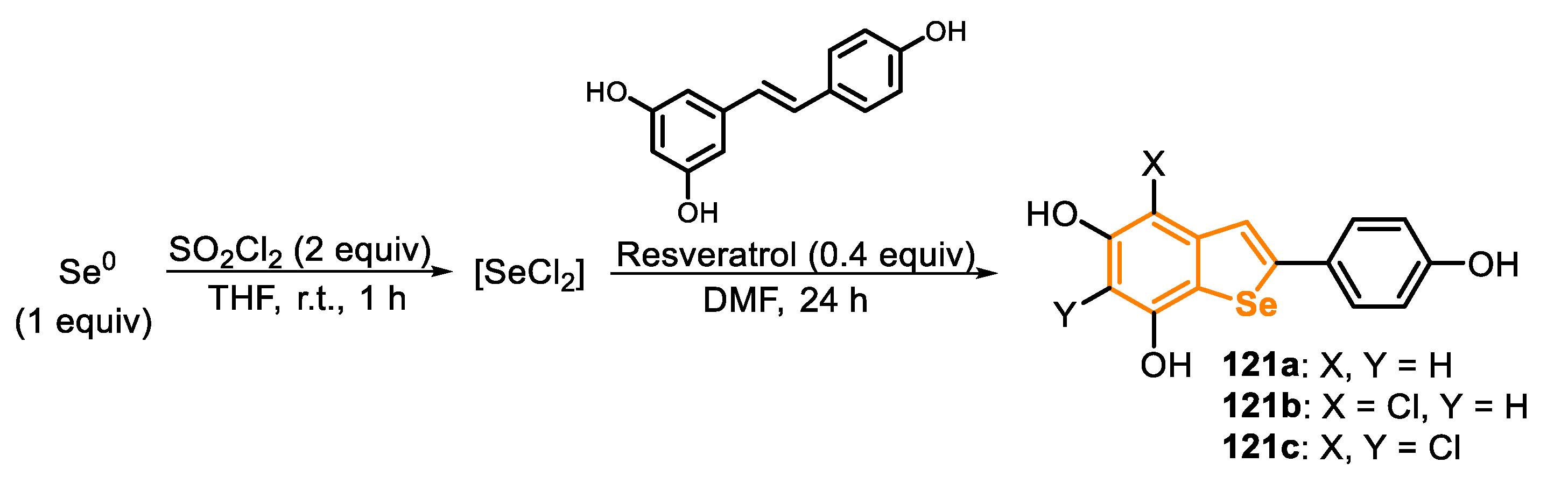
Since then, these protocols have paved the way for the development of several transformations in organic synthesis. For instance, this was the key step in the recently-described synthesis of benzo[b]selenophene-based analogues of the resveratrol dimers viniferifuran and (±)-dehydroampelopsin B (Scheme 39) [147]. The 5-endo-dig cyclization was carried out under microwave irradiation, in just 75 min, using I2 as an electrophile and DCM as a solvent, affording the 3-iodo-selenophene 53 in 96% yield. This benzo[b]selenophene was the precursor of the intermediate 54, obtained after two consecutive Suzuki–Miyaura coupling reactions. Finally, the demethylation of compound 54 was performed with BBr3 (6 equiv) in DCM, giving the Se-analogue of viniferifuran 55 (34% yield) and the demethylation/cyclization product (±)-dehydroampelopsin B Se-analogue (14% yield), after 3 days of reaction. The authors reported that the cyclization product 56 was favored (45% yield) by adding BBr3 (15 equiv) and aqueous HBr.
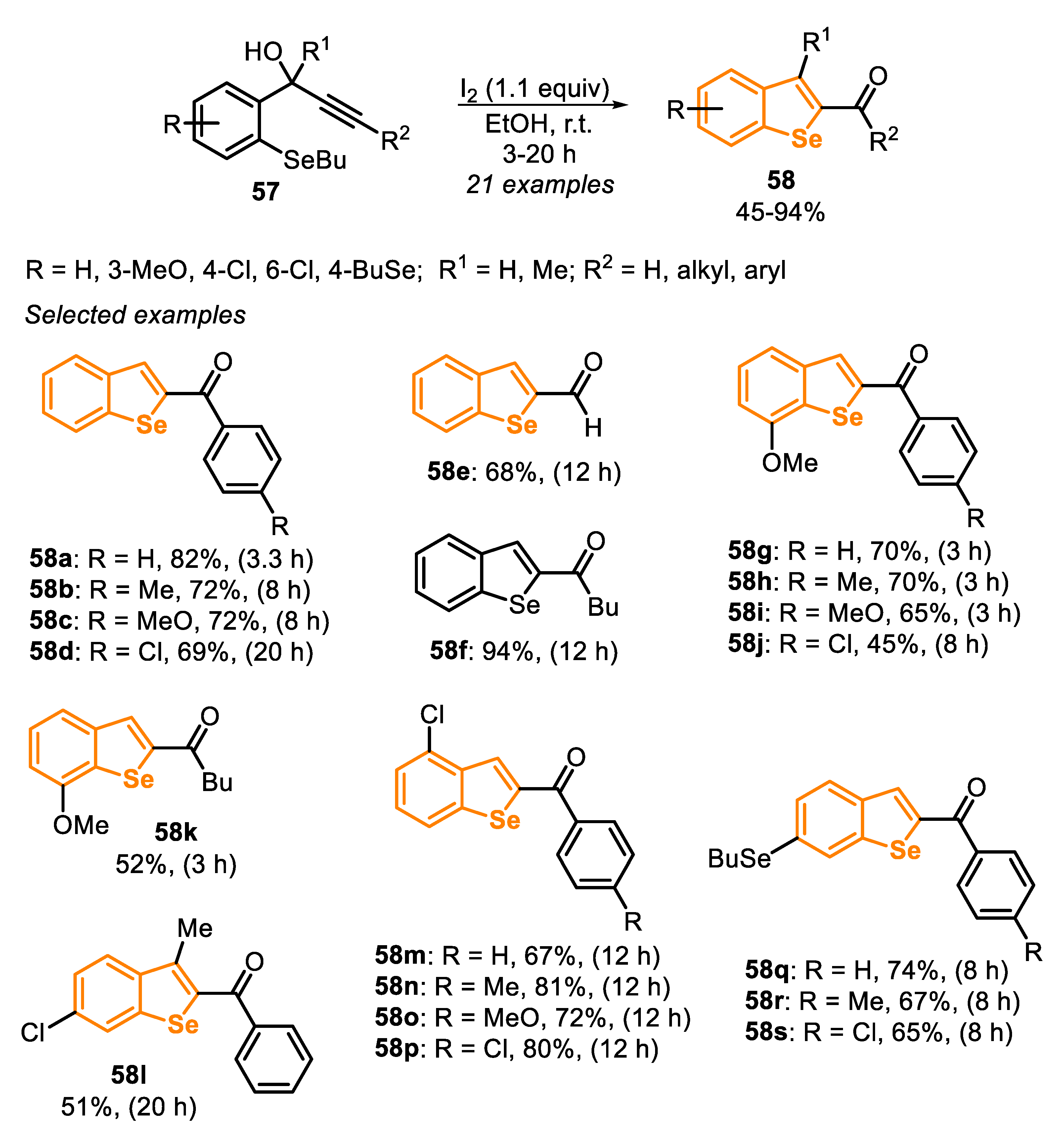
A close-related protocol was described in 2015 [148], through the intramolecular cyclization of [2-(butylselanyl)phenyl]propynols 57 (related to type C precursor), using I2 (1.1 equiv) at room temperature and under aerobic conditions (Scheme 40). Under these conditions, a study on the reaction scope was carried out, using several electron-rich and electron-deficient substrates, allowing for the synthesis of twenty-one 2-acylbenzo[b]selenophenes 58 analogues in moderate to good yields (45–94%). The protocol was general and efficient for a variety of [2-(butylselanyl)phenyl]propynols 57 containing aryl or alkyl groups directly bonded to the triple bond. A good result was also obtained, starting from terminal alkyne, giving the product 58e at 68% yield after 12 h. Different electron-donating and electron-withdrawing substituents directly connected to the central aromatic ring (fused to selenophene) were explored in the reaction, and no significant electronic effect was observed.
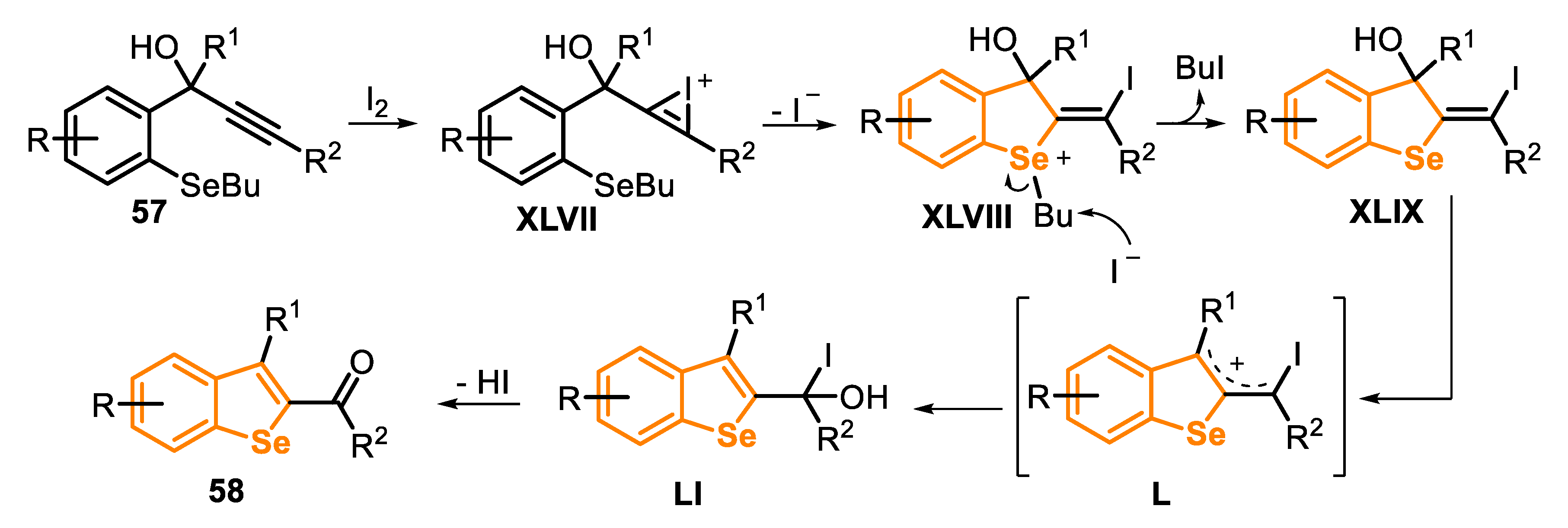
The reaction mechanism involves initially the formation of the iodonium ion XLVII, which is formed by the addition of I2 to the C-C triple bond (Scheme 41). The anti-nucleophilic attack of the selenium atom at the activated iodonium intermediate produces the salt XLVIII, via a 5-exo-dig cyclization. The removal of the alkyl group via SN2 displacement by the iodide anion, present in the reaction mixture, generates the vinylic iodide XLIX and butyl iodide. Then, a 1,3-migration of hydroxy, via the formation of the allylic cation L, gives the alcohol LI, which affords the 2-acylbenzo[b]selenophenes 58 after the elimination of HI (Scheme 41).
In recent years, the use of ortho-butylselanyl-substituted 1,1-dibromoalkenes 59 as precursors to access benzo[b]selenophenes 60 has shown great advances, through the application of two different strategies: the use of I2 and of TM-based catalysis (Scheme 42).
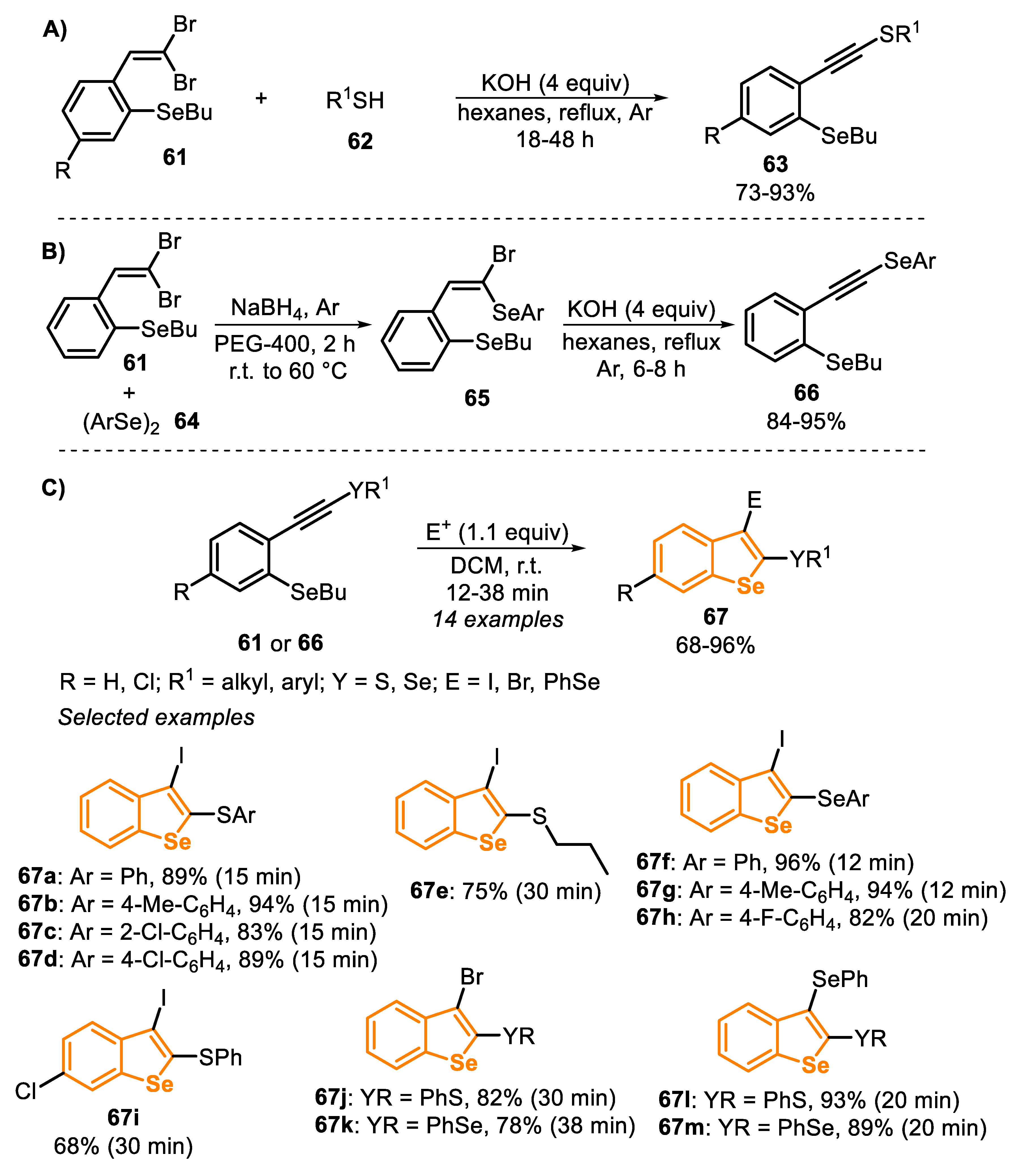
In 2017, some of us [149] described a protocol that is close-related to the one previously addressed using I2 to promote the cyclization of type C precursors, which are obtained in situ from ortho-butylselanyl-substituted 1,1-dibromoalkenes 61. The versatility of the compound 61 was explored in the synthesis of the intermediate thioalkynes 63, by the reaction with aryl or alkyl thiols (1.2 equiv) in the presence of KOH (4 equiv.) and hexanes (degassed) as a solvent, under reflux temperature in argon atmosphere, by 18 to 48 h (Scheme 43A). In order to obtain the selenoalkynes 66, a two-step protocol was performed: initially, the o-butylselanyl-substituted 1,1-dibromoalkene 61 was submitted to the reaction with diaryl diselenides 64, in the presence of NaBH4 and PEG-400 as the solvent. After 2 h, at 60 °C under argon atmosphere, the respective (E)-1-bromo-1-arylselenoalkenes 65 were obtained. In the second reaction, the crude product was submitted to a dehydrobromination using KOH (1 equiv) in refluxing hexanes for 6 to 8 h (Scheme 43B).
The type C precursors 61 and 66 were reacted in the presence of I2 (1.1 equiv) and using DCM as a solvent to prepare a broad scope of benzo[b]selenophenes 67 (Scheme 43C). The reactions were carried out in short reaction times (12–38 min), and the desired products 67a-i were obtained in good to excellent yields (68–96%). Other electrophilic species, such as Br2 and PhSeBr, were successfully used in substitution to iodine (compounds 67j-m).
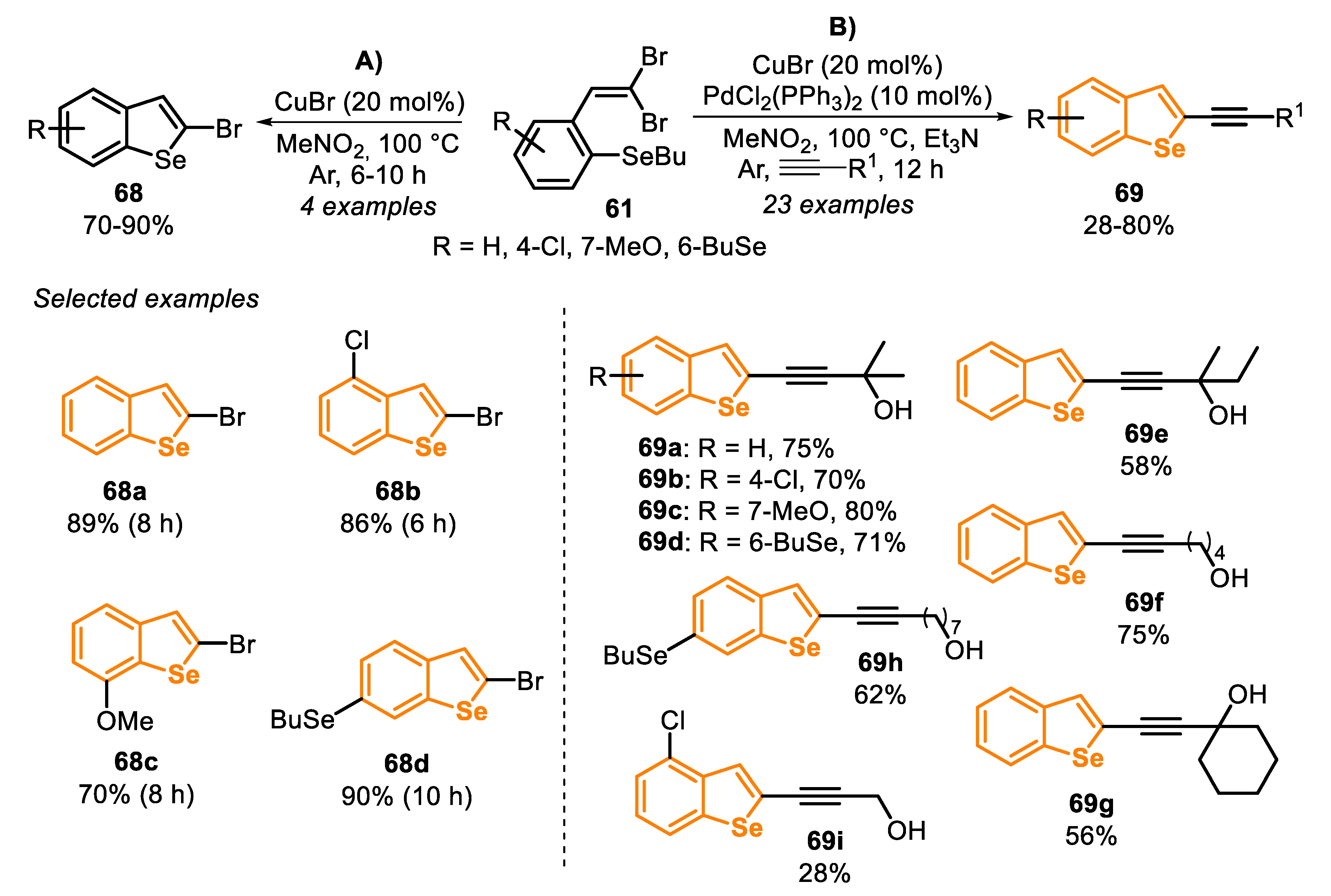
In the same year, the synthesis of benzo[b]selenophenes derivatives 68 and 69, through the Cu(I)-catalyzed annulation of ortho-butylselanyl-substituted 1,1-dibromoalkenes 61 was described [150]. The reaction was carried out in the presence of CuBr (20 mol%) in MeNO2 as a solvent at 100 °C, giving four unprecedented 2-bromobenzo[b]selenophenes 68 in 70% to 90% yields (Scheme 44A). Due to the efficiency of this protocol, the authors extended their studies to the synthesis of 2-alkynylbenzo[b]selenophenes 69, starting from o-substituted 1,1-dibromoalkenes 61, through a sequential cyclization/Sonogashira cross-coupling strategy (Scheme 44B). In this one-pot procedure, the 2-bromoselenophenes 68 were generated in situ, as above, and then reacted with propargyl alcohols in the presence of PdCl2(PPh3)2 (10 mol%) and Et3N. A total of twenty-three 2-alkynylbenzo[b]selenophenes 69e-g were prepared in 28–80% yields after 12 h of reaction. The presence of bulky groups on the alkyne caused a slight decrease in the reaction yield, affording the products 69 moderate yields. The method was not sensitive to the presence of electron-donor and electron-withdrawing groups in the pendant phenyl of the 1,1-dibromoalkenes 61, and the respective products 69 were obtained in moderate to good yields, except the compound 69i, derived from 2-propyn-1-ol, which was obtained in only 28% yield. This protocol did not work with other alkynes, (different from propargyl alcohols), such as phenylacetylene and 1-pentyne, affording, in these cases, the products 69 together an inseparable mixture of homocoupling byproducts.
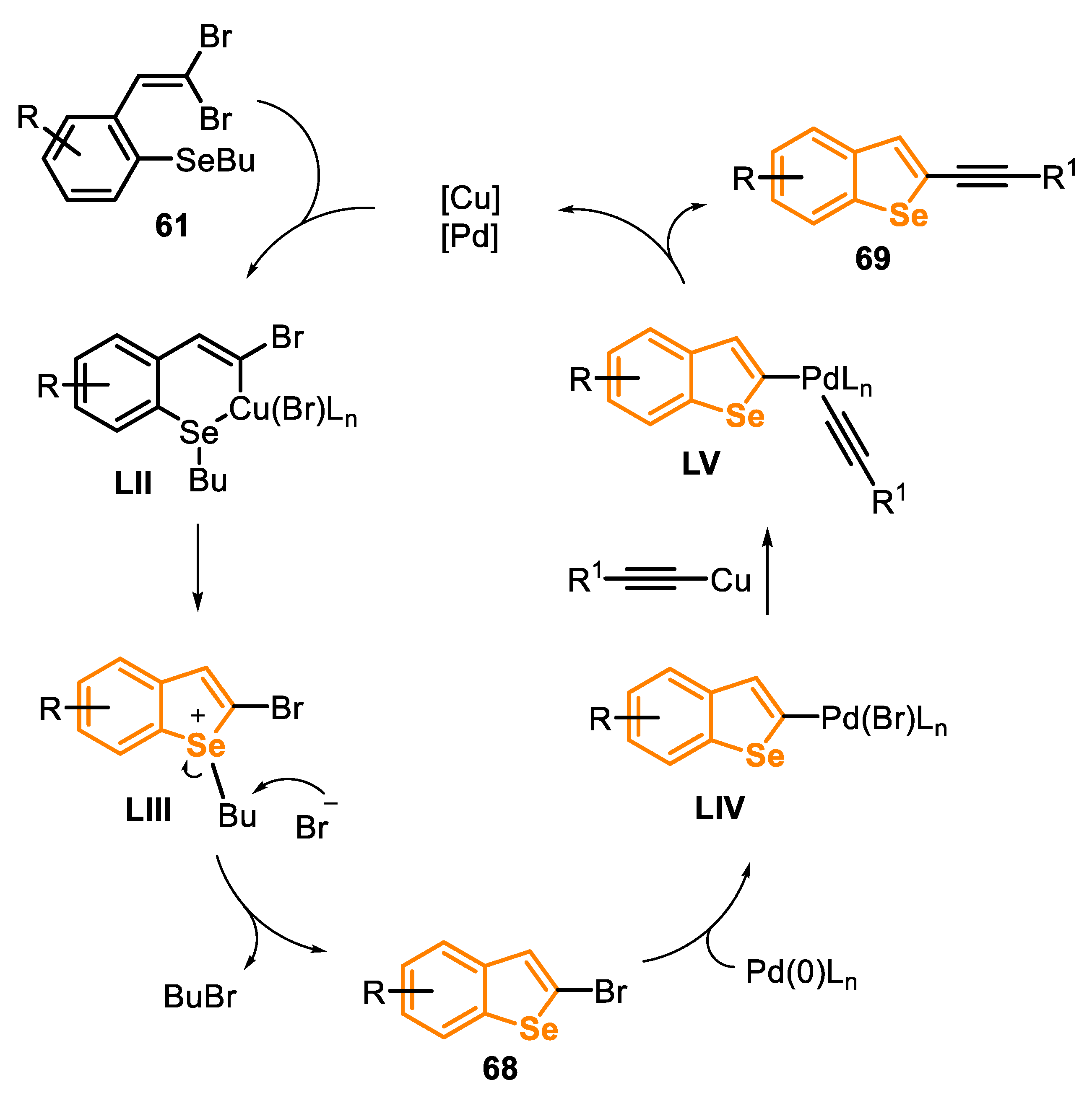
The proposed mechanism of the reaction starts with the oxidative addition of copper to the ortho-substituted 1,1-dibromoalkene 61 to give the 2-bromobenzo[b]selenophene 68 via an intramolecular Ullmann reaction process, followed by nucleophilic substitution on the selenium atom. The oxidative addition of 2-bromobenzo[b]selenophene 68 to the Pd(0) species affords the Pd(II) intermediate LIV, which reacts with the copper-activated alkyne to give the intermediate LV. Then, the species LV undergoes a reductive elimination, generating the product 69 and releasing the catalysts for a new cycle (Scheme 45).
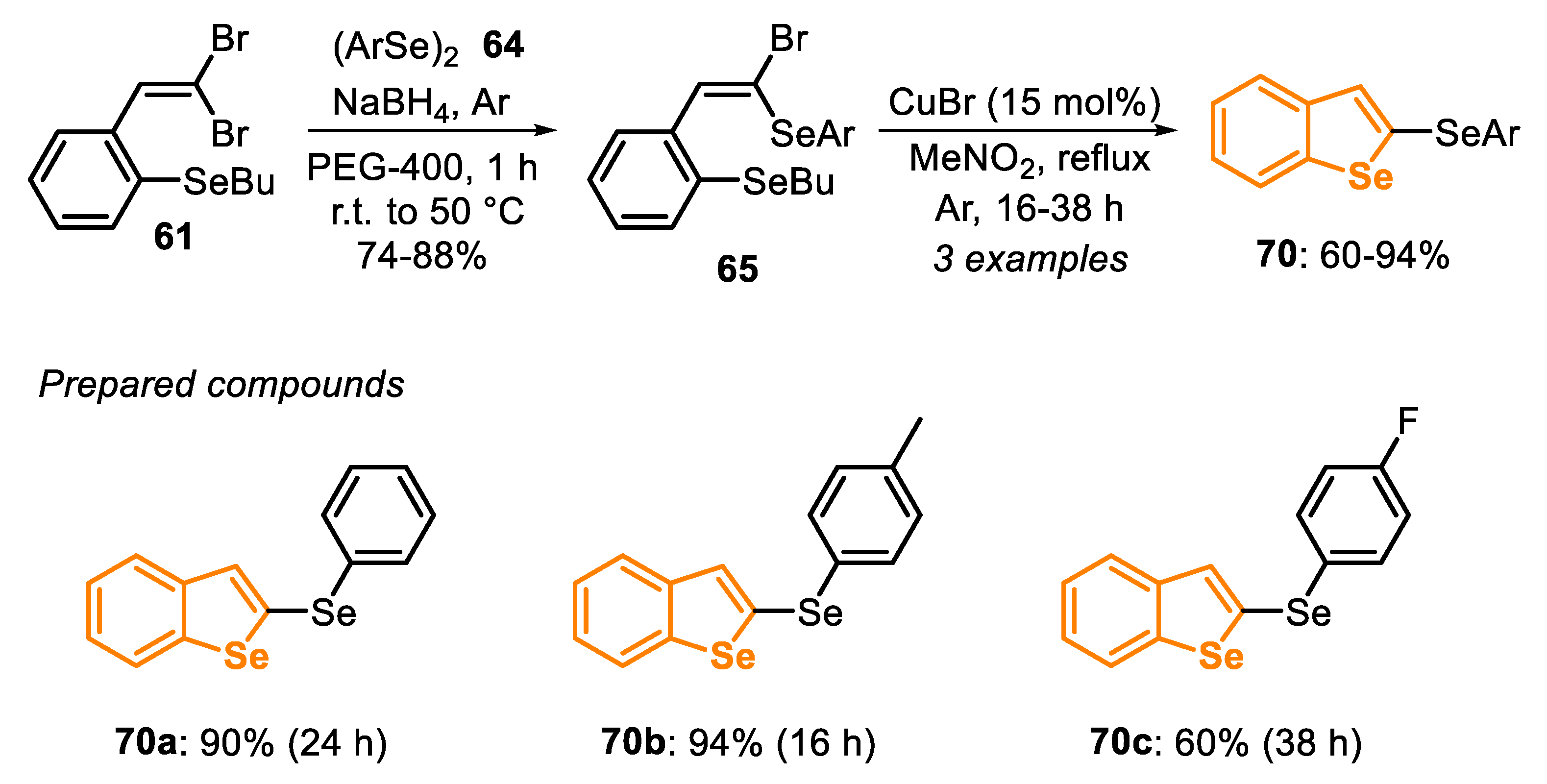
In 2019, Cu(I)-catalyzed annulation of ortho-SeBu-substituted (E)-1-bromo-1-arylselenoalkenes 65 (obtained as described in Scheme 43B) was described in [151]. The best condition for this annulation was obtained using CuBr (15 mol%) as the catalyst in MeNO2 as the solvent under argon atmosphere at reflux temperature for 16–38 h (Scheme 46). Using this condition, three 2-arylselanyl-benzo[b]selenophenes 70 were obtained in 60% to 94% yields. The presence of electron-withdrawing groups linked to the aromatic ring of the diselenide 64 (R = 4-F) negatively affected the reaction efficiency, giving the product 70c in 60% yield after 38 h, while the electron-rich p-tolyl derivative (R = 4-Me) was more reactive, affording the selenophene 70b in 94% yield after 16 h (Scheme 46).
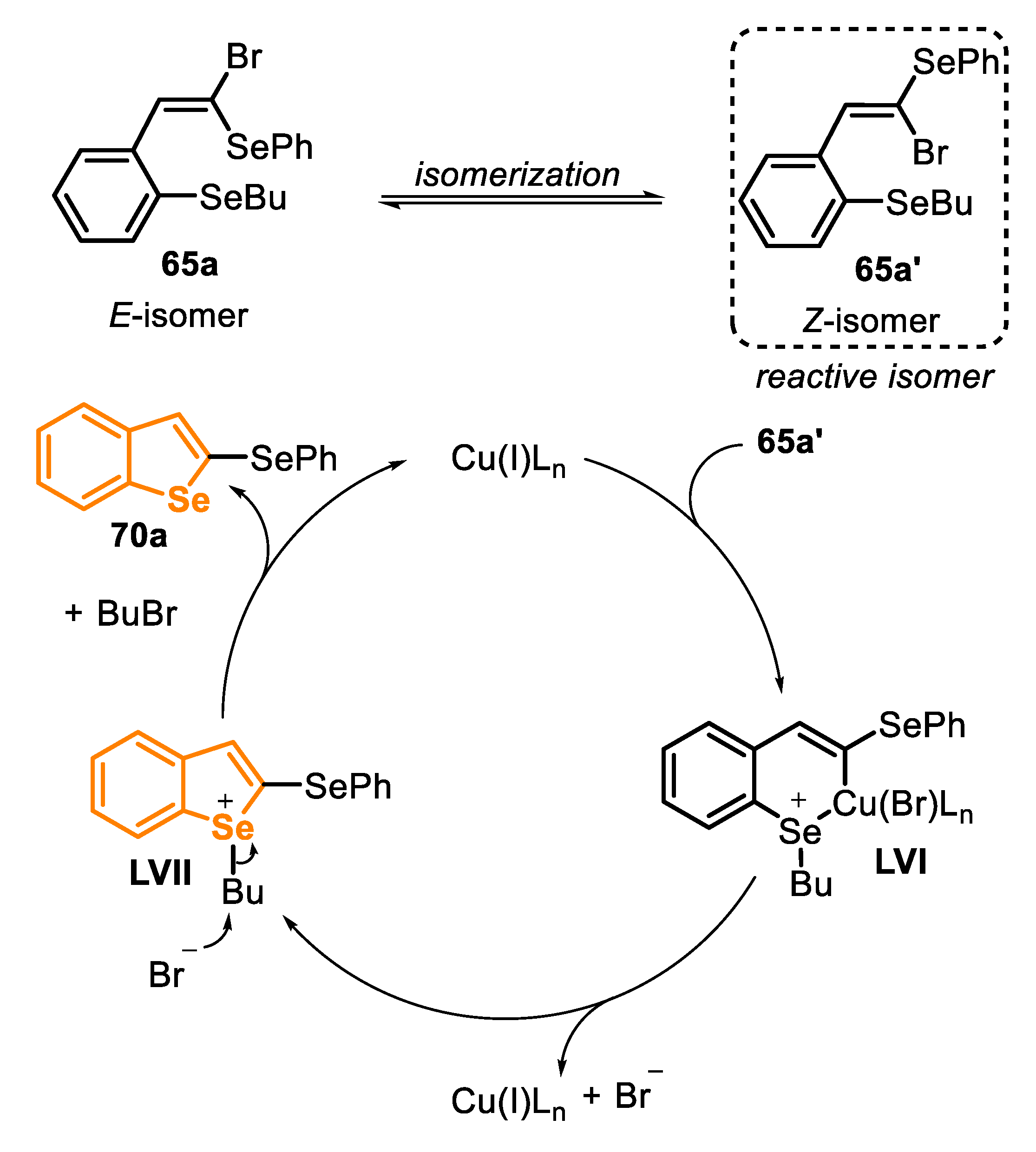
The key reaction step in intramolecular cyclization is the conversion of the E-isomer 65a to the Z-isomer 65a’, which has a more suitable configuration to undergo an oxidative addition to the coordination sphere of the Cu(I) species. This hypothesis was confirmed by carrying out a control experiment using pure (E)-1-bromo-1-(phenylselanyl)-2-phenylethene under the standard reaction conditions. In this case, after 24 h, 45% of the E-isomer was isomerized to the Z-one (determined by GC/MS analysis). After the coordination of the Cu(I) species, which gives the intermediate LVI, an Ullmann-type coupling generates the intermediate LVII, releasing to the reaction medium, the copper catalyst and bromide. Finally, the bromide acts as a nucleophile, attacking the butyl group through an SN2 mechanism, giving the product 70a and eliminating 1-bromobutane as a byproduct (Scheme 47).
Another strategy to access benzo[b]selenophenes was described in 2019 [152]. In this protocol, the Oxone® was used as an alternative oxidant for the generation of Se-based electrophile in situ, through an oxidative cleavage of the Se-Se bond of diorganyl diselenides.
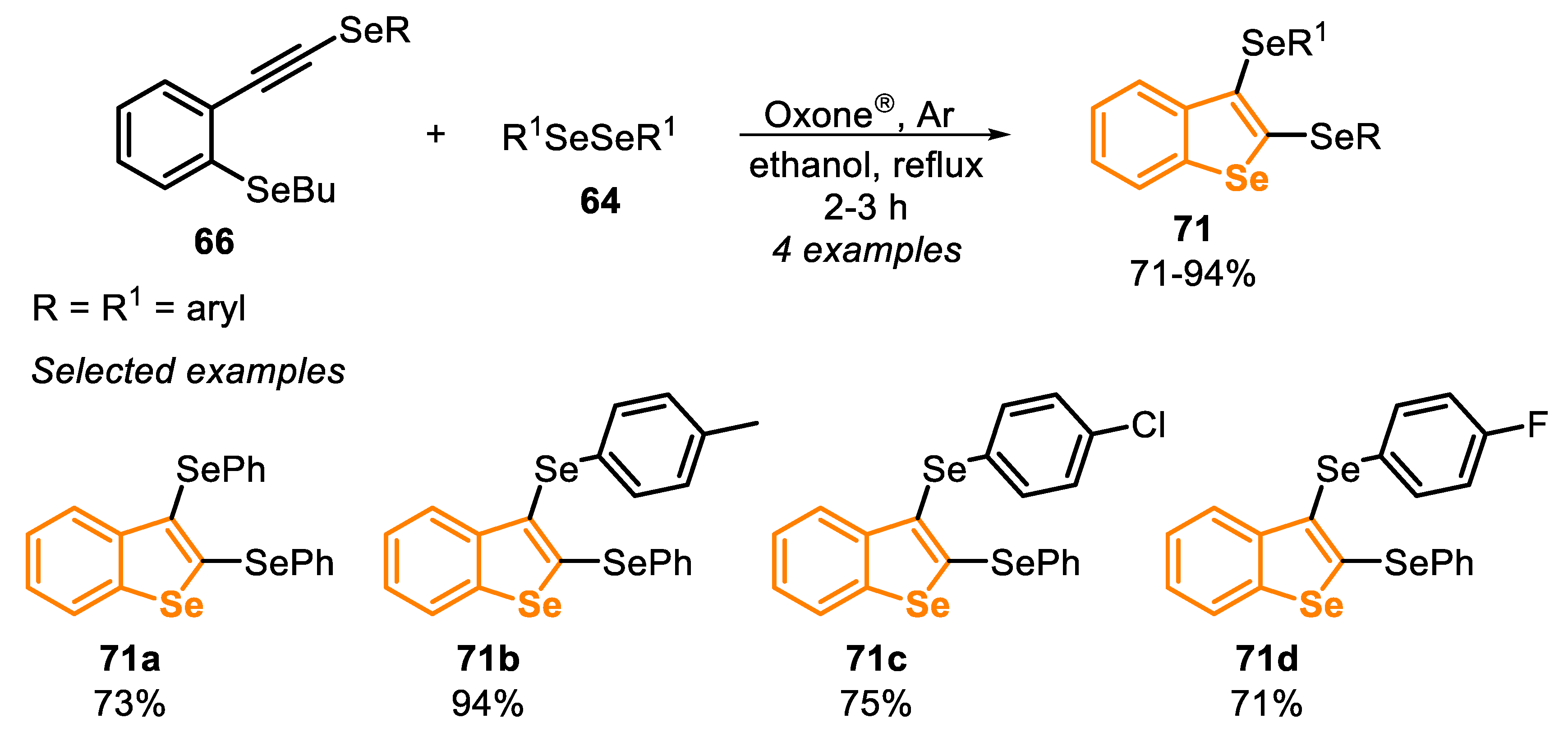
The electrophilic cyclization of ortho-SeBu selenoalkynes 66 (type C precursors) was promoted by the system Oxone®/diorganyl diselenide 64, affording the respective 2,3-bis-selanyl-benzo[b]selenonophenes 71 in good to excellent yields (71–94%) with short reaction times (2–3 h) (Scheme 48). The scope was extended to different ortho-functionalized chalcogenoalkynes, accessing eleven benzo[b]thiophenes and seven benzo[b]furans derivatives.
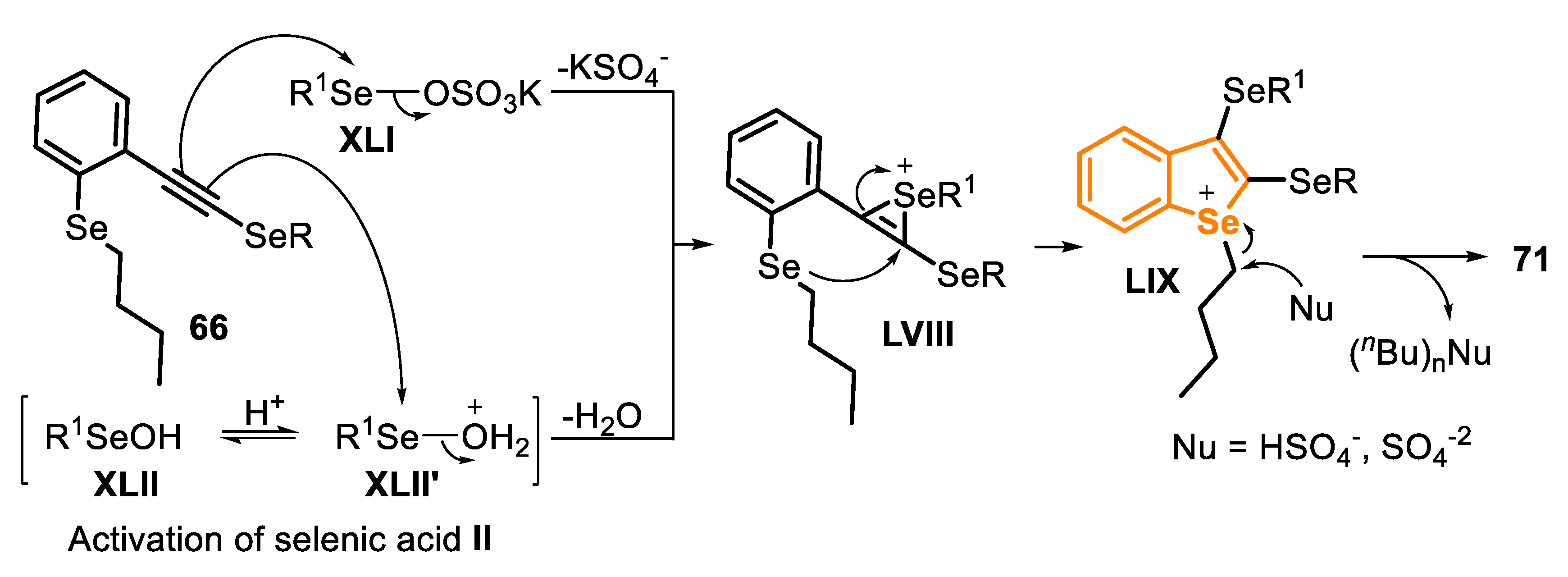
The mechanism, which involves the formation of the Se-based electrophilic species from the system Oxone®/diorganyl diselenide, was already exposed in the previous session (Scheme 36). In the cyclization step, the C(sp) of the type C precursor reacts with the active species XLI and XLII’, generated in situ, to give the intermediate seleniranium LVIII (activation step). Then, the intermediate LVIII undergoes an intramolecular anti-attack by the Se atom (a 5-endo-dig cyclization), producing the intermediate LIX. Finally, the butyl group attached to the selenium atom is removed via an SN2 reaction, by a nucleophile, to generate the product 71 (Scheme 49).
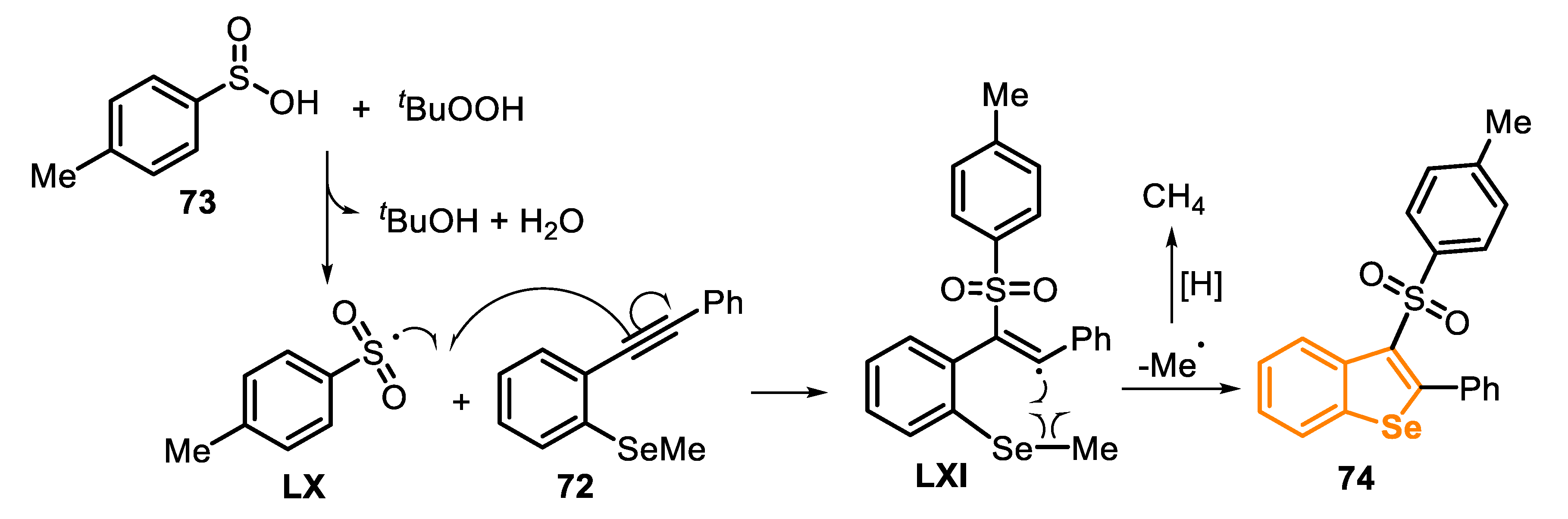
Recently, two important protocols using type C precursors in radical reactions to prepare benzoselenophenes were developed [153,154]. The first method was described in 2017, through a TBHP-promoted radical cyclization between ortho-alkynylaryl selenides 72 (a type C precursor) and sulfinic acids 73 (Scheme 50) [153]. The protocol required a high load of TBHP (80 mol%) and MeCN as a solvent, being the resulting mixture stirred at 100 °C for 1 h. By this procedure, six different 3-(arylsulfonyl)benzoselenophenes 74 were prepared in moderate yields (52–65%). The method was shown to be general and was not sensible to the presence of electron-withdrawing and electron-donor groups attached to the aromatic rings of both the ortho-alkynyl arene and the sulfinic acid portions (Scheme 50).
The mechanism of the synthesis of 3-(arylsulfonyl)benzoselenophene derivatives 74 involves an initial combination of TBHP and sulfinic acid 73, giving the sulfinyl radical LX, through a single electron transfer (SET) pathway, which selectively attacks the C(sp) bond of 72 to afford the vinyl radical LXI. Subsequently, the vinyl radical LXI reacts with the SeMe moiety, in a 5-exo-trig cyclization mode, leading to the product 74 and the elimination of methyl radical. Furthermore, the methyl radical is trapped by a hydrogen in the reaction medium (Scheme 51).
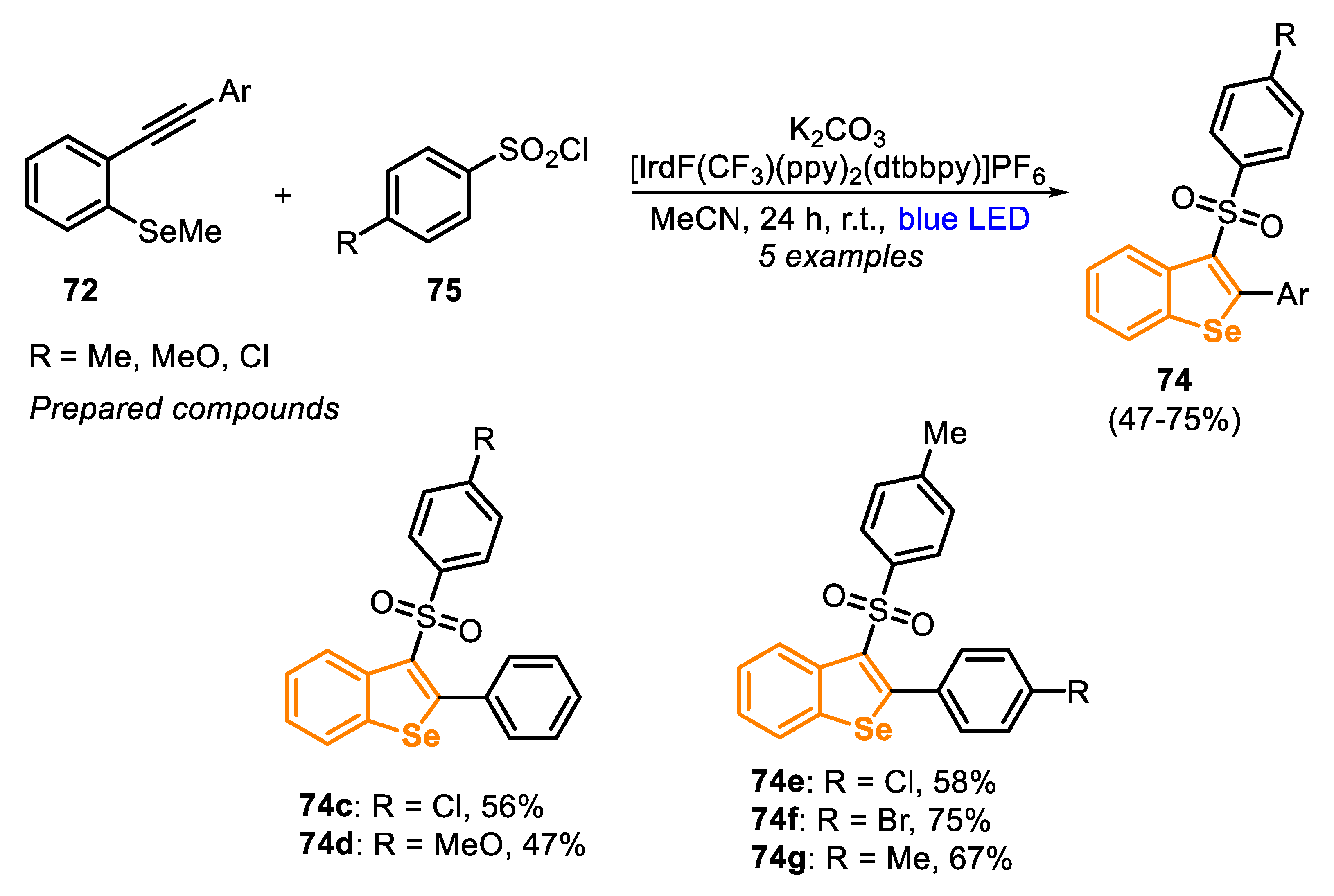
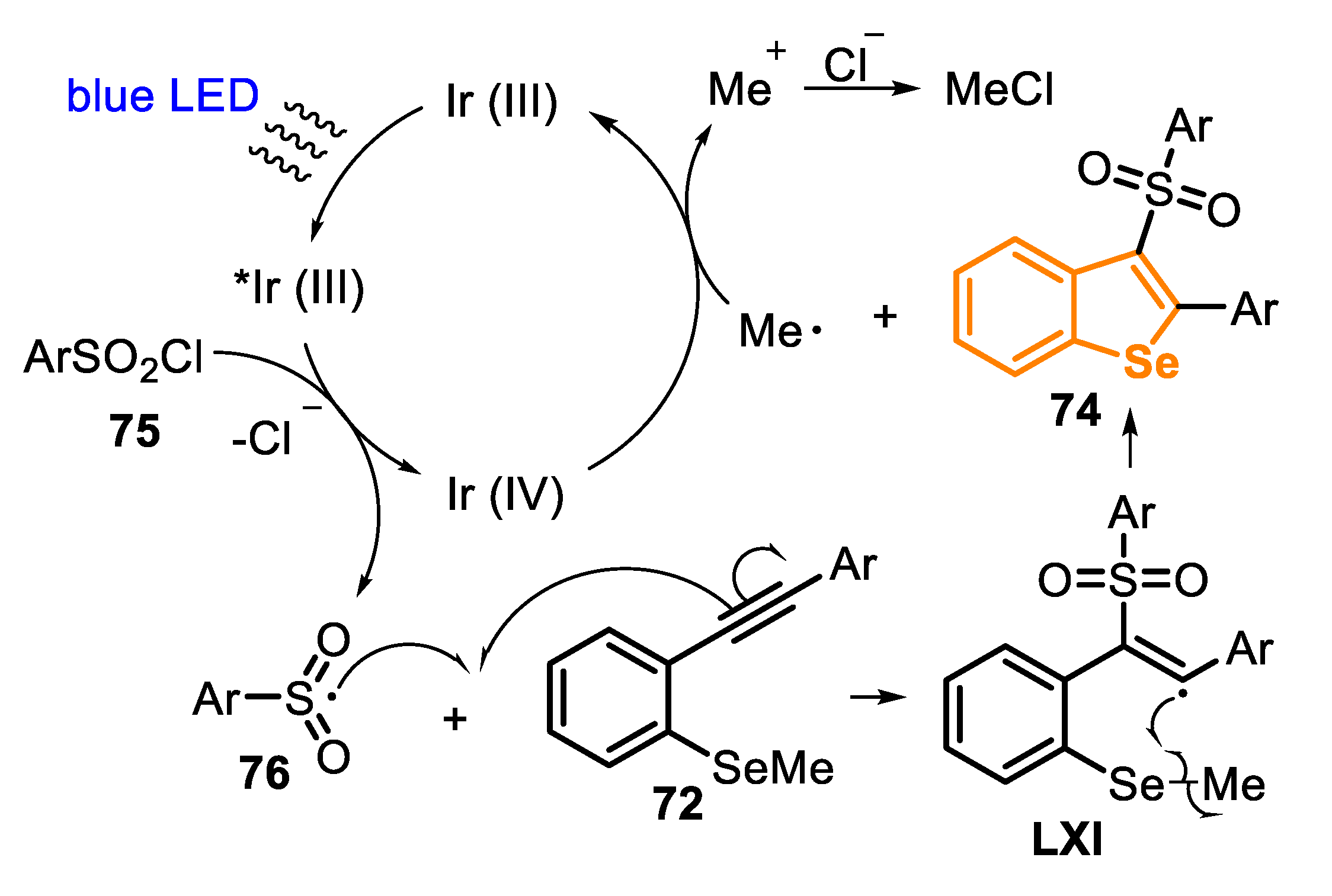
In 2018, the same group [154] described an alternative procedure to prepare the 3-(arylsulfonyl)benzoselenophene derivatives 74, through the visible light photoredox-catalyzed cascade annulation of ortho-alkynylaryl selenides 72 (type C precursor) with sulfonyl chlorides 75 (Scheme 52). The reaction was performed in the presence of an Ir-based photocatalyst (2 mol%) and K2CO3 (2 equiv), in MeCN at room temperature and under blue LED (5 W) irradiation, affording five different functionalized selenophenes 74 in 47–75% yield.
The reaction mechanism is very similar to the annulation previously described in Scheme 51, except in the radical formation step. According to the authors, under the visible light irradiation, the Ir(III)-photocatalyst is converted to the excited state *Ir(III), through a metal-to-ligand charge-transfer (MLCT) phenomena. In the sequence, the excited species *Ir(III) undergoes an oxidative quenching in the presence of sulfonyl chloride 75, through an SET process, releasing the sulfonyl radical 76 into the reaction medium and affording the oxidized Ir(IV) species. Then, an addition of the sulfonyl radical 76 to the alkynyl moiety on the molecule 72 takes place to give the vinyl radical LXI, which performs a radical intramolecular attack in the SeMe moiety (a 5-exo-trig cyclization), affording the product 74 and releasing a methyl radical species. Finally, the methyl radical (Me•) reduces the Ir(IV), regenerating the Ir(III) photocatalyst and releasing a methyl cation (Me+), which is converted to MeCl (Scheme 53).
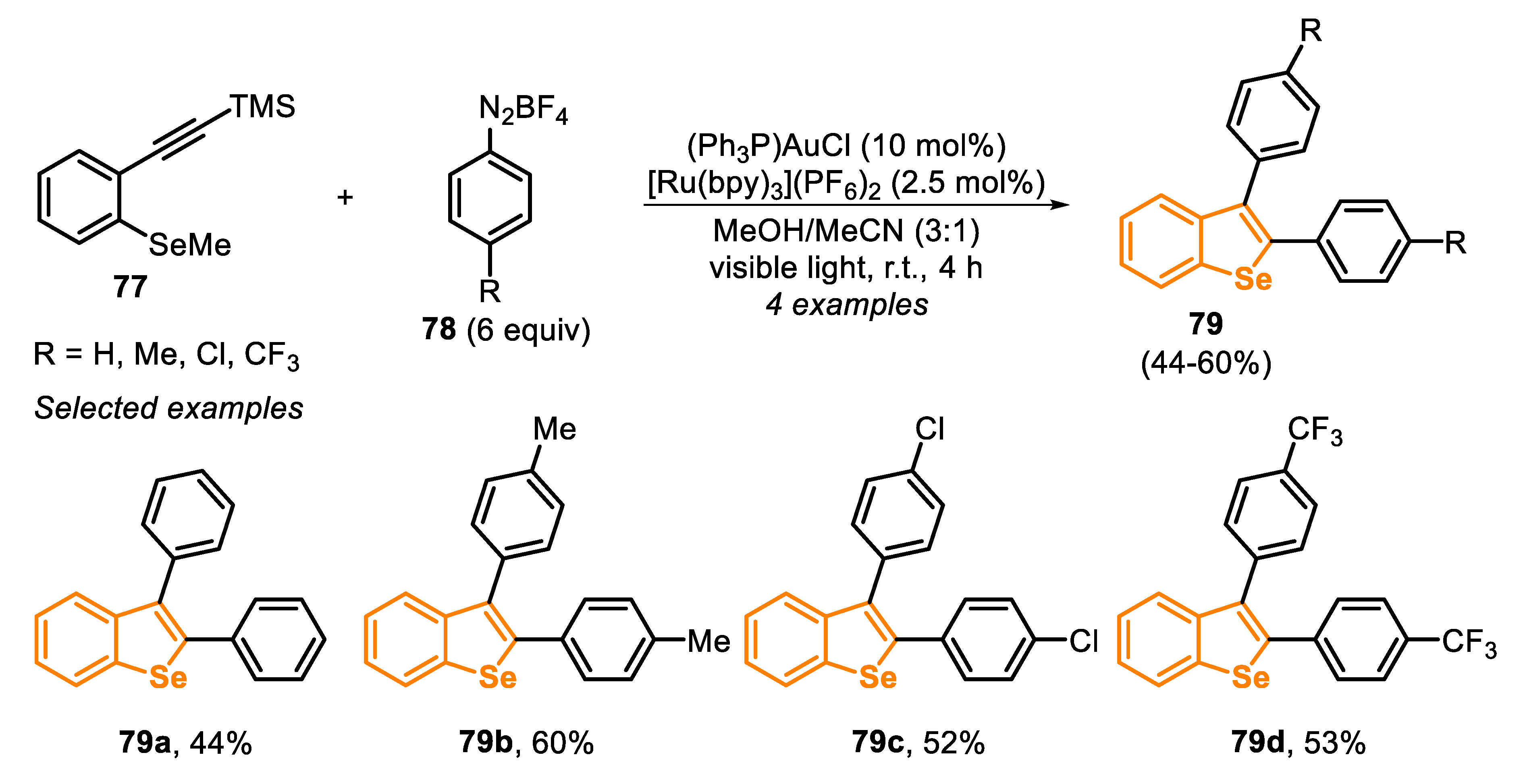
Another visible light-mediated electrophilic cyclization to prepare benzoselenophenes was developed in 2017 [155]. In this protocol, the authors employed the type C precursors, generated in situ by a gold-catalyzed sila-Sonogashira-type coupling, which can be further trapped by an Se-based nucleophile, with a concomitant second arylation, through a domino reaction sequence. In this protocol, a mixture of TMS-(ethynyl) arene 77, arenediazonium salt 78, an Au-based catalyst and a Ru-based photocatalyst in MeOH/MeCN (3:1) as the solvent, was irradiated by visible light (21 W fluorescent light bulb) for 4 h, yielding the respective products 79 in moderate yields (Scheme 54).
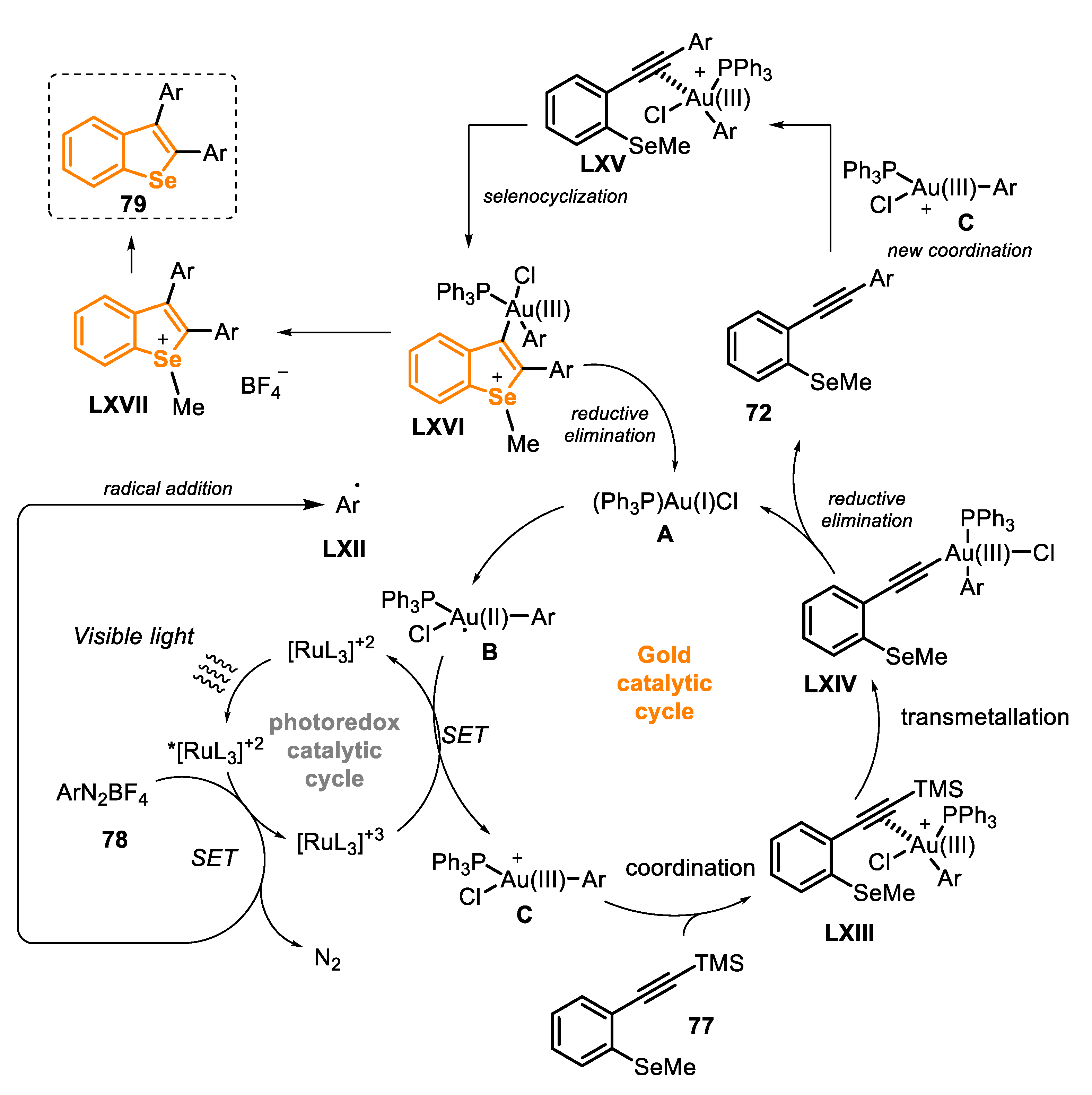
The reaction mechanism involves two simultaneous catalytic cycles, the Ru(II)-photoredox and the Au(I)-catalysis (Scheme 55). Initially, the Ru(II) complex is irradiated by the visible light, affording the excited long-lived Ru(II)* species, which, in the presence of the diazonium salt 78, undergoes an oxidative quenching, being converted to the Ru(III) species, releasing into the reaction medium N2 and one equivalent of the aryl radical LXII (the photoredox catalytic cycle). The aryl radical LXII reacts with the Au(I) precatalyst A to be converted to the unstable radical organogold(II) intermediate B. The Au-centered radical B is able to reduce Ru(III) to the Ru(II) photocatalyst, through an SET process, being converted to the cationic organogold(III) derivative C, a strong electrophile. Then, the intermediate C, in the presence of TMS-(ethynyl) arene 77, is converted to the intermediate LXIII, through the alkyne coordination with the Au complex, which undergoes a transmetallation of the C(sp)-Si bond, giving the Au(III) intermediate LXIV. Finally, LXIV undergoes a reductive elimination, releasing the precursor 77 and regenerating the Au(I) catalyst A to restart a new reaction cycle (the gold catalytic cycle). Then, the compound 72 generated in situ, coordinates with the highly electrophilic Au(III) species C, affording the intermediate LXV, which undergoes an intramolecular seleno-cyclization, through a 5-endo-dig reaction, to access the cyclic intermediate LXVI. Finally, a reductive elimination regenerates the Au(I) catalyst and delivers the cationic intermediate LXVII, which is converted to the desired product 79.
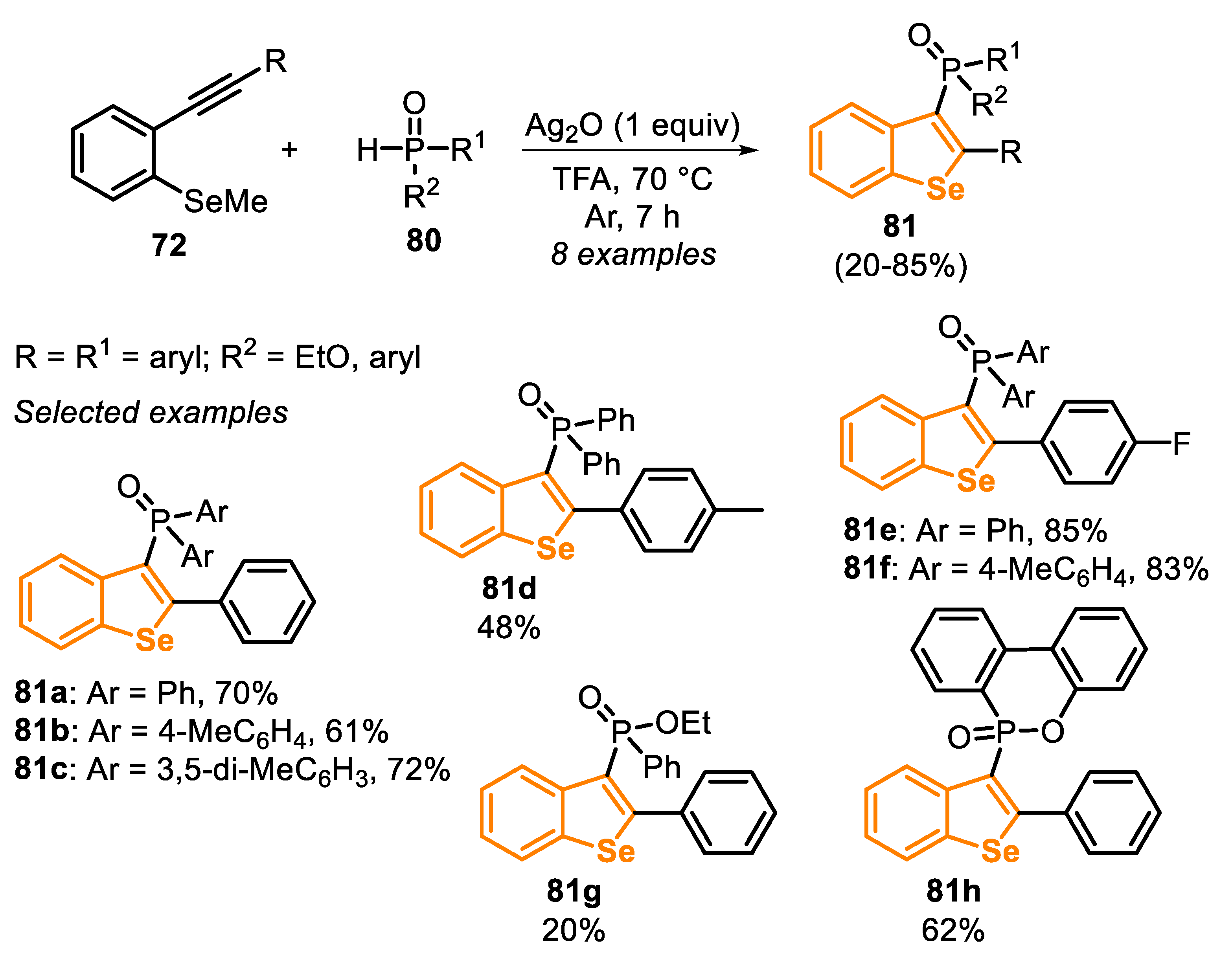
In 2019, a new protocol for the construction of benzo[b]selenophenes through an Ag-promoted radical cyclization between the type C precursor 72 and secondary phosphine oxides 80 was developed (Scheme 56) [156]. This protocol allowed us to extend the synthesis to a wide scope of benzoselenophene derivatives 81a-c using phosphines bearing aryl groups (Ph, 4-MeC6H4, 3,5-di-MeC6H3) in good yields (61–72%). The influence of electron-donor (R = 4-MeC6H4) and electron-withdrawing substituents (R = 4-FC6H4) in the pendant aromatic ring of the arylalkynes (type C precursors) was evaluated. The electron-rich system was less reactive than the electron-deficient and the neutral ones, and the product 81d was obtained in 48% yield, while 81e and 81f were isolated in 85% and 83% yields, respectively. Moreover, the reactivity of differently substituted phosphine oxides 80 was accessed, with ethyl phenylphosphinate and 9,10-dihydro-9-oxa-10-phosphaphenanthrene 10-oxide (DOPO) derivatives affording the respective benzoselenophenes 81g and 81h in 20% and 62% yields.
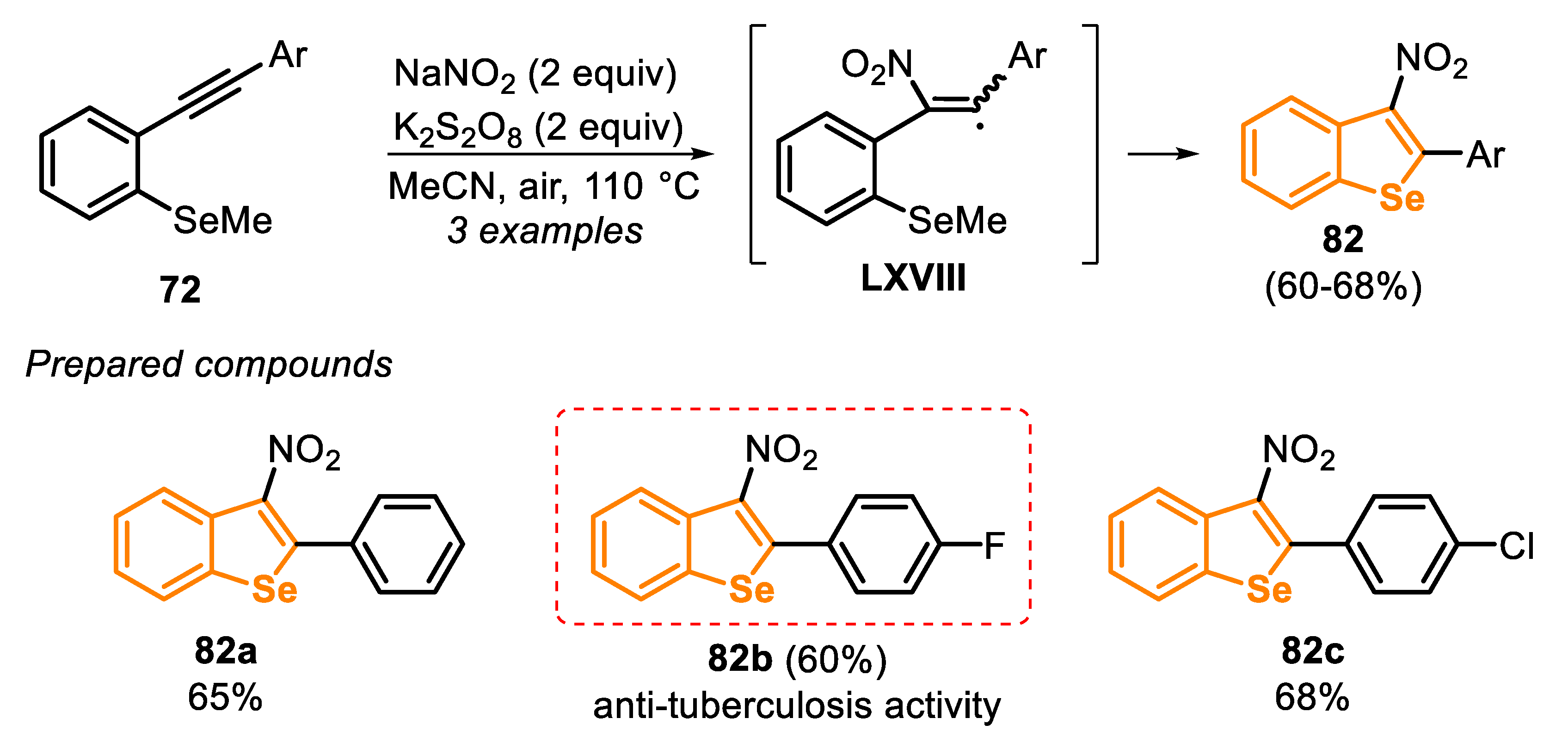
In this year (2020), a similar radical protocol was described via a 5-exo-trig cyclization of type C precursors 72, being the reaction promoted by K2S2O8 (2 equiv) and NaNO2 (2 equiv) as oxidants, in the presence of MeCN as a solvent [157]. The method was mainly used to prepare benzo[b]thiophenes (19 examples, 46–90% yields) and was extended to prepare selenium analogues (only three examples). After stirring the reaction mixture for 12 h at 110 °C under open air, 3-nitrobenzo[b]selenophenes 82a-c were obtained in moderate yields (60–68%). The protocol was applied to neutral (Ar = phenyl) and electron-deficient (Ar = 4-FC6H4 and 4-ClC6H4) aryl groups, and no electronic effect was observed (Scheme 57).
This was the first example of using highly unstable 2-nitrovinyl radicals in the formation of C-S and C-Se bonds, circumventing the problems usually faced in classical methods, including the application of harsh reaction conditions, by employing strong acids, low yields and poor regioselectivity. However, the reaction of the thio-analogue type C precursors with the TMS-ethynyl and alkylethynyl moieties did not afford the desired products, evidencing a limitation of the method. The reaction mechanism was proposed after functional density theory (DFT) calculations and control experiments, confirming the presence of the vinyl radical intermediate LXVIII. In addition, the products were tested for their anti-tuberculosis activity in vitro, and the fluoro-containing 3-nitrobenzoselenophene 82b showed a potent anti-tuberculosis activity against drug-resistant Mtb strains, with an MIC90 of 2 μg mL−1.
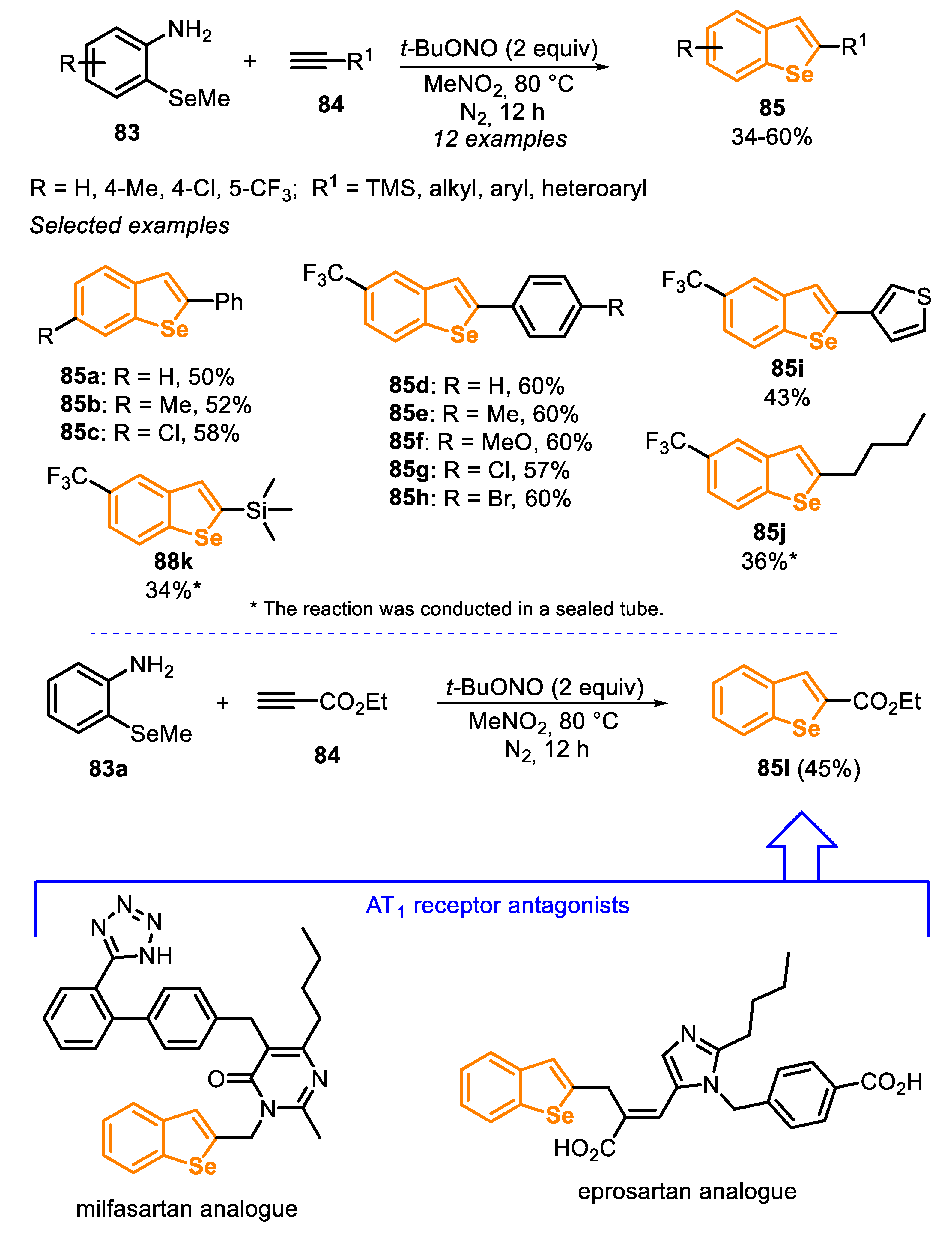
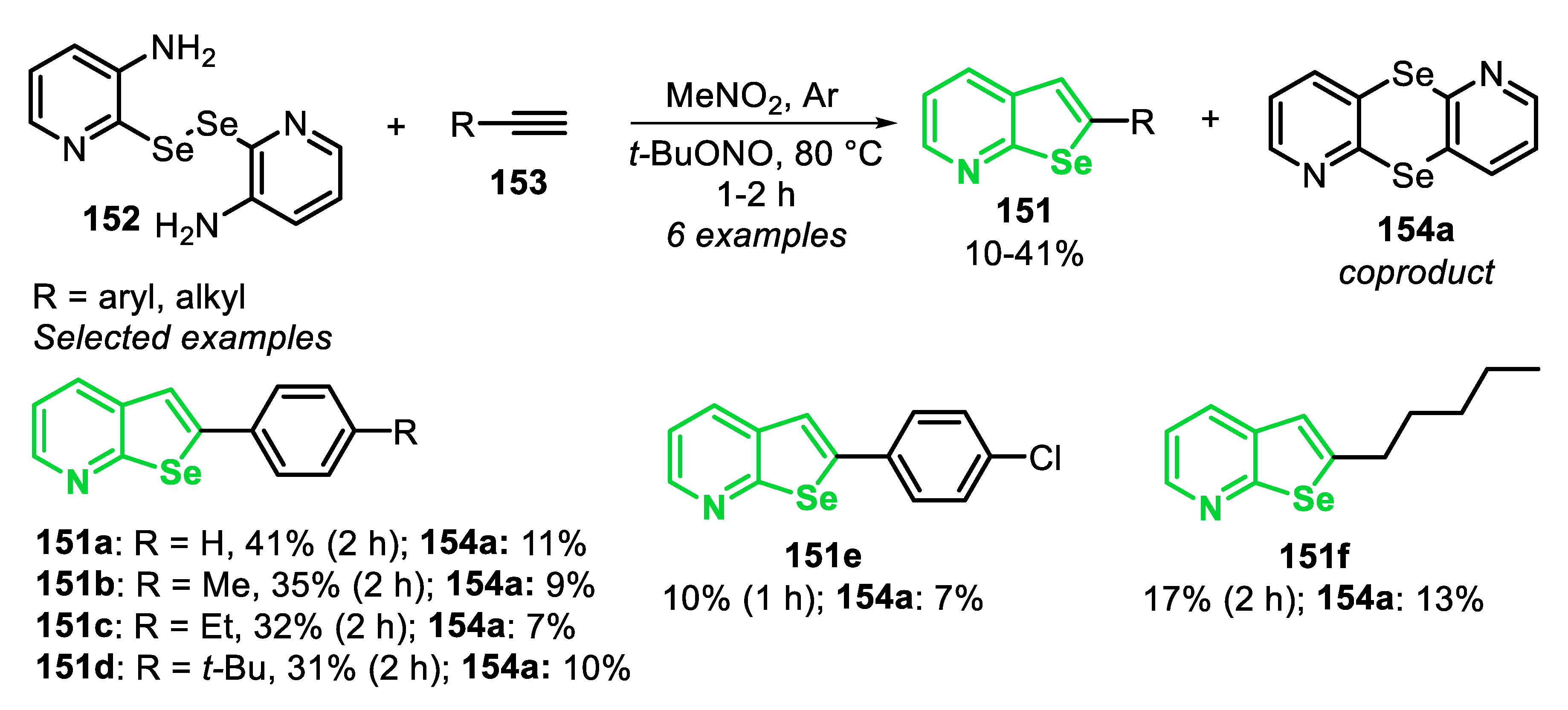
Closing our discussion on the reactivity of type C precursors in the synthesis of benzoselenophenes, a new strategy to access benzo[b]selenophenes 85 from ortho-methylselanyl-arylamines 83 and alkynes 84 is presented (Scheme 58) [158]. The protocol presented the advantage of generating aryl radical intermediates from arylamines 83 and tert-butyl nitrite (t-BuONO), avoiding the use of the difficult to prepare and unstable diazonium salts. t-BuONO acts in the diazotization reaction of ortho-methylselanyl-arylamines 83, affording the reactive radical species in situ. The optimal procedure involves the reaction between ortho-methylselanyl-arylamines 83 and alkyne 84 (3 equiv) in the presence of t-BuONO (2 equiv) as a nitrosating agent and MeNO2 as the solvent, at 80 °C for 12 h, under nitrogen atmosphere. The methodology was regioselective and the products 85 were obtained in low to moderate yields (34–60%). Apparently, the method was not sensible to the electronic effects of substituents in the para-position of the aryl substituted alkynes 84 nor in the arylamines 83, as shown for the yields obtained for the benzo[b]selenophenes 85d–h vs. 85b-c. In addition, the authors used different terminal alkynes 84f–h, including 3-ethynylthiophene, hexyne, ethynyltrimethylsilane, obtaining the respective products 85i–k in satisfactory yields. The scope of the reaction was extended to several thio-analogues to prepare benzothiophenes in good yields. Ethyl propiolate 84i, together with ortho-methylselanyl-benzylamine 83a, were used in the preparation of benzoselenophene 85l, a key intermediate in the synthesis of milfasartan and eprosartan analogues.
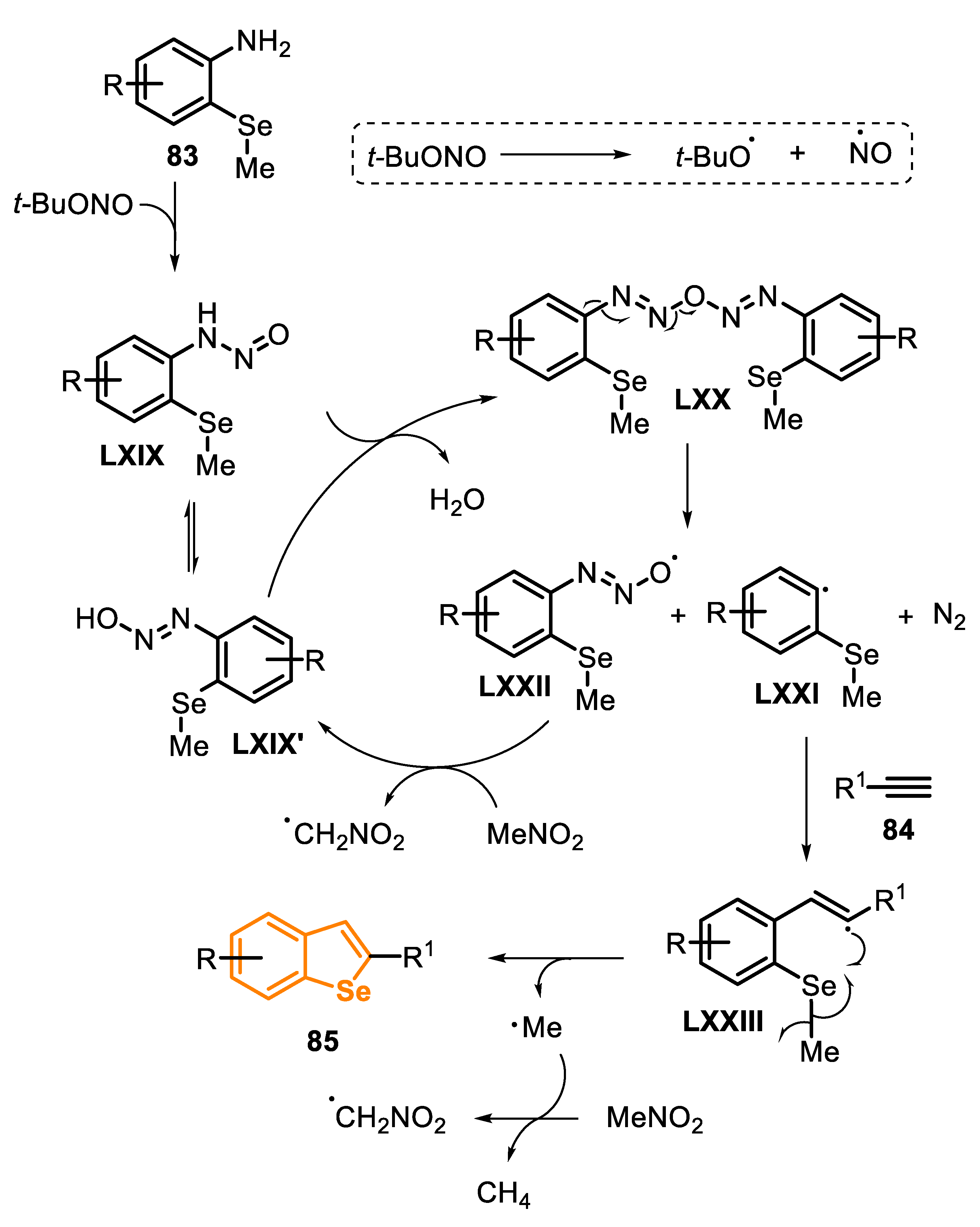
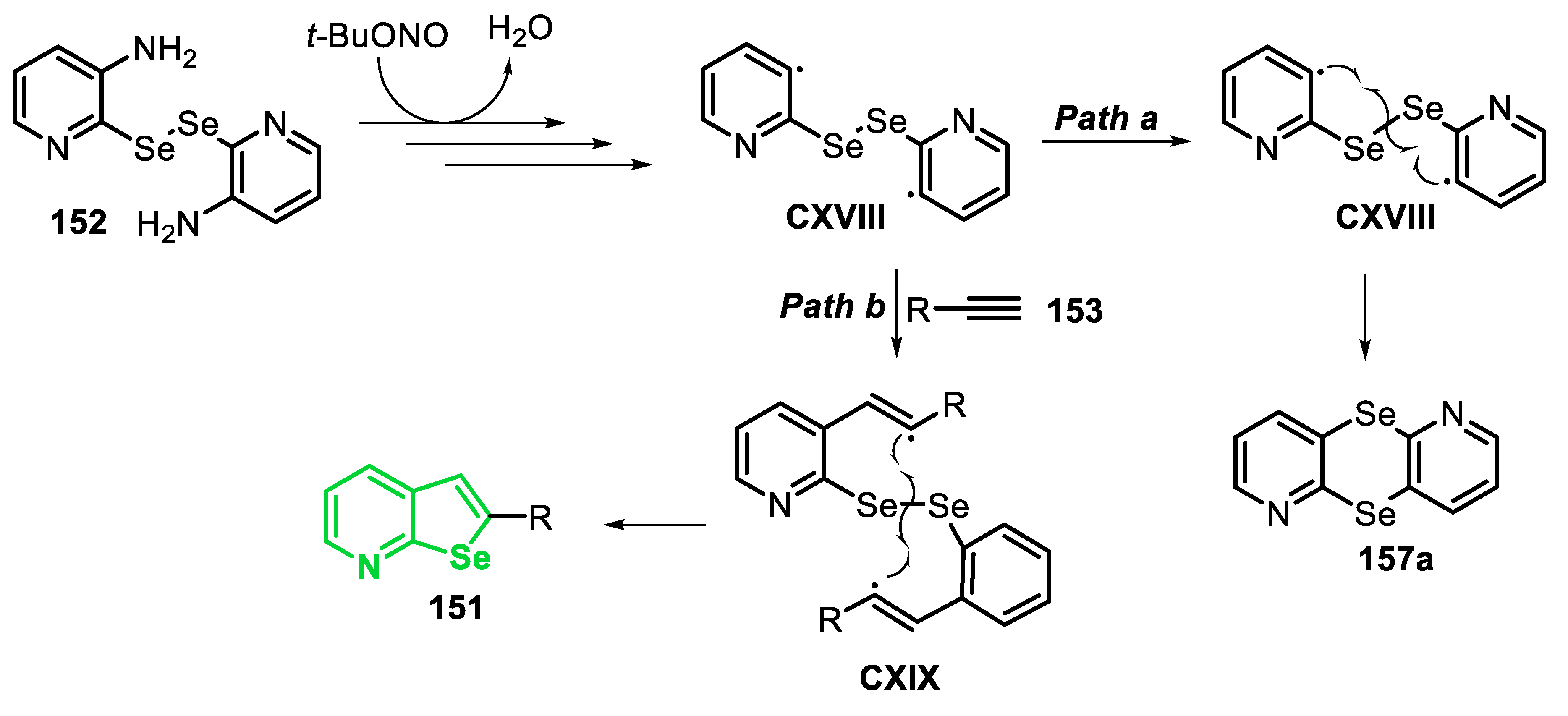
After some control experiments, a mechanism for the intermolecular radical cascade reaction was proposed (Scheme 59). Initially, arylamine 83 reacts with t-BuONO to generate nitrosamine LXIX, which undergoes a self-condensation reaction to generate the diazo anhydride LXX. The N-O homolysis of LXX provides the aryl radical LXXI, along with the azoxy radical LXXII, and releases molecular nitrogen. Then, the radical LXXII abstracts a hydrogen atom from the solvent, in order to generate the diazo hydroxide LXIX’ (easily interconverted into nitrosamine LXIX), which can react again to give the aryl radical LXXI. The addition of LXXI to alkyne 84 leads to the vinyl radical LXXIII, which reacts via an intramolecular homolytic substitution, by the Se atom, to give the benzo[b]selenophene 85, followed by the elimination of methyl radical (Me•), which can react via an H-abstraction, from the solvent, giving CH4.
3.2. Starting from Type D Precursors
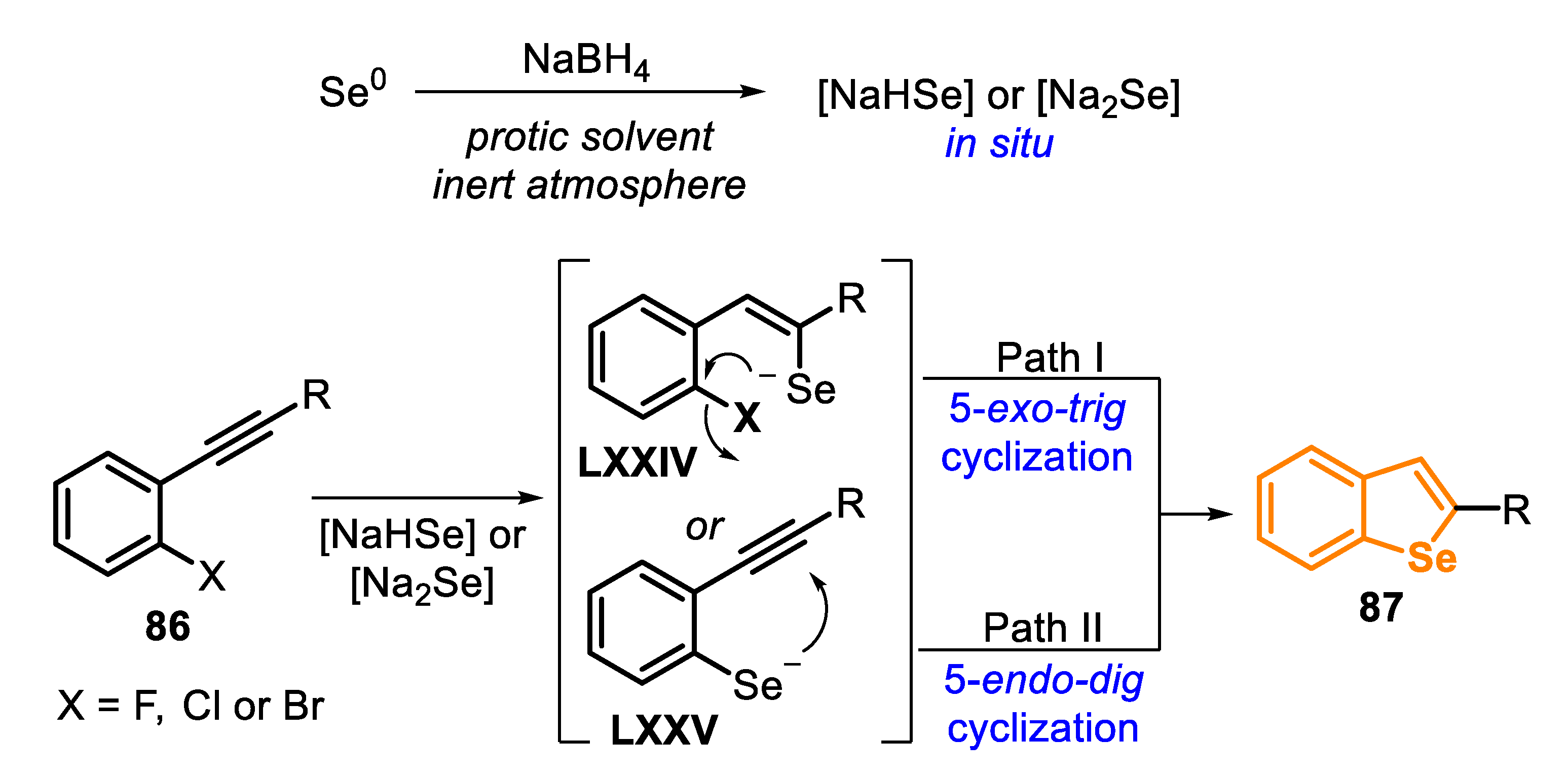
In the last few decades, ortho-halo-substituted ethynylarenes 86 (type D precursors) have been frequently used as starting material in the synthesis of benzoselenophenes 87. The most common protocol involving these precursors involves the generation of Se-based nucleophilic species in situ, using sodium borohydride (NaBH4) and protic solvents. Some parameters, including stoichiometry, solvents and the presence of base, may directly affect the identity of the formed reactive species [159].
In general, the mechanism for the synthesis of benzoselenophenes 87 using nucleophilic Se species, and type D precursors 86 can follow two different paths (Scheme 60). One possible pathway involves firstly the formation of the intermediate LXXIV, after an attack by NaHSe or Na2Se (pre-formed in situ) to the triple bond of 86, which is followed by an intramolecular SNAr reaction (Path I). However, the process can also follow a nucleophilic substitution (SNAr) of the selenide anion, by the X group, generating the intermediate LXXV, which undergoes an intramolecular seleno-cyclization by the reaction of the selenium anion to the triple bond (Path II).
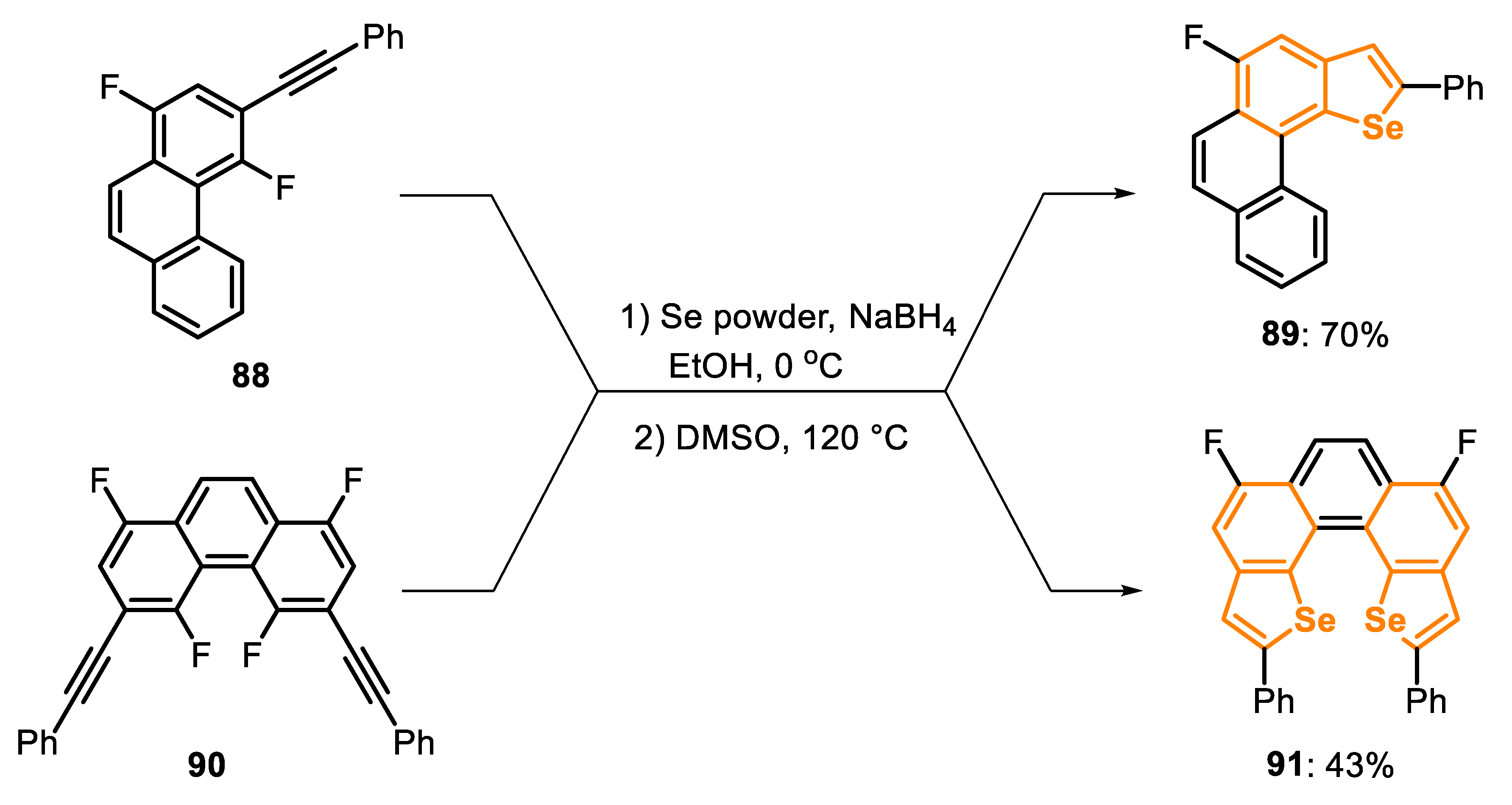
These reaction pathways have been widely applied over the last few years and can be used in several organic transformations, even for the construction of complex structures. For example, in this year the synthesis of Se-helicenes 89 and 91 derivatives was reported [160]. In this one-pot protocol, the Se-based nucleophilic species were firstly generated (in situ) through the reaction of Se powder with NaBH4 at 0 °C for 40 min. In the sequence, the respective ortho-fluoro-ethynylarene 88 and 90 and DMSO were added to the reaction flask, and the temperature was increased to 120 °C. After 18 h, the respective products 89 and 91 were obtained in 70% and 43% yields, respectively (Scheme 61).
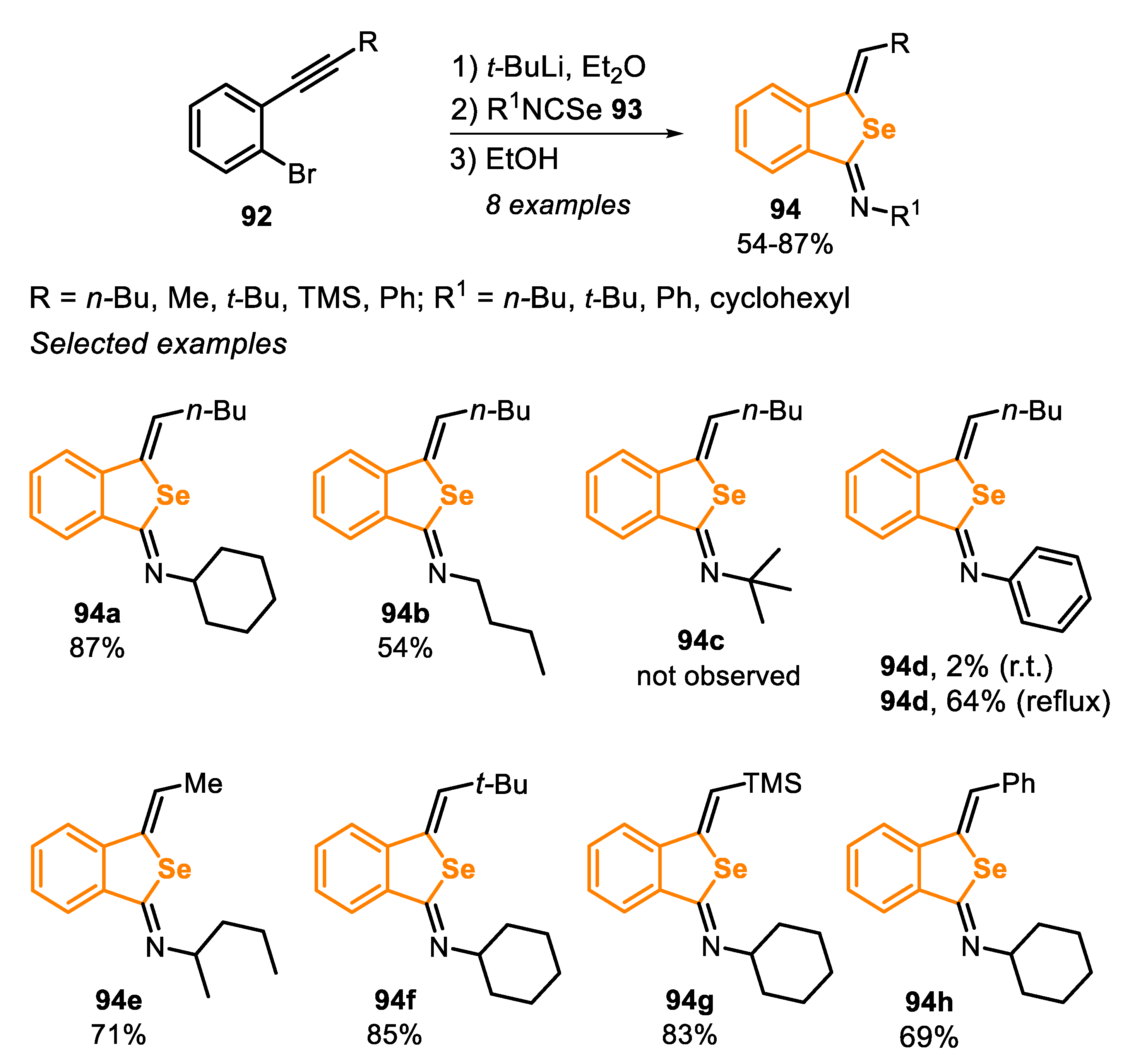
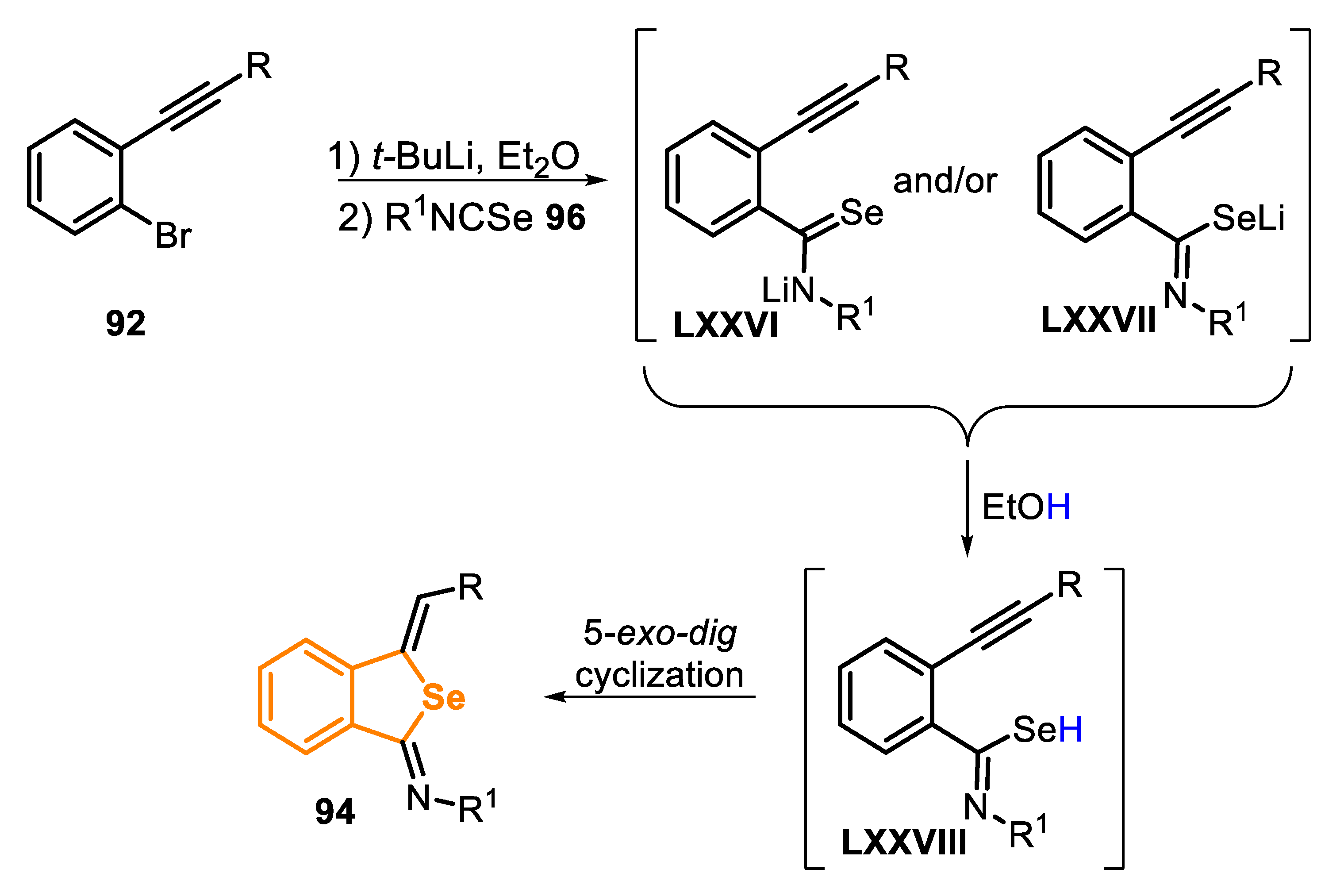
Type D precursors normally lead to the formation of benzo[b]selenophenes; however, benzo[c]selenophenes can also be obtained by the lithiation of ortho-bromo-ethynylarene 92 with t-BuLi in Et2O, followed by the addition of isoselenocyanates 93 and EtOH as a proton source (Scheme 62) [161]. Through this method, it was possible to prepare eight different 3-methylidenebenzo[c]selenophenes 94 in moderate to good yields (54–87%). In this work, the authors used different o-bromo-ethynylarenes 92 with alkyl, phenyl and trimethylsilyl groups linked to the triple bond, that were reacted with alkyl and phenyl isoselenocyanates 93. Phenyl isoselenocyanate (R1 = C6H5) was less reactive than the alkyl analogues, and reflux temperature was necessary to afford the respective product 94d acceptable yields (64% vs. 2% yield at room temperature). The method was not applicable to the bulky tert-butyl isoselenocyanate 93c; in this case, a complex mixture was obtained instead of the expected benzo[c]selenophenes 94c (Scheme 62).
Aiming to elucidate the reaction mechanism, a reaction was carried out in the absence of a proton source (EtOH), but no product was formed, and the starting isoselenocyanate 93 was recovered. Based on this, it was proposed that the reaction passes by the formation of selenol intermediate LXXVIII through the reaction of EtOH with intermediates LXXVI and LXXVII. Once formed, the key selenol LXXVIII reacts by a selective 5-exo-dig intramolecular seleno-cyclization, giving the respective benzo[c]selenophene 94 (Scheme 63).
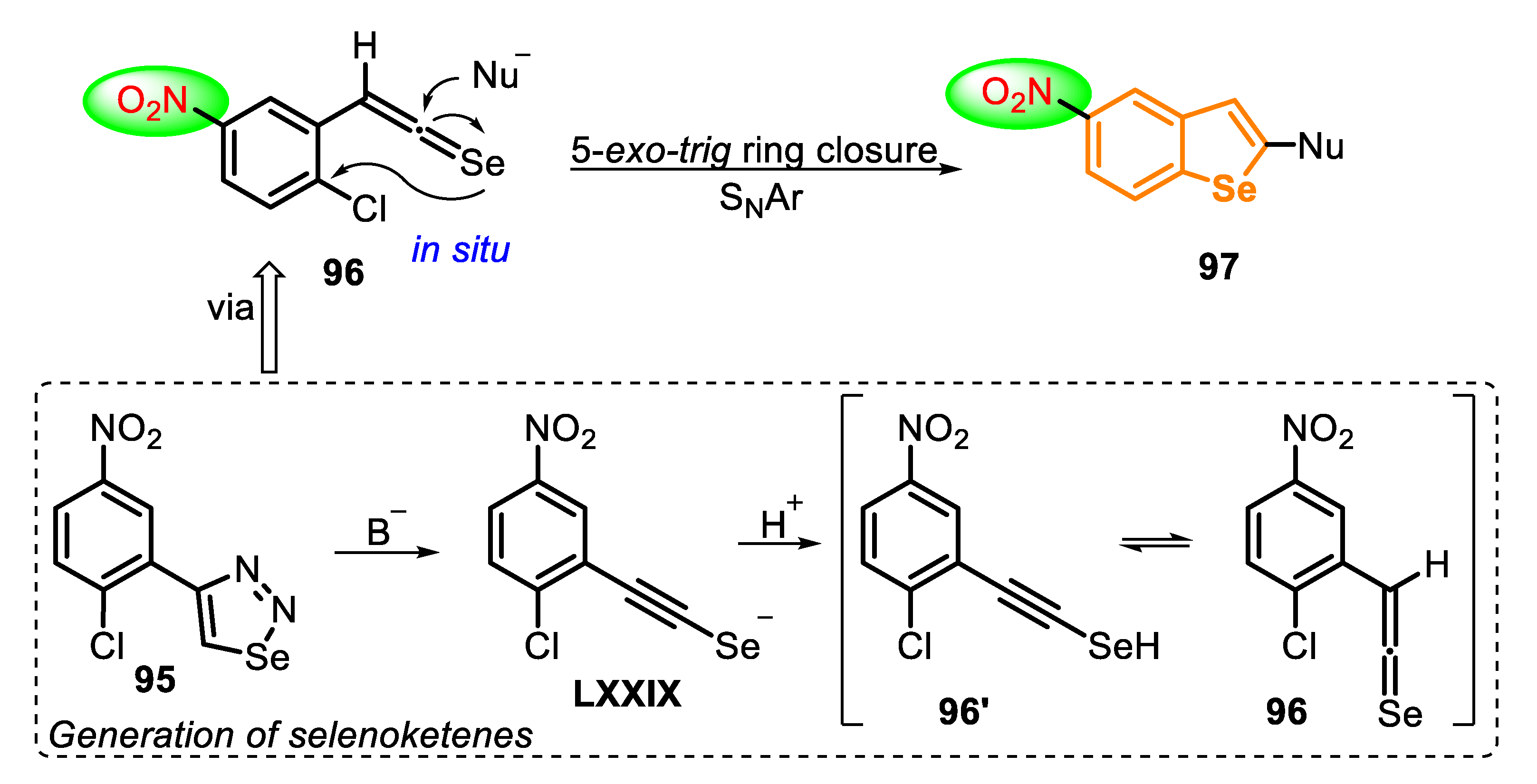
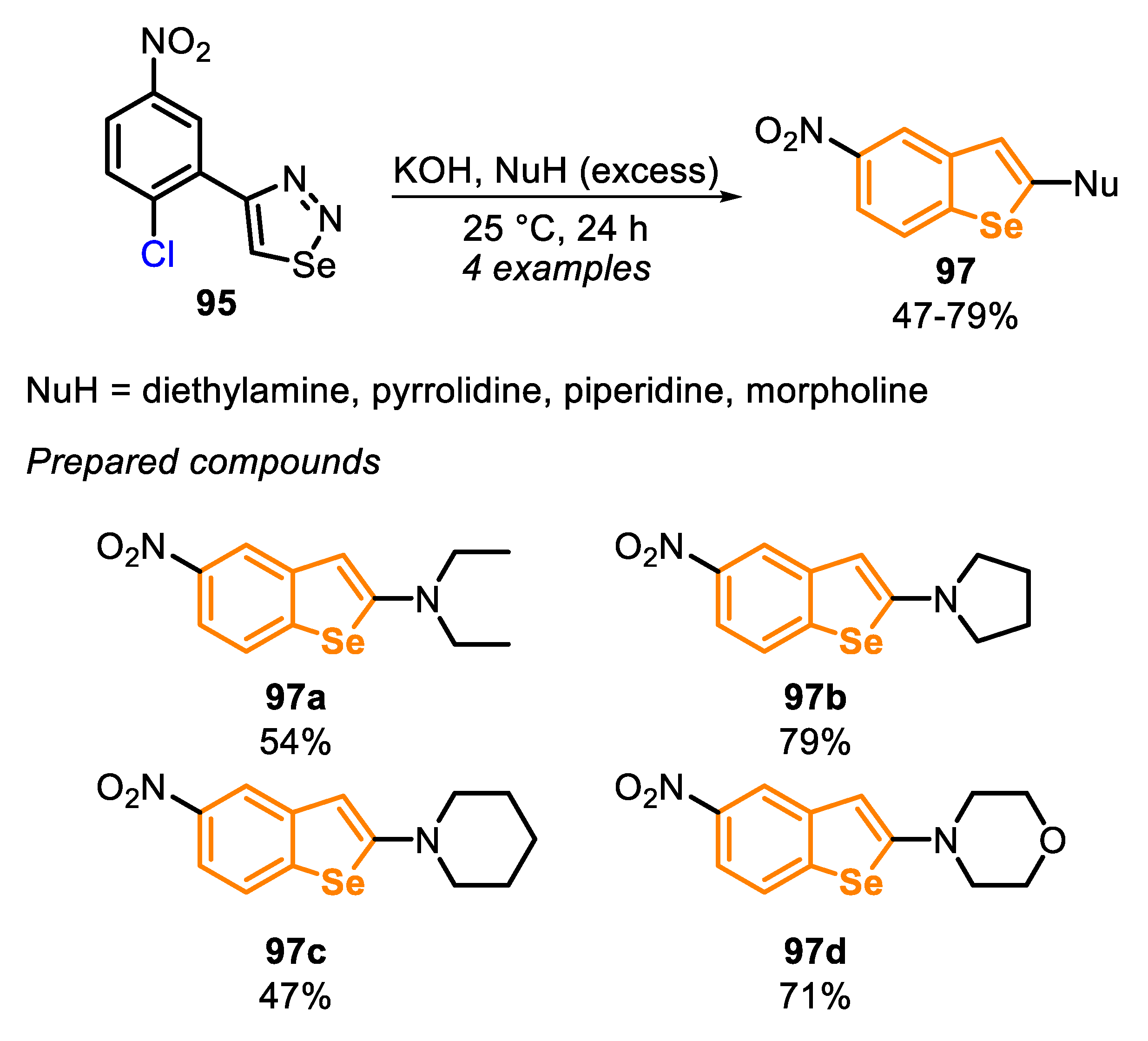
In 2013, a new approach, based on an intramolecular SNAr cyclization to obtain benzo[b]selenophenes 97, was developed [162]. The protocol is grounded in a ring closure of ortho-chloro-arylselenoketenes 96, by a 5-exo-trig cyclization, using different secondary amines both as nucleophile and solvent (Scheme 64). The selenoketene precursor 96 is generated in situ from the decomposition of 4-substituted 1,2,3-selenadiazoles 95 (with elimination of N2). The treatment of 95 with a strong base afforded the alkynylselenolate LXXIX (confirmed by trapping using MeI), that is converted to a mixture of alkyneselenol 96′ and tautomeric selenoketene 96, after protonation.
Using this strategy, four nitro-substituted benzo[b]selenophenes derivatives 97 were prepared in moderate to good yields (47–79%). Initially, 4-(2-chloro-5-nitrophenyl)-1,2,3-selenadiazole (95) were treated with KOH for the formation of the selenoketene 96 in situ. In the sequence, a secondary amine was added, and the resulting mixture was stirred at 25 °C for 24 h (Scheme 65).
The presence of the NO2 group at the para position to the chlorine is mandatory for the reaction success, once it favors the intramolecular SNAr reaction, due to the strong electron-withdrawing effect. No cyclized product was observed starting from 4-(2-chlorophenyl)-1,2,3-selenadiazole, even at 130 °C, just accessing selenoamide derivative after the final acidic treatment. The protocol was expanded to the synthesis of benzo[b]thiophene derivatives, starting from 4-(2-chloro-5-nitrophenyl)-1,2,3-thiadiazoles instead the 1,2,3-selenadiazole analogues.
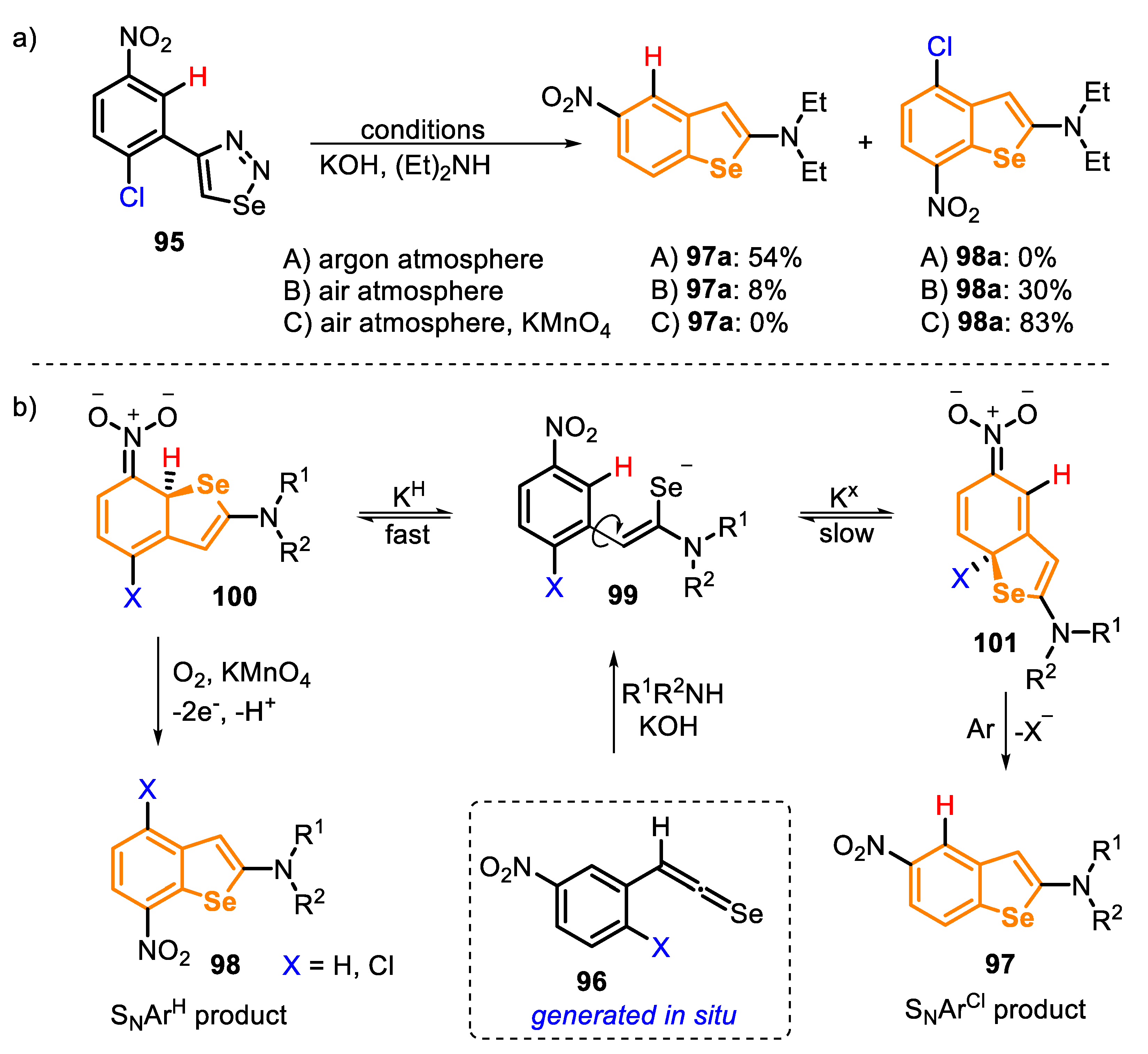
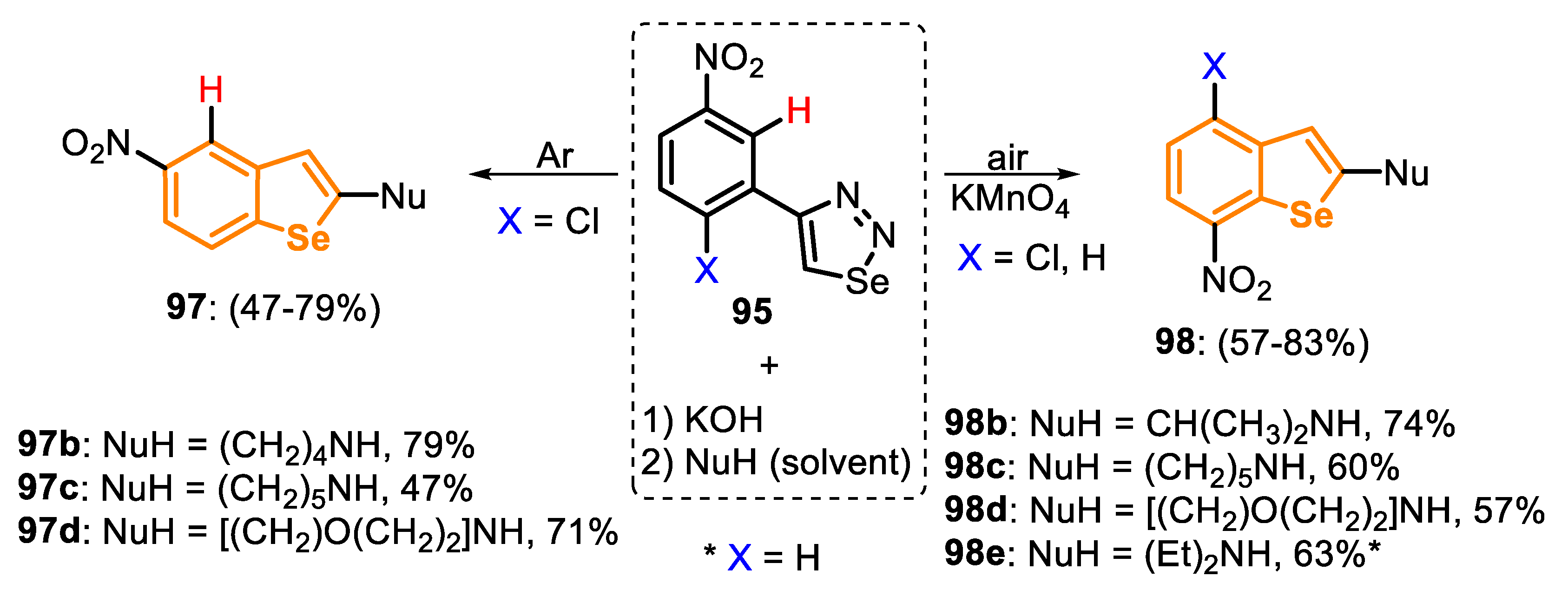
In 2013, an interesting study on the 5-exo-trig cyclization of selenoketenes 96, obtained in situ from the treatment of 4-(2-chloro-5-nitrophenyl)-1,2,3-selenadiazole (95), was reported, as described above [163]. The authors discovered that the reaction course can be completely changed by adding a strong oxidant in the reaction medium. When the reaction described in Scheme 65 was performed under an air atmosphere, a mixture was created between the SNArCl 97a and SNArH 98a products, which were isolated in 8% and 30% yields, respectively (Scheme 66, condition B). This observation indicates that the formation of the σH-adduct is faster than the σCl one. The reaction was directed to the exclusive formation of the σH-adduct, through the addition of a strong oxidant (KMnO4), by an oxidative nucleophilic substitution of hydrogen, giving the product 98a in 83% yield (Scheme 66, condition C).
The high regioselectivity achieved in the reaction allowed us to access four benzoselenophene derivatives via SNArCl (97: 47–79% yields) and five via SNArH (98: 57–83% yields), using different secondary amines as nucleophiles. One example was conducted starting from 4-(5-nitrophenyl)-1,2,3-selenadiazole (X = H), which, after the generation of the ketene intermediate, reacted with EtNH2 in the presence of KMnO4/air, affording the respective selenophene 98e in 63% yield (Scheme 67).
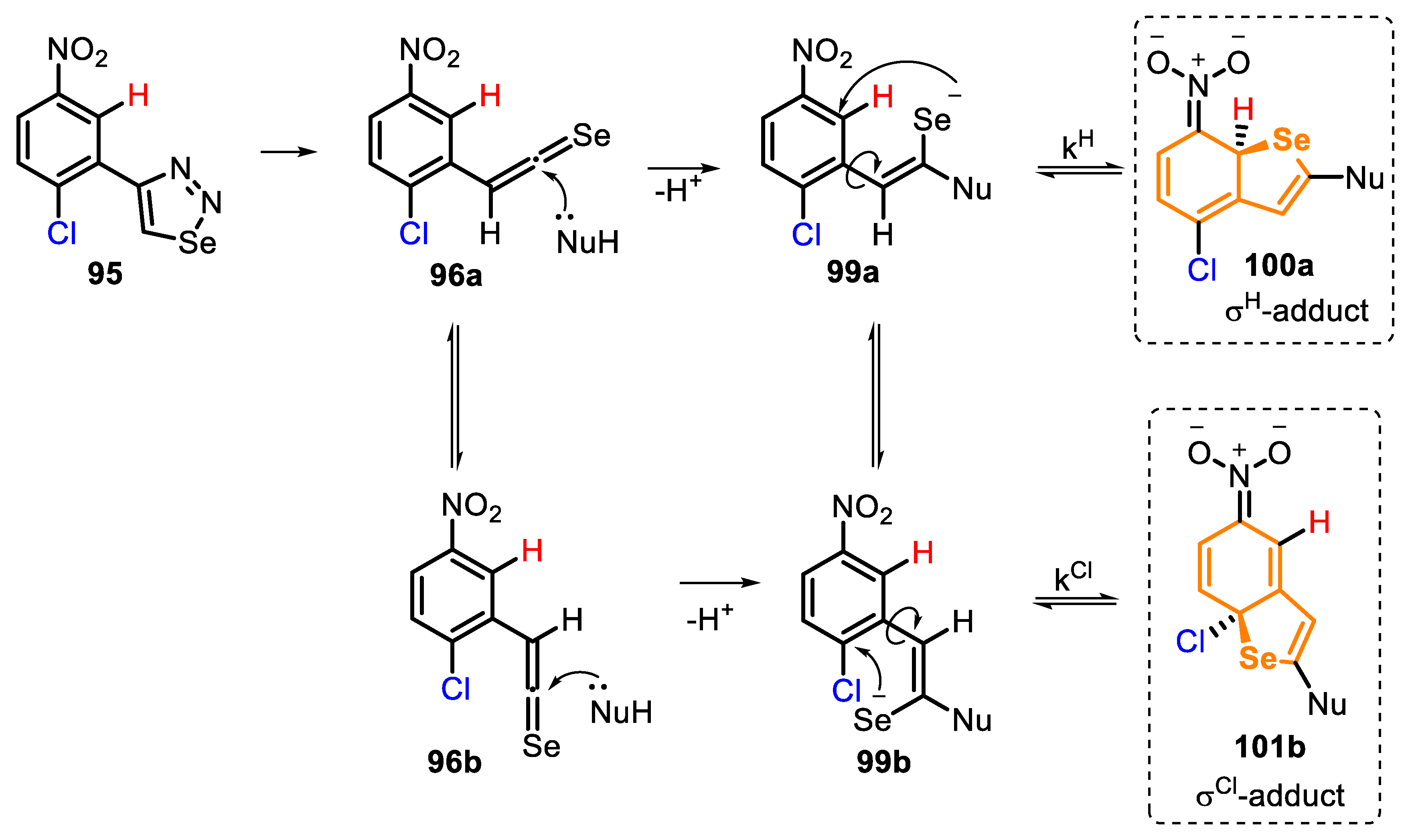
According to the authors, the selenoketene intermediate 96 can exist in two rotational conformers, 96a and 96b. In each case, the sequential nucleophilic attack occurs on the opposite side of the bulky aryl ring, affording the conformeric eneselenolates 99a and 99b, which, after the intramolecular attack from selenium anion, lead to the respective σH- and σCl-adducts 100a and 101b (Scheme 68).
3.3. Starting from Type E Precursor
A third usual strategy to prepare benzoselenophenes is through the cyclization of type E precursors. This approach involves electrophilic aromatic substitution reactions (SEAr), employing different selenium derivatives and arylalkynes (the type E precursors).
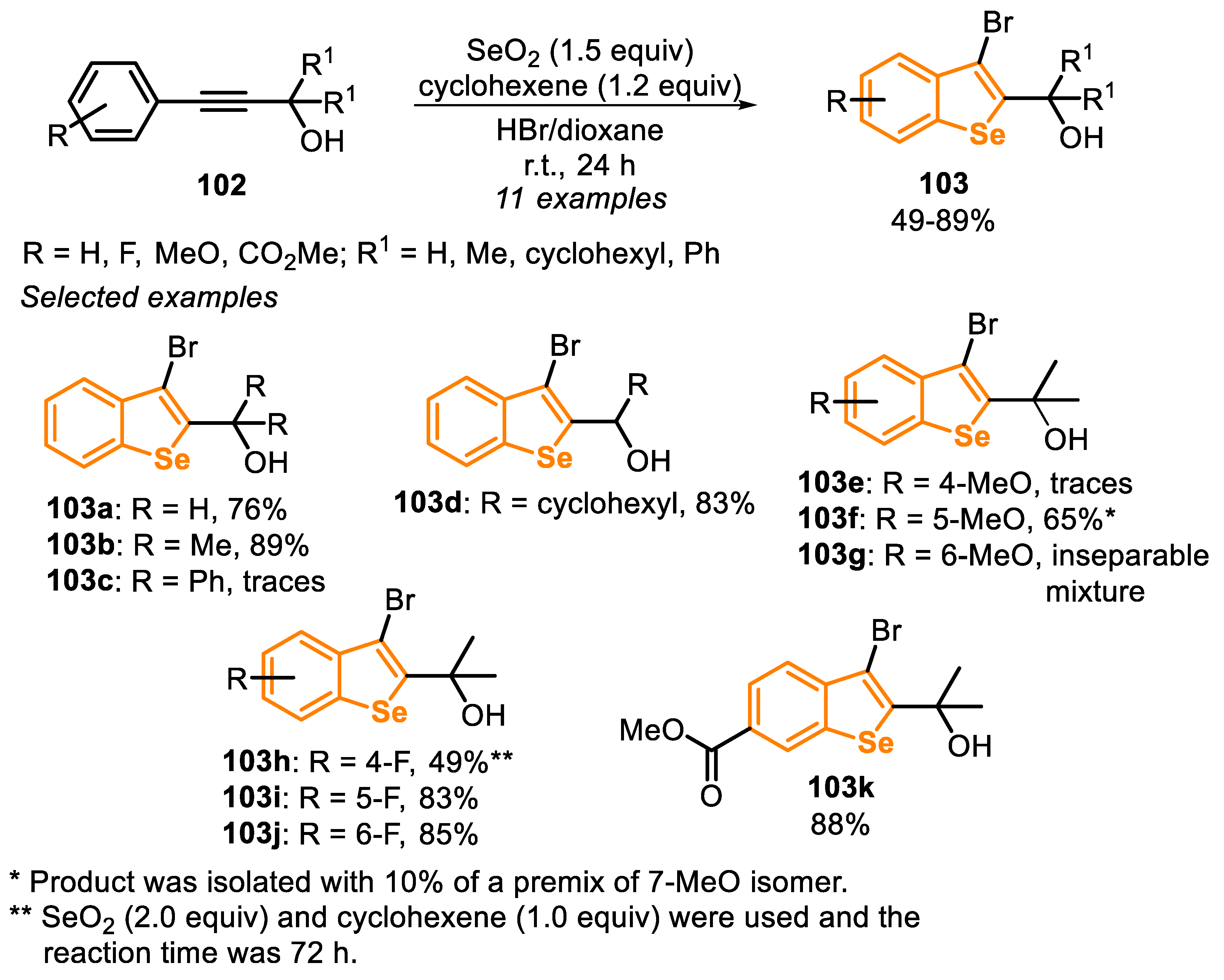
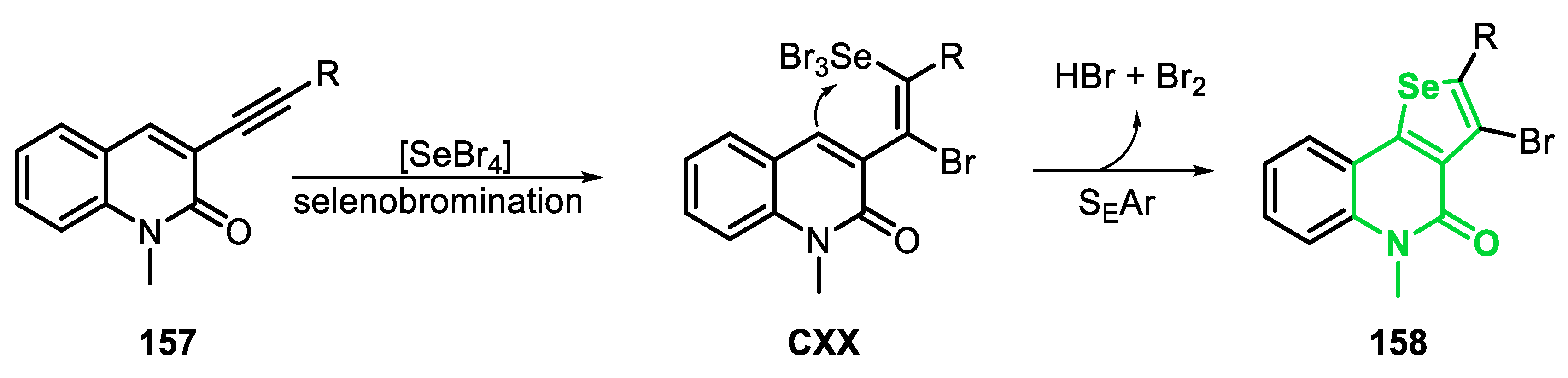
In 2014, an important protocol based on the selenobromination of type E precursors 102 was reported [164]. The best condition for this transformation involves stirring a mixture of SeO2 (1.5 equiv) in dioxane as a solvent in the presence of 48% HBr (to generate selenium(IV) bromide in situ) with cyclohexene (1.2 equiv) as an additive, at room temperature for 24 h (Scheme 69). Under these conditions, a study on the reaction scope was carried out using several electron-rich and electron-deficient substrates, allowing for the synthesis of eleven 3-bromo-benzo[b]selenophenes analogues 103 in moderate to good yields (49–89%). The reaction was efficient using different substituted phenylpropargyl alcohols 102; however, when phenyl groups were bonded to the propargylic carbon (R1 = C6H5), only traces of the respective product 103c were observed, which can be explained by steric hindrance imparted by the phenyl groups.
In general, SEAr reactions are strongly dependent on the nature of the substituents in the aromatic ring, being favored using directing groups (DG). This dependence is evident when the methoxyl group (MeO) was present in the aromatic ring of arylpropargyl alcohol 102. In this case, only traces of product 103e (R = 2-MeO) were observed (the dibromo derivative was the major product), while an inseparable mixture was obtained in the attempt to reach the product 103g (R = 4-MeO) and the corresponding dibromo derivative. However, the product 103f (R = 3-MeO) was obtained in 65% yield, but in an isomeric mixture (10% of 7-methoxy isomer). In general, the presence of strong electron-withdrawing group favored the transformation. For instance, products 103i (R = 3-F), 103j (R = 4-F) and 103k (R = 4-CO2Me) were obtained in 83%, 85% and 88% yields, respectively. The presence of a fluorine in the ortho-position of the phenyl ring in 102 (R = 2-F), caused a decrease in the reactivity, and the product 103h was obtained in 49% yield, after 72 h of reaction (Scheme 69).
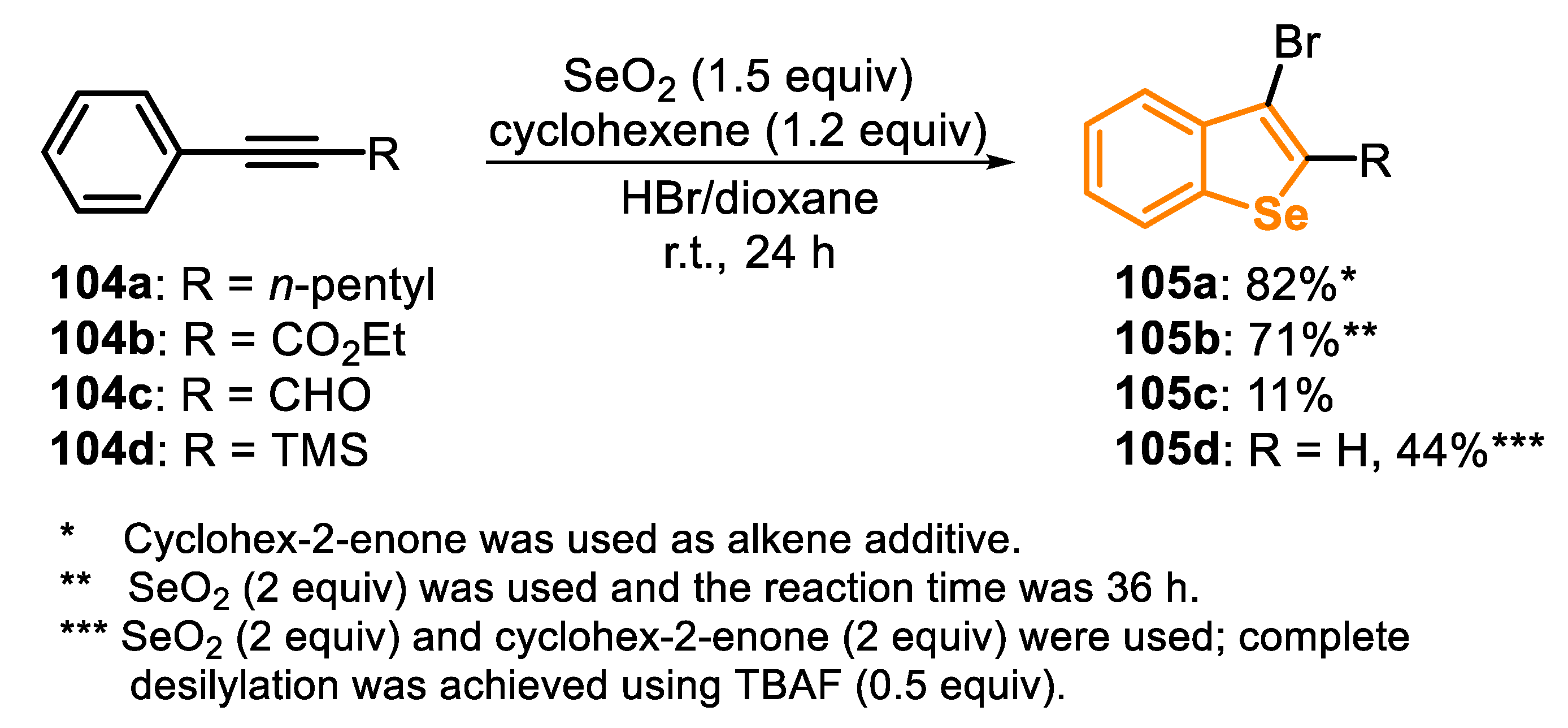
The reaction with SeO2 was extended to other internal aryl alkynes 104: hept-1-ynylbenzene (104a), ethyl phenylpropiolate (104b), 3-phenylpropiolaldehyde (104c) and trimethyl(phenylethynyl)silane (104d). Good results were obtained using 104a and 104b, and the respective benzoselenophenes 105a and 108b were obtained in 82% and 71% yields after small modifications of the optimal conditions used with the propargyl alcohols 102. The aldehyde 104c, however, afforded the respective product 105c in only 11% yield, while the TMS-protected alkyne 104d generated the deprotected benzoselenophene 105d, in 44% yield, which was desilylated in situ by the addition of TBAF (0.5 equi) in the reaction medium (Scheme 70).
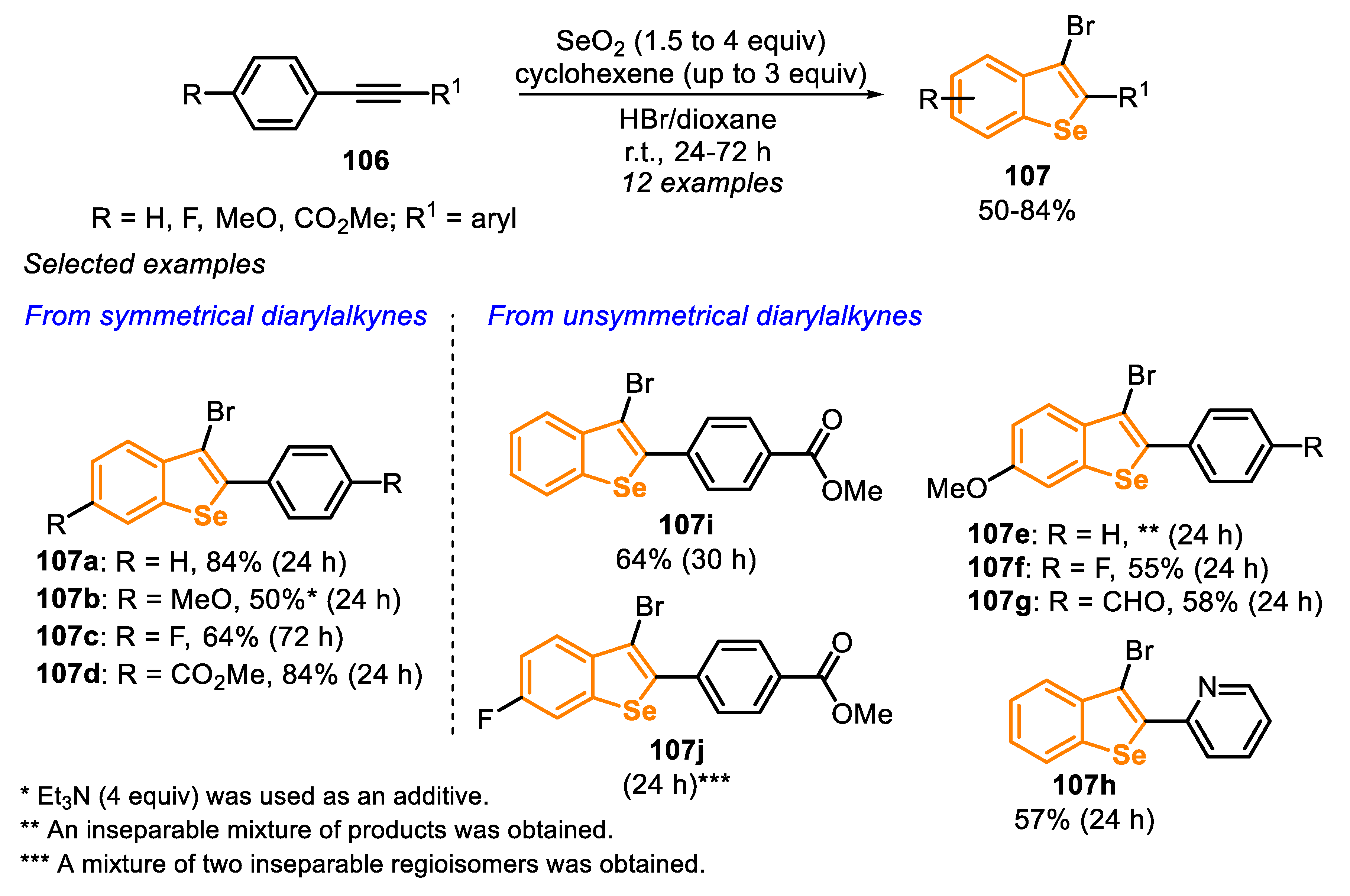
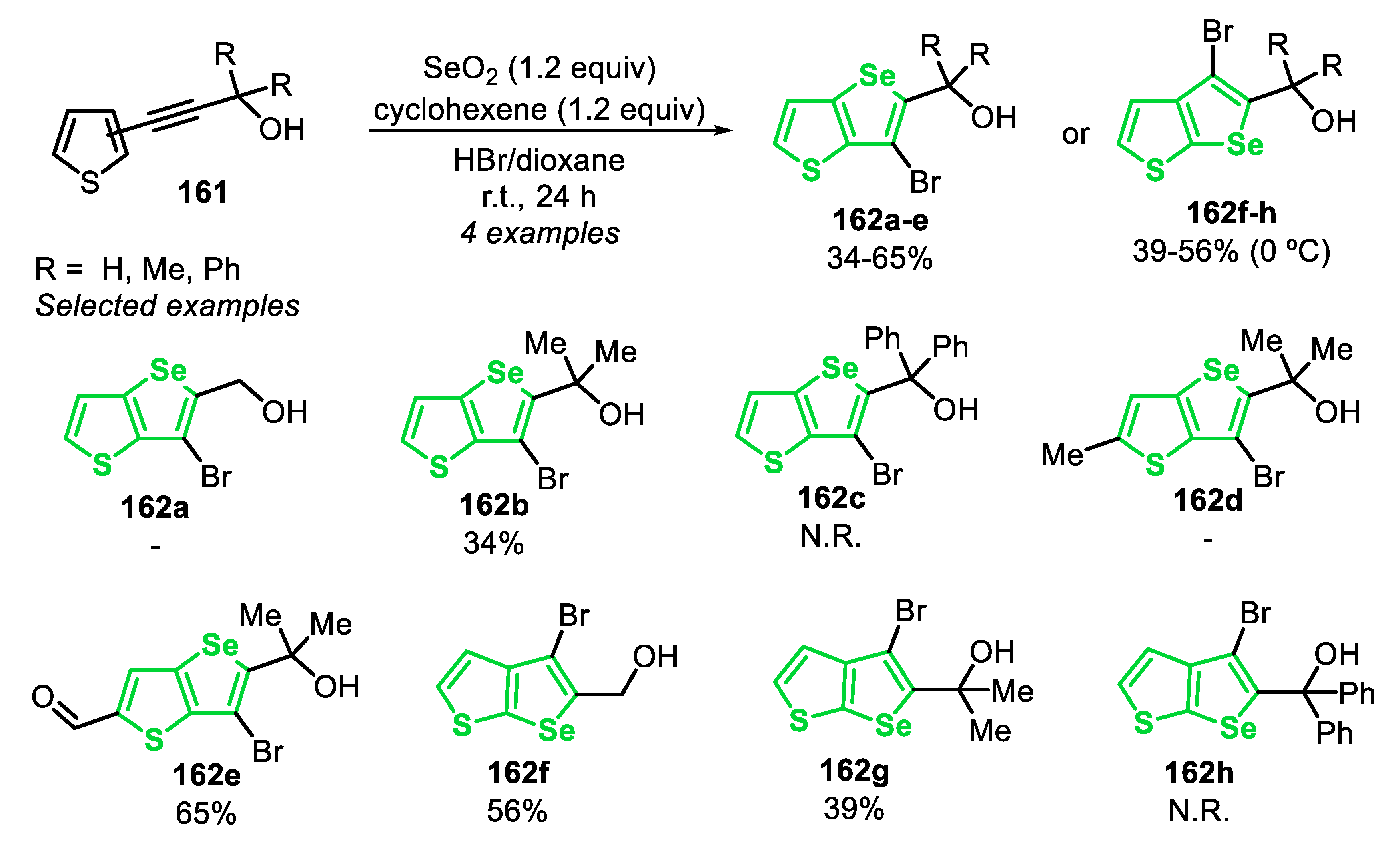
In the following year (2015), additional outcomes on the SeO2-mediated cyclization of type E precursors were described (Scheme 71) [165]. The authors used different symmetrical and unsymmetrical diarylalkynes 106, demonstrating the strong influence of different substituents attached to the aromatic ring. This effect was more substantial when unsymmetrical substituted diarylalkynes 106 were used. For example, starting from alkyne 106e (R = 4-MeO; R1 = C6H5), the bromination of the triple bond was observed, including a low regioselectivity, resulting in a complex mixture of inseparable products, without the presence of a measurable amount of the expected benzoselenophene 107e (Scheme 71). A different behavior was observed when electron-withdrawing groups were attached in the para-position of the pendant phenyl ring, such as in 106f (R1 = 4-FC6H4) and 106g (R1 = 4-CHOC6H4), and the respective benzoselenophenes 107f and 107g were obtained in 55% and 58% yields. The electronic effect, however, is not predictable, once the substrate 106j, bearing two electron-withdrawing groups in distinct aromatic rings (R = 4-F; R1 = 4-CO2MeC6H4), was not a suitable substrate for the reaction. In this case, the bromination of the C-C triple bond was not observed; however, the transformation showed a poor regioselectivity, leading to the formation of both possible regioisomers and an inseparable mixture of compounds (Scheme 71, compound 107j).
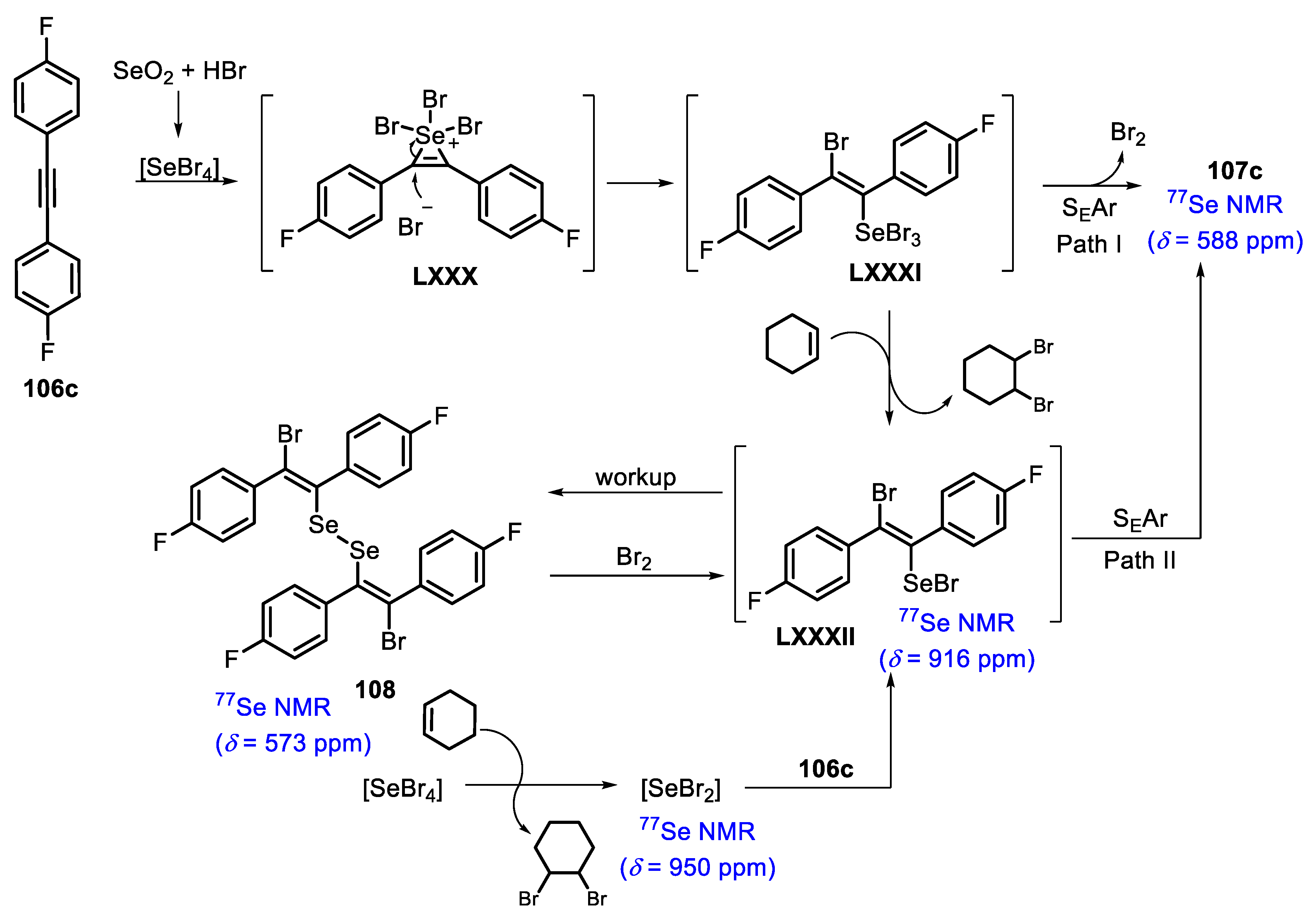
To collect insights into the mechanism of the reaction, the authors chose the synthesis of the difluoro-substituted derivative 107c, once it is a slow reaction (72 h) and allows the different steps to be monitored by 19F and 77Se NMR spectroscopy. Initially, SeBr4 adds to the C-C triple bond of 106c, giving the cationic seleniranium intermediate LXXX. This selenobromination step was crucial to achieve the regioselectivity in the unsymmetrical substrates, in which the nucleophilic attack of the bromide anion occurs at the carbon with the lowest electron density. Thus, a more polarized C-C triple bond leads to more pronounced regioselectivity. Based on this, the reaction can follow two different pathways. Firstly, the authors suggest that the cyclization step passes by the formation of the intermediate LXXXI, due to the fast formation of the cyclized product 107c, in the absence of an alkene additive. This could be explained by the fact that more electrophilic Se(IV) LXXXI species are involved in the SEAr step, in comparison to the intermediate Se(II) LXXXII. The bromine equivalent that was released during the cyclization process reacts with the starting material 106c by the bromination of the C-C triple bond, giving the byproducts (Scheme 72, Path I). In the second possibility, the reaction proceeds via the intermediate LXXXII (detected by 77Se NMR), obtained by the reaction between the intermediate LXXXI and cyclohexene, which is directly converted into the product 107c via a SEAr (Scheme 72, Path II). Additionally, besides being formed from the intermediate LXXXI, the intermediate LXXXII can also be obtained by the direct addition of SeBr4 to the alkene additive, before the addition of substrate 106c, affording SeBr2 (identified by 77Se NMR). When the crude reaction mixture was quenched with brine and AcOEt, the diselenide derivative 108 was formed (isolated in 42% yield), after a disproportionation of the intermediate LXXXII and subsequent Se-Se bond formation. This observation provided substantial evidence of a stereospecific anti-1,2-addition in the selenobromination step. In another experiment, the diselenide 108 was able to be converted to the intermediate LXXXII by the oxidative addition of Br2 (1 equiv), also following the path II (Scheme 72).
In addition to this protocol, in 2016, a general method to prepare functionalized 2- and 3-arylbenzo[b]selenophenes (109 and 110, respectively), through a 3,2-diaryl migration starting from the same precursor 111 was developed (Scheme 73) [31]. This approach overcame the regioselectivity limitations faced previously, which restricted the scope of the direct cyclization of unsymmetrical diarylalkynes. An advantage of this 1,2-aryl shift in the rearrangement step is the elimination of the debromination/bromination steps. A large and complex study was carried out to evaluate the redox properties, free-radical-scavenging ability, and cytotoxicity of the prepared compounds against cancerous cell lines. Based on the experimental results, the structure–activity relationships (SARs) of these compounds were evaluated.
3.4. Starting from Type F Precursors
In 2013, the synthesis of benzoselenophenes 114 through a Cu-catalyzed reaction of ortho-alkenylaryl iodide 113 (a type F precursor) with selenium powder and K2CO3, in N-methyl-2-pyrrolidone (NMP) as the solvent at 120 °C was described (Scheme 74) [166]. Under these reaction conditions, twelve different benzoselenophenes 114a-g, bearing electron-donating and electron-withdrawing substituents at the C6-position, were prepared in moderate to good yields. When the 6,7-methylenedioxy derivative 113h was employed, the corresponding benzoselenophene 114h was obtained in 61% yield. This protocol was also suitable to prepare the seleno[2,3-b]quinoline 114i and the selenopheno[2,3-b]thiophene 114j in good yields. Finally, benzoselenophenes 114k and 114l, bearing different substituents on the C2- and C3-positions, were obtained in the poorest yields.
Similarly, in recent years, several protocols have been described for the synthesis of fused benzoselenophene derivatives through TM-catalyzed tandem cyclization reactions, employing ortho-haloarene heterocycles and elemental selenium. In this context, in 2016, the Cu-catalyzed tandem cyclization of 2-(2-iodophenyl)imidazo[1,2-a]pyridines 115 with selenium, for the synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 116 via Ullmann-type Se-arylation and C(sp2)-H selenylation was described (Scheme 75) [167]. The reaction was performed using CuI (10 mol%) as the catalyst, under aerobic conditions in DMSO at 130 °C. The efficiency and versatility of this protocol were demonstrated by employing different 2-(2-iodophenyl)imidazo[1,2-a]pyridines 115 as substrate. Substrates 115 bearing neutral (R = H), electron-donating (R = Me and MeO) and electron-withdrawing (R = F, Br, CO2Me and CF3) groups at the C6 position of the imidazopyridine ring were suitable substrates, affording the corresponding benzoselenophene-fused imidazopyridines 116a-g poor to good yields (42–87%). The procedure was also applicable to the synthesis of 6-bromo-2-(2-iodophenyl)imidazopyridine 115d (78% yield), and the highly reactive bromine moiety remained intact. Additionally, 2-(2-iodophenyl)imidazopyridines 115 with methyl groups at the C7- and C8-positions afforded the corresponding coupling products 116h and 116i in 88% and 82% yields, respectively. Reactions starting from C5-substituted 2-(2-iodophenyl)imidazo[1,2-a]pyridines 115j and 115k afforded the products 116j and 116k in low yields after 20 h of reaction, demonstrating the high sensitivity of the reaction to steric effects.
The proposed mechanism for the tandem Se-arylation between 2-(2-iodophenyl)imidazopyridine 115 and Se powder involves, in the first step, the formation of the intermediate LXXXIV via the intermediate LXXXIII, by a Cu-mediated electrophilic substitution of the iodo-imidazopyridine 115 with Se (Scheme 76). After, the intermolecular oxidative addition of Cu(II) to the Ph-I bond, LXXXIV forms the metallacycle LXXXV. Finally, a reductive elimination of the intermediate LXXXV leads to benzoselenophene-fused imidazopyridine 116 and regenerates the Cu catalyst. Another possible route involves the Cu-catalyzed reaction of 115 with Se powder to form the complex LXXXVI, via an oxidative addition of 115 in the CuI coordinating sphere. Then, an oxidative cyclization of LXXXVI occurs, affording the metallacycle LXXXV, which is converted to the desired product 116 through a reductive elimination.
In 2017, the synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 118 through a Cu-catalyzed direct selenylation of readily available 2-(2-bromophenyl)imidazo[1,2-a]pyridines 117 was described [168]. The reaction involves a regioselective cleavage of C(sp2)-Br and C(sp2)-H bonds using Se powder (2 equiv) as the selenium source, in DMSO as a solvent at 130 °C for 30 h under air conditions (Scheme 77). The reaction scope was explored by employing several C6-substituted imidazo[1,2-a]pyridines bearing neutral, electron-donating and electron-withdrawing substituents, affording the desired benzoselenophene-fused imidazopyridines 118a-d in good yields. Similar results were obtained when disubstituted 2-(2-bromophenyl)imidazo[1,2-a]pyridines were employed as substrates (118e-n). Additionally, 6-(2-bromophenyl)imidazo[2,1-b]benzo[d]thiazole was found to be a suitable substrate in this transformation, giving the selenylated product 118o in 50% yield.
The mechanism of the reaction starts with the oxidation of the Cu(I) catalyst to the Cu(II) species in the presence of atmospheric oxygen. In the sequence, the Cu(II) species promotes an SET reaction, oxidizing the 2-(2-bromophenyl)imidazo[1,2-a]pyridines 117 to the cation radical LXXXVII and releasing Cu(I) to the reaction medium. Then, the intermediate LXXXVII is deprotonated to afford the vinyl radical intermediate LXXXVIII, which reacts with elemental selenium to give the selenium free radical species LXXXIX. Subsequently, the radical LXXXIX undergoes an intramolecular cyclization to generate the radical intermediate XC. Finally, a Cu(I)-mediated bromine abstraction of the radical intermediate XC takes place, releasing the fused benzo[b]selenophene 118, one equivalent of the bromide ion and the Cu(II) species (Scheme 78).
In 2018, another approach for the synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 118 was reported [169]. This protocol involves a Co-catalyzed reaction of 2-(2-bromophenyl)imidazo[1,2-a]pyridine 117 and potassium selenocyanate (KSeCN, 1.5 equiv) as the Se source, in the presence of N-chlorosuccinimide (NCS, 1.5 equiv), Cs2CO3 (1.5 equiv), 1,10-phenanthroline (10 mol%), CoC2O4 (10 mol%) and MeCN as the solvent. The reaction mixture was stirred at 130 °C for 5 h in a sealed tube (Scheme 79). The study on the reaction scope was firstly performed by employing different activated and unactivated 2-(2-bromophenyl)imidazo[1,2-a]pyridines 117, giving the desired products 118a-h in good yields (75-85%). The benzo[b]selenophene-fused imidazo[2,1-b]thiazole 118i could be prepared in 74% yield, starting from a properly substituted imidazo[2,1-b]thiazole 117i.
The first step in the proposed mechanism is the reaction between the 2-(2-bromophenyl)imidazo[1,2-a]pyridine 117 and N-selenocyanate-succinimide, which was formed in situ from the reaction of NCS with KSeCN, to give SeCN-substituted intermediate XCI (Scheme 80). Then, the intermediate XCI coordinates to the active cobalt catalyst, followed by an intramolecular addition to the Se-CN bond, providing the intermediate XCII. Then, the aryl halide displaces CN−, affording the intermediate XCIII, which undergoes a reductive elimination to regenerate the active cobalt catalyst, creating the desired product 118.
In 2016, a Cu-catalyzed intramolecular phenylselenation of 2-(2-iodophenyl)-1H-indoles 119 in the presence of elemental selenium for the synthesis of the benzoselenopheno[3,2-b]indoles 120 was described [170]. The optimal conditions involve the use of CuO (10 mol%) as a catalyst and DMSO as the solvent at 110 °C, under a N2 atmosphere (Scheme 81). The protocol tolerates the presence of several substituents in the indole aromatic ring: neutral (R = H), electron-donating (R = Me and MeO) and electron-withdrawing (R = CF3O, F, Cl and Br) groups were able to afford the corresponding fused-benzoselenophenes 120a-g in good to excellent yields (80–95%). Additionally, disubstituted 2-(2-iodophenyl)-1H-indoles 119 afforded the respective benzoselenopheno[3,2-b]indoles 120h–j in excellent yields (96–97%). The 2-(2-iodophenyl)-1H-indole 119k (R = 7-Me); however, was less reactive, and the respective benzoselenophene 120k was obtained in 61% yield.
3.5. Other Approaches to Prepare Benzoselenophenes
In addition to the use of types C, D, E and F precursors, there are other approaches for the synthesis of benzoselenophenes, starting from different reagents—for example, the use of resveratrol, a natural compound isolated from a broad range of plants, and present in red wines [31,171]. In these works, the authors promoted the direct selenenylation of resveratrol using SeCl2, wich was prepared in situ from Se(0) and SO2Cl2 in THF. The variation in the SO2Cl2 ratio resulted in different chlorinated or non-chlorinated benzo[b]selenophenes (Scheme 82, compounds 121a–c). The authors also report that the structural modification in the Resveratrol scaffold afforded derivatives with promising pharmacological properties.
In 2016, the synthesis of diarylannulated selenides through the selenium-iodine exchange of diaryliodonium salts 122 was reported [172]. The protocol involves the stirring a mixture of the iodonium salt, elemental selenium (3 equiv) and t-BuOK (4 equiv) as a strong base in DMSO as the solvent at 80 °C for 2 h, under a N2 atmosphere. A broad range of diaryliodonium salts 122, bearing neutral, electron-rich and electron-deficient substituents were efficiently used as starting materials, giving the respective selenophenes 123 in good to excellent yields. The protocol was not sensitive to steric hindrance effects in the diaryliodonium salts, allowing for the synthesis of diarylannulated selenides 123i–l with conjugated groups at the 3- and 4-positions. The optimal conditions were used in the synthesis of the BTBS material OFET 123m (53% yield); however, using 2 equivalents of base at 100 °C (Scheme 83).
In 2017, the I2-catalyzed selenocyanation of styryl bromides 124 with potassium selenocyanate, affording benzo[b]selenophenes 125 was described [173]. The best conditions for the annulation reaction was found as the stirring a mixture of the bromide 124 and KSeCN (1.2 equiv) in the presence of I2 (20 mol%) and DMSO as the solvent at 110 °C, under an argon atmosphere, for 48 h. Under these conditions, a total of twenty-six examples of benzoselenophenes 125 were prepared in moderate to excellent yields (45–95%). Electron-rich styryl bromides 124 substituted with methoxy, dimethoxy, trimethoxy, ethoxy, butyloxy, pentyloxy, dodecyloxy, and allyloxy groups on the aromatic ring were used as substrates in the reaction (Scheme 84).
The proposed mechanism starts with the reaction of the styryl bromide 124 with I2 to give a cycloiodonium ion XCIV, which is attacked by the nucleophile (-SeCN), affording the intermediate XCV. Then, the corresponding trans-styryl selenocyanate 126 is formed via an E2 mechanism, releasing I2 to the catalytic cycle. The next step is dependent on the presence of the electron-donor group (DG) and consists in a homolytic cleavage of the selenocyanate 126, to generate the Se-centered radical XCVI. Thanks to the DG attached to the arene, the stabilized cyclic intermediate XCVII is generated. Finally, an oxidation-deprotonation occurs, affording the respective benzo[b]selenophene 125 (Scheme 85).
In 2017, the synthesis of multifunctionalized dibenzoselenophenes 128 through the hexadehydro-Diels-Alder (HDDA) reaction of tetrayne systems 127 with diaryl diselenides 64 was described [174]. The best results were obtained when a mixture of tetrayne 127, and diaryl diselenide 64 (1.1 equiv), in toluene, was stirred at 90 °C for 24 h (Scheme 86). This approach is free of metals, catalysts, and additives, such as bases or oxidants, or directing groups, and worked well with a range of substrates. In general, the protocol was efficient, being suitable for tetraynes bearing neutral (H), electron-donating (Me, Et, i-Pr) and electron-withdrawing (F, Cl) substituents in the aryl ring. A total of twenty-four new fused dibenzoselenophenes 128 were prepared in good to excellent yields (74–87%). Good results were obtained, starting from bis-(o-tolyl) diselenide (Ar’ = 2-MeC6H4), and the products 128n-s were isolated in 75–87% yields (Scheme 86).
A plausible reaction mechanism was proposed for the synthesis of dibenzoselenophenes 128, which consists in a cascade hexadehydro-Diels-Alder (HDDA) reaction, followed by a radical based C-H activation reaction. Initially, the HDDA reaction of tetrayne 127 produces an aryne as intermediate (XCIX), which subsequently reacts with the radical Ar’Se• (generated under heating) to give the intermediate C. Then, there is a homolytic α-C-H bond cleavage into the aryl group in the intermediate C, followed by the abstraction of an hydrogen by the free radical (Ar’Se•), giving the compound 128 (Scheme 87).
In 2019, a three-component cycloaddition reaction for the synthesis of benzoselenophenes 131 was developed [175]. The protocol involves the reaction of substituted indoles 129 with acetophenones 130 and Se powder, under metal-free conditions. After a systematic reaction optimization study, the best condition was established as being the stirring of a mixture of the acetophenone 130 and the indole 129 in the presence of elemental selenium (1.5 equiv), IBr (1.2 equiv) as the oxidant agent, in NMP as the solvent at 140 °C for 8 h, under O2 atmosphere. A study on the reaction scope was carried out, driving the formation of a diversity of thirty-nine new bis-heteroaryl compounds 131 containing both indole and benzoselenophene moieties in moderate to very good yields (Scheme 88). The optimized methodology was shown to be compatible with a range of substituted acetophenones 130 and indoles 129 bearing electron-releasing and electron-withdrawing groups. In general, the electronic effects did not affect the reaction efficiency to access the products 131. Substrates bearing strong electron-withdrawing groups (e.g., CF3, NO2) were also compatible with the reaction conditions. However, the presence of an ester group in the aromatic ring of both, the acetophenone 130 or the indole 129 caused the total failure of the reaction, and the expected products 131ad-af were not observed. The protocol was also satisfactorily scalable to 6 and 5 mmol, affording the products 131a and 131g in 68% and 61% yields, respectively.
The proposed mechanism involves, initially, a dehydrative condensation of 2-phenyl-1H-indole 129a and acetophenone 130a, giving the intermediate 3-vinyl-indole 132a. Then, the allyl radical intermediate CII is generated from 3-vinyl-indole 132a via an SET oxidation, performed by IBr. In the sequence, the intermediate CII reacts with elemental selenium to give the Se-centered radical intermediate CIII, which is converted to the intermediate CIV, through an intramolecular cyclization. Then, a new SET oxidation converts the radical intermediate CIV to the product 131a (Scheme 89).
4. Fused Selenophenes and Benzoselenophenes
4.1. Starting from Type C Precursor
In recent years, several protocols for the synthesis of selenophenes and benzoselenophenes fused to other heterocycles, including chromenes, thiophenes, quinolines and indoles, among others, have been reported. These compounds are mainly obtained by electrophilic cyclization reactions of type C precursors, using commercially available halogenated selenium electrophiles or selenium and tellurium electrophilic species, prepared in situ from the respective diorganyl dichalcogenides using Fe-based salts.
In this context, the synthesis of 3-substituted selenonophene[3,2-c]chromenes 134 through the electrophilic cyclization of 3-alkynyl-4-chalcogen-2H-chromenes 136 using I2, PhSeBr and BuTeBr3 as electrophilic sources, and DCM as the solvent at room temperature was described (Scheme 90) [176]. Under these reaction conditions, the electrophilic cyclization of substrates 133 bearing aryl or alkyl groups at the triple bond, using I2 as electrophilic source, gave the products 134a, 134b and 134d in good yields (75–84%). In addition, substrate 133, bearing bulky alkyl and propargyl alcohols groups, afforded the desired products 134c and 134e in lower yields (65% and 50%, respectively). Besides that, the protocol was explored using different substituents in the chromene ring, such as naphthyl and aryl ones, producing the desired fused selenophenes 134f-h in good yields (74–86%). When the cyclization reaction was carried using the BuTeBr3 and PhSeBr as electrophiles, the corresponding 3-chalcogen-selenophene[3,2-c]chromene 134i and 134j were obtained in similar yields after 2 h and 15 min, respectively.
The reactivity of the 3-iodo-selenophene[3,2-c]chromenes 134 was evaluated in the Pd-catalyzed cross-coupling reaction. Thus, the 3-iodo-selenophene[3,2-c]chromenes 134a reacted satisfactorily with terminal alkynes and boronic acid, to give the Sonogashira and Suzuki-type products 135 and 136 in 50% and 80% yields, respectively (Scheme 91).
In 2013, a FeCl3/diorganyl dichalcogenides-promoted intramolecular cyclization of 2-organochalcogen-3-alkynylthiophenes 137, for the synthesis of selenophene[2,3-b]thiophenes 138, using DCM as the solvent at room temperature and under air atmosphere was developed (Scheme 92) [177]. The scope of the reaction was firstly evaluated by using alkyl and diaryl diselenides bearing neutral, electron-donor and electron-withdrawing substituents in the aromatic ring, producing the desired 3-organoselanyl-selenophene[2,3-b]thiophenes 138a–d in moderate to good yields (65–83%). When dialkyl and diaryl ditelluride were used as substrates, similar results were obtained. Dibutyl dichalcogenides were less reactive than the diaryl analogues (Scheme 92, products 138d and 138f vs. 138a–c and 138e). This behavior was attributed by the authors to the presence of β-hydrogen atoms in the butyl group bonded to the chalcogen atom, which may favor a decomposition via a telluroxide or selenoxide syn-elimination during the purification or work-up process. The 3-Alkynylbenzothiophenes were less reactive in the cyclization reaction, affording the products 138j–l in 62%, 45% and 54% yields, respectively. A competitive cyclization reaction between the nucleophilic oxygen from an o-methoxyl substituent in the aryl group of the alkyne and a selenium nucleophile directly bonded to the thiophene ring was not observed. For instance, the selenophene 138m formed by a Se-cyclization, was the only product observed in the reaction of the alkyne 137m, with the complete absence of furan derivatives.
The reactivity of 3-organochalcogenyl-selenophene[2,3-b]thiophenes 138 was explored in the Suzuki Pd-catalyzed (YR1 = TeBu) and Li-Se exchange (YR1 = SePh) reactions (Scheme 93). Under the Suzuki reaction conditions, 4-butyltelluro-selenophene[2,3-b]-thiophene 138f underwent the cross-coupling reaction smoothly, by the exposure to aryl boronic acids bearing electron-rich and electron-deficient substituents, affording the desired products 139a and 139b in 74% and 63% yields, respectively. In addition, the 4-phenylselanyl-selenophene[2,3-b]thiophene 138j was submitted to the Li-Se exchange reaction, with butyllithium in THF at −78 °C, giving the lithium intermediate, which was trapped with 4-chlorobenzaldehyde, affording the secondary alcohol 140 a 63% yield after 2 h.
In 2014, was described the intramolecular hetero-cyclization and cyclopalladation of selenoalkyne-substituted propargyl imines 141, for the synthesis benzoselenophene[2,3-c]pyridinones 142 [178]. In order to promote this reaction, the palladations of ortho-selenoanisole perfluoro propargyl imines 141 was performed using a stoichiometric amount of dichoro-bis(triphenylphosphine)palladium(II) in dry THF, for 2 h at 0 °C, producing the respective benzoselenophene-based palladacycles 142a-c in 70–85% yields (Scheme 94). A plausible reaction mechanism for the formation of benzoselenophene-based palladacycles involves the coordination of the palladium catalyst to the nitrogen atom, followed by an interaction of the triple bond to the palladium atom. After, the electron-rich selenium atom attacks the activated triple bond, with concomitant removal of the methyl group via an SN2 nucleophilic displacement, which promotes it by the chloride anion present in the reaction medium, to give the product 142 (Scheme 94).
These palladacycles were then subjected to the insertion of diphenyl acetylene (DPA) 143 into the Pd-C bond, in refluxing toluene for 12 h, affording three different benzoselenophene[2,3-c]pyridinones 144a-c in 61–71% yields (Scheme 95).
The proposed reaction mechanism to prepare the fused benzoselenophene derivatives 144 involves, firstly, the generation of the seven-membered ring intermediate CVI, after the coordination of the C-C triple bond to the Pd coordinating sphere (intermediate CV). Then, the insertion into the Pd-C bond occurs, where the Pd atom interacts with the alkynyl carbon followed by heterolytic Pd-C cleavage and ring closure to form the intermediate CVII. The formation of benzoselenophene[2,3-c]pyridinones 144 rather than the expected pyridinium salt may be attributed to the presence of the CF3 group bonded to the pyridinium ring in CVII. The carbon atom attached to CF3 is more electrophilic because of the highly electronegative nature of CF3 group, which in turn is susceptible to a nucleophilic attack by the hydroxyl ion to the intermediate CVIII. Finally, the intermediate CIX undergoes an oxidation to form the carbonyl group in 144 (Scheme 96).
In 2018, the synthesis of selenopheno[2,3-b]quinoline 146 through the iodo-cyclization reaction of 3-alkynyl-2-(methylseleno)quinolines 145 in DCM at room temperature for 6 h, using I2 or NIS as an electrophilic source, was reported [179]. Under these reaction conditions, the corresponding fused selenophenes 146a and 146b were synthetized in 54% and 75% yields, respectively, by the iodocyclization of 3-alkynyl-2-(methylseleno)quinolines 145a (R = H) and 145b (R = Br), using N-iodosuccinimide (NIS) as an electrophilic source. Similarly, 2-substituted selenopheno[2,3-b]quinolines, 146c and 146d, bearing phenyl and phenylethynyl groups, were obtained in 86% and 87% yields, respectively, using I2 as an electrophile (Scheme 97).
To investigate the reaction mechanism of the iodocyclization of 3-alkynyl-2-(methylseleno)quinolines 145, the authors performed density functional theory (DFT) calculations. Initially, the mechanism involves the addition of electrophile NIS or I2 to the C-C triple bond of the 3-alkynyl-2-(methylseleno)quinoline 145, producing the three-membered cyclic intermediate iodonium CX or CXI. In the sequence, an anti-attack of the Se atom to the activated triple bond of CX or CXI occurs, producing the selenonium salt CXII or CXIII, via a 5-endo-dig cyclization (Scheme 98). Subsequently, an SN2 displacement of the methyl group, linked to the Se atom by the nucleophile present in the reaction medium, creates the selenopheno[2,3-b]quinoline 146 and MeI or N-methylsuccinimide as byproducts.
A similar protocol was developed this year for the synthesis of 3-iodo-selenophene-fused indoles 148, through an intramolecular electrophilic cyclization of 3-organoselanyl-2-alkynylindoles 147 using I2 in THF, at room temperature for 3 h (Scheme 99) [180]. The versatility of this protocol was firstly evaluated by the reaction of alkynylindoles 147 bearing neutral, electron-rich, and electron-deficient aryl groups, as well as the naphtyl derivative, producing the 3-iodo-selenophene-fused to indoles 148a-f in moderate to good yields. In these reactions, lower efficiency was observed starting from ortho-methoxy- and ortho-chloro-substituted arylalkynes, probably due to steric effects, and 148d and 148e were obtained in 58% and 60% yields, respectively. When heteroaryl groups directly bonded to the alkyne were employed as substrates, the desired cyclized products 148g and 148h were afforded in 39% and 55% yields, respectively. Good results were obtained starting from n-butyl- or n-hexyl-substituted alkynes, affording the fused selenophenes 148i and 148j in 79% and 78% yields, respectively. The reaction did not work when the N-methyl indole 147k was employed as a substrate, and the expected cyclized product 148k could not be obtained.
The mechanism of the formation of 3-iodo-4H-selenopheno[3,2-b]indole 148 involves the initial activation of the C-C triple bond of the substrate 147, via the formation of the iodonium ion CXIV, which was obtained by the coordination of the triple bond to the I2. In the sequence, an anti-nucleophilic attack of the Se atom to the activated triple bond gives the selenium salt CXV, through a selective intramolecular 5-endo-dig cyclization. The iodide anion, present in the reaction mixture, promotes the SN2 displacement of the butyl group bonded to selenium, affording the desired fused selenophene 148 and butyl iodide (Scheme 100).
In the same work, the authors described the synthesis of 3-(alkylselanyl)-selenophene-fused indoles 149 through the cyclization reaction of 3-butylselanyl-2-alkynylindoles 147 using a selenium electrophilic species generated in situ. In this reaction, a mixture of the substrate 147, FeCl3 and dibutyl diselenide in DCM was stirred at room temperature for 3 h. By this protocol, 3-butylselanyl-2-alkynylindoles bearing pendant naphthyl, neutral, electron-poor and electron-rich aryl groups in the alkyne were converted to the respective 3-butylseleno-selenonophene-fused indoles 149a-d in 64%–70% yields. Good results were obtained starting from N-protected 3-(butylselanyl)indole and other dialkyl diselenides, and compounds 149e-g were prepared in 55%–67% yields (Scheme 101).
4.2. Starting from Type D Precursors
Recently, some of us [181] described the synthesis of 2-organylselenopheno[2,3-b]pyridines 151 by the insertion of Se-based nucleophilic species into 2-chloro-3-(organylethynyl)pyridines 150, followed by an intramolecular cyclization reaction. The nucleophilic selenide was generated in situ by the reduction of elemental selenium using the NaBH4/PEG-400 system (Scheme 102). The experimental procedure for the synthesis of selenophenes fused to pyridines 151 consists of two steps: firstly, Se powder (0.3 mmol) and NaBH4 (0.9 mmol) are poured into a vessel containing PEG-400 (2 mL), under an argon atmosphere at 50 °C, and are stirred at this temperature for approximately 0.5 h (the color of the mixture changes from black to white). Then, in a second step, 2-chloro-3-(organylethynyl)pyridine 150 (0.25 mmol) is added, and the temperature is raised to 100 °C for an additional 2 h. The generality of this protocol was evaluated in the reaction of differently substituted 2-chloro-3-alkynylpyridines 150, including those with electron-rich and electron-poor arenes and alkyl groups. In a general way, the desired fused-selenophenes 151a-e were obtained in moderate to good yields (55–77% yield). One exception was the 2-chloropyridine 150f, containing a propargyl alcohol at the 3-position, which afforded the corresponding selenopheno[2,3-b]pyridin-2-ylmethanol 151f in only 31% yield after 1 h.
After some control experiments, the authors were able to propose a mechanism for the synthesis of 2-organylselenopheno[2,3-b]pyridines 151. The first step is the attack of the pre-formed NaHSe to the C-C triple bond of the alkynylpyridine 150, generating the intermediate CXVI, followed by an intramolecular SNAr reaction (Path I). However, the reaction may also go through another pathway, which consists in a nucleophilic attack (SNAr) of the selenium anion to the 2-chloroalkylpyridine 150, followed by an intramolecular hydroselenation reaction of the triple bond (Path II) (Scheme 103).
In addition, some of us [182] reported the synthesis of 2-organylselenopheno[2,3-b]pyridines 151, starting from bis(3-amino-2-pyridyl)diselenide 152 and aryl- or alkylacetylenes 153, in the presence of t-BuONO as a nitrosating agent in MeNO2, at 80 °C under argon atmosphere for 2 h (Scheme 104). The scope of the protocol was investigated by employing different aryl- and alkylacetylenes 153 in the reaction with bis(3-amino-2-pyridyl)diselenide 152 under the optimized conditions. Thus, when the reaction was performed using arylacetylenes with electron-donating groups at the para-position of the aromatic ring, the respective fused selenophenes 151b-d were obtained in similar yields than that using the neutral phenylacetylene (151a). In contrast, the presence of the electron-withdrawing Cl-substituent at the aromatic ring caused a fast consumption of the starting material; however, the product 151e was obtained in only 10% yield, due to the formation of many byproducts. When heptyne was employed as substrate, the respective 2-pentylselenopheno[2,3-b]pyridine 151f was obtained in 17% yield after 2 h. In all cases 1,6-diazaselenanthrene 154a was formed as a coproduct in yields ranging from 7% to 13%.
The proposed mechanism of the synthesis of the fused selenophenes 151 initiates with the reaction between pyridylamine 152 and tert-butyl nitrite, resulting in the radical species CXVIII. From the intermediate CXVIII, the reaction can follow two different paths, which depend on the interaction with the acetylene. In Path a, the reaction occurs without the presence of acetylene 153, through two intramolecular radical reactions to afford 1,6-diazaselenanthrene 154a. Alternatively, the reaction could follow the Path b, with the reaction between the radical intermediate CXVIII and the acetylene 153, affording the vinyl radical CXIX, which reacts by an intramolecular homolytic substitution at the Se atom, leading to the final product 151 (Scheme 105).
To demonstrate the synthetic applicability of the prepared 2-phenylselenopheno[2,3-b]pyridines 151a, this compound was reacted with Br2, in dry chloroform at 0 °C, affording, after 1 h, 3-bromo-2-phenylselenopheno[2,3-b]pyridine 155 in 92% yield, which is a suitable substrate in several classic cross-coupling reactions, such as Suzuki–Miyaura, Sonogashira and Negishi. In addition, the compound 151a reacted with diphenyl diselenide, in the presence of Oxone®, in AcOH at 100 °C to give the 2-phenyl-3-(phenylselanyl)selenopheno[2,3-b]pyridine 156 in 85% yield after 12 h (Scheme 106).
4.3. Starting from Type E Precursors
In 2014, the synthesis of selenophenes fused to quinolines 158 and 160 through the selenobromination of alkynilquinolones 157 and 159, respectively, was described [183]. This reaction was promoted by selenium tetrabromide in dioxane as the solvent, at room temperature for 24 h. The selenium(IV) species was generated in situ from SeO2 and HBr (Scheme 107). The optimal conditions were applied in the selane-Friedel-Crafts cyclization of 3-alkynylquinolones 157b-c to afford the respective selenophene[3,2-c]quinolones 158b and 158c in 77% and 75% yields. When the 3-alkynylquinolone 157a, containing the hydroxyl group, was employed as a substrate, the expected product 158a was not formed. Better results, however, were obtained starting from the 4-alkynylquinolone isomers 159, for which the same conditions were afforded the selenopheno[2,3-c]-quinolones 160a-c in 75–99% yield, including the fused selenophene 160a containing the hydroxyl group that was selectively formed in a nearly quantitative yield. In the same work, the compounds were tested for their cytotoxic activity in vitro against several tumor cells lines. The 3-Bromo-5-methyl-2-(piperidinylmethyl)selenopheno[3,2-c]quinolone (158b) presented a pronounced cytotoxic activity against the human uterine sarcoma MES-SA.
The proposed mechanism involves a cyclization reaction, followed by two sequential processes. The first step involves an anti-addition of selenium tetrabromide to the C-C triple bond of the 3-alkynylquinolone 157, affording the selenotribromide intermediate CXX. Then, an intramolecular cyclization through a SEAr mechanism occurs, giving the selenopheno[3,2-c]quinolones 158, while one equivalent of HBr and Br2 is released. The formation of selenopheno[2,3-c]quinolones 160 from the 4-alkynylquinolone 158 proceeds by the same mechanism (Scheme 108).
In 2014, a close-related procedure was developed, the selenobromination of thienylpropargyl alcohols 161, using cyclohexene as an additive, for the synthesis of selenopheno[3,2-b]- and selenopheno[2,3-b]thiophenes 162, when the reaction is conducted at room temperature or at 0 °C, respectively (Scheme 109) [164]. The selenium(IV) bromide was prepared in situ by the reaction of SeO2 with an appropriate amount of concentrated HBr. The reaction using the unsubstituted thienylpropargyl alcohol 161a (R = H) produced a complex mixture of products besides the expected selenophene 162a, including the corresponding dibromo-derived and the bromination of the α-position of the thiophene ring. The cyclization of dimethyl-substituted thienylpropargyl alcohol 161b (R = Me) led to the formation of the selenopheno[3,2-b]thiophene derivative 162b in 34% yield. The cyclization product 162c was not detected, probably due to the steric hindrance of the phenyl groups in the starting material. A low reactivity was also observed when the α-methyl-substituted thienylpropargyl alcohol 161d was the substrate, and only a trace amount of 162d was observed by GC/MS, with the corresponding dibromo derivative being formed as the main product. The presence of the strongly electron-withdrawing formyl group in the pendant thienyl ring positively influenced the reaction, and the fused selenophene 162e was afforded a 65% yield, without formation of the corresponding dibromo derivative. The selenopheno[2,3-b]thiophenes 162f and 162g were obtained in 56% and 39% yields, respectively, while the diphenyl-substituted selenophene (162h) was not formed, reinforcing the influence of steric effects in the reaction.
This strategy to generate SeBr4 in situ was used in 2015 by the same group [165], in the synthesis of selenophenothiophenes 164, through the cyclization of diheteroarylalkynes 163. The cyclization reactions were regiospecific in all cases, affording the corresponding selenophenothiophene derivatives 164a–g in modest yields (28–40%). For the synthesis of 164a (R = 2-pyridinyl) and 164b (R = 3-pyridinyl), the optimal reaction conditions were achieved when 1.2 equiv. of SeO2 and 1.5 equiv. of cyclohexene were used in the presence of HBr in dioxane at room temperature. The low yields observed in all the cases could be attributed to the competing α-bromination of the thiophene ring. In the reaction starting from bis(thiophen-2-yl)ethyne, the expected product 164c was obtained in 33% yield. In this case, cyclohex-2-enone was used as a bromine scavenger, reducing the α-bromination of the thiophenes. In the case of unsymmetrically substituted derivative 163d (R = 5-thiophenaldehyde, R1 = Me), the α-positions of the thiophene ring are blocked, preventing their bromination. However, a large excess of SeO2 (4 equiv) and cyclohexene (3 equiv) were necessary to afford the corresponding cyclization product 164d in only 38% yield after 24 h of reaction. It was observed that the bromination of the C-C triple bond of the starting dihetarylalkyne 163d was quite pronounced. This approach was suitable to prepare the selenopheno[2,3-b]thiophene derivatives 164e and 164f, which were obtained in 39% and 32% yields after 24 h. The cyclization of 2,5-bis(pyridin-3-ylethynyl)thiophene (163g) was a successful example of bis-cyclization, giving 164g in 40% yield after 96 h. This reaction was performed in the presence of 3 equivalents of SeO2 and 2.4 equivalents of cyclohexene (Scheme 110).
In 2018, the synthesis of 3-selenophen-3-yl-1H-indoles 166 through the aromatic electrophilic cyclization of propargyl indoles 165, promoted by SeCl2, was described [184]. The electrophilic selenium species was prepared in situ by the reaction between SO2Cl2 and selenium powder in DMF at room temperature under an argon atmosphere. The versatility of this protocol was evaluated by using a diversity of propargyl indoles 165, containing substituents at the terminal triple bond, at C-2, C-5, and at the nitrogen atom of the indole core, as well as the other two substituents at the propargylic position. Regarding the aryl group directly bonded to the alkyne, neutral, electron-rich, and electron-poor groups were able to give the desired products 166a–c in good yields (88–91%) and short reaction times (5–30 min). The replacement of the aryl group by a less reactive aliphatic one caused a remarkable decrease in the reaction efficiency, affording the fused-selenophene 166d an only 22% yield after 30 min. The reactivity of the substrate was not affected by modifications in the indole core at the C-5 position, and selenophenes 166e (R = Br), 166f (R = CN) and 166g (R = MeO) were obtained at 88–92% yields, without any evident electronic effect. A similar good reactivity was observed when the 2-Me-indole derivative 165k was employed, and the corresponding product 166k was obtained in 89% yield. In contrast, the nature of the N-substituent was relevant to the reaction performance; while N-methyl and N-benzyl indoles were suitable substrates, affording the respective products in good to very good yields, no reaction was observed when the N-Boc derivative was used, and the selenophene 166i (R = Boc) did not form, even after 24 h of reaction. Unsubstituted (N-H free) indole 165j afforded the product 166j in 84% yield after 15 min at 0 °C. The low temperature was necessary to avoid the formation of coproducts, observed at room temperature. The presence of an electron-donating group at the para-position of the aromatic ring of the propargylic position in 165m (R5 = 4-MeOC6H4) adversely affected the reaction performance, and the respective selenophene 166m was obtained in 75% yield after 1 h. Interestingly, when a methyl group was present at the propargylic position, such as in 165n (R5 = Ph, R6 = Me), the respective fused selenophene[2,3-b]indole 166n was obtained in 81% yield after 45 min (Scheme 111).
The first step of the mechanism of the synthesis of selenophenes 166 is the electrophilic addition of SeCl2 to the C-C triple bond of the propargyl indole 165, affording the seleniranium intermediate CXXI (Scheme 112). This triggers a cationic cascade process that could undergo at least three different evolutions, including a chloride-mediated and two intramolecular (routes A1 and A2) nucleophilic seleniranium ring-openings. In route A1, a 1,2-sigmatropic rearrangement of the aryl group (R2) results in the tertiary allyl carbocation intermediate CXXII, stabilized by the pendant 3-indolyl moiety. Alternatively, the intermediate CXXI could be involved in a 1,2-indolyl shift, with the aid of the lone pair of electrons of the indole nitrogen, affording the polysubstituted spirocycle CXXIII (route A2). The ring-opening of the seleniranium and the C-C bond formation, through SN2 attack of the indole C-3 position to the electron-deficient center, is a concerted process, which would avoid passing through a highly reactive vinyl cation species. Once formed, the intermediate CXXIII undergoes a ring-opening of the strained cyclopropane motif, affording the carbocation intermediate CXXIV. The migratory aptitudes of the migrating moieties are typically correlated to their electron-richness and their ability to stabilize a positive charge in the transition state/intermediate. When R2 is an aromatic substituent, the reaction mechanism would occur by Path a, by losing a proton from the intermediate CXXIV, creating the selenodiene CXXV, which is suitable for the cyclization to the cationic intermediate CXXVI to afford, finally, the trisubstituted selenophene products 166a-m. Considering this, it is plausible that the presence of an alkyl R3 substituent may fail to provide a satisfactory stabilization to the carbocation intermediate CXXVI, resulting in lower product yields (such as 166d). In contrast, the formation of the fused selenophene[2,3-b]indole 166n suggests the involvement of a slightly different route (Path b). The carbocation intermediate CXXIV would lose a proton to give the selenodiene CXXVII a geometric isomer of CXXV. Then, the selenodiene CXXVII undergoes an intramolecular nucleophilic attack by the indole ring, affording the carbocation intermediate CXXVIII that, after deprotonation, affords the selenophene 166n.
In the same work, the authors evaluated the behavior of other aromatic substituents in the 1,2-shift process that initiates the cascade reaction toward substituted selenophenes. Thus, using 3-methyl-substituted 1,3,3-triphenyl-1-butynes 167 as starting materials in the reaction with SeCl2, under the previously optimized conditions, the corresponding fused-selenophenes 168a–c were obtained in moderate to good yields (Scheme 113). These results confirmed that other aromatic rings are also able to promote the required initial 1,2-shift and suggest that the reaction could have a broader scope. Further, the isolation of 168c as a mixture of products resulted from the migration of the phenyl and 4-chlorophenyl moieties, reinforced the conjecture that the migration aptitude is related to the electronic richness of the potentially migrating groups. These outcomes indicate that the formation of the benzo[b]selenopheno[3,2-d]selenophenes 168 occurs via the selenophene intermediate CXXIX, isostructural to the trisubstituted selenophenes 166a–m. The intermediate CXXIX reacts with another equivalent of SeCl2 by a SEAr, resulting in the chloroseleno intermediate CXXX, that undergoes an intramolecular cyclization to afford, after releasing HCl, the selenophene 168 (Scheme 113).
4.4. Other Approaches to Prepare Fused Selenophenes
Other efficient approaches for the synthesis of fused selenophenes 170 have been recently developed. For instance, the double intramolecular cyclization reaction of ortho-diynyl/triynyl chalcogenides 169, using Se-based electrophilic species, generated in situ by the reaction between dialkyl diselenides and iron salts or Oxone® (Scheme 114).
In this context, the synthesis of benzo[b]furan/thiophene/selenophene-fused selenophenes 172 through the double intramolecular 5-endo-dig cyclization of butyl[2-(phenylbuta-1,3-diynyl)phenyl]-chalcogens 171 was described (Scheme 115) [185]. The reaction was carried out in the presence of FeCl3·6H2O (2 equiv) and diorganyl diselenides (1.75 equiv) in refluxing DCM under air atmosphere for 4–24 h. The versatility of the protocol was firstly evaluated by using different dialkyl diselenides, giving the desired fused selenophenes 172a–d in good yields. Several alkyl groups were used; however, butyl proved to be the more reactive one. These standard reaction conditions were also compatible with several 1,3-diynes bearing electron-rich and electron-poor aryl groups and 3-thyenyl, affording the respective products 172e–g and 172i in 67–74% yields. A lower yield was obtained when an alkyl 1,3-diyne was employed, probably due to the absence of π-bonds next to the alkyne moiety, which decreases the reactivity for the electrophilic attack at the C-C triple bond. The nucleophilic effect of a heteroatom other than oxygen was analyzed by the introduction of sulfur, selenium, and nitrogen atoms at the 2-position of the aryl group bonded to the alkyne. The cyclization reaction of sulfur and selenodiynes with dibutyl diselenide gave the fused selenophenes 172j–o in 60–89% yields after 4–15 h. The cyclization of 1,3-diynes ortho-functionalized with nitrogen atom as a nucleophile, however, gave a mixture of byproducts.
In the same work, an interesting study using unsymmetrical 1,3-diynes bearing two potential nucleophiles was conducted; considering the differences in reactivity of the nucleophiles, the reaction could lead to different products of cyclization (Scheme 116). In the competitive cyclization reaction between the nucleophilic oxygen at the ortho-position of the aryl group and the sulfur or selenium at the ortho-position of the second aryl group, the selenophenes 172s and 172t were exclusively obtained, due to the higher nucleophilicity of sulfur and selenium compared to that of oxygen. In contrast, the cyclization of 1,3-diyne 171 involves the competition between selenium and sulfur nucleophiles and gave the product 172u, from the sulfur nucleophilic attack, in 74% yield. In this case, although the nucleophilicity of the selenium atom is higher than that of the sulfur atom, the steric hindrance of the butyl group was determinant to the observed selectivity.
The proposed mechanism for the synthesis of the fused selenophenes 172 involves, firstly, the reaction between the iron salt and the dialkyl diselenide, promoting the heterogeneous cleavage of the Se-Se bond to give a butylselanyl cation and butylselenolate anion (Scheme 117). Then, the iron(III) coordination with one selenium atom from dibutyl diselenide activates the other one towards nucleophilic attack by the alkyne giving the seleniranium ion CXXXI. After an intramolecular nucleophilic anti-attack by the pendant o-selenium atom to the activated seleniranium ion CXXXI, the selenonium salt CXXXII and butylselenolate anion are formed. Then, the BuSe- attacks the butyl group bonded to the selenium atom in the intermediate CXXXII, affording the 2-alkynyl benzoselenophene CXXXIII. In the sequence, a second similar cyclization process occurs with benzoselenophene CXXXIII to give the final product 172.
In 2019, the use of the FeCl3/Bu2Se2-promoted cascade cyclization of ortho-diynyl benzyl chalcogenides 173 for the synthesis of isochromene-fused selenophene derivatives 174 was described (Scheme 118) [186]. A total of nineteen fused selenophenes 174 were prepared by stirring a mixture of the diyne 173, FeCl3 and dibutyl diselenide in refluxing DCM under an argon atmosphere for 0.5–24 h. It was observed that ortho-diynyl benzyl selenide 173a, containing a pendant unsubstituted phenyl group on the terminal alkyne (R1 = Ph) gave the respective selenophene 174a in 77% yield after 1 h, while the presence of substituent at the ortho- (R1 = 2-Me-C6H4 and 2-Cl-C6H4) or para-positions (R1 = 4-MeOC6H4 and 4-FC6H4) of the phenyl caused a decrease in yields of products 174b-e, which were obtained in 40%–62% yields. No reaction occurred when the naphthyl group was bonded on the terminal alkyne, or when dioxolane and naphthyl were present as substituent groups at the benzyl aryl ring. The use of oxygen as a nucleophile instead of selenium was evaluated. Although less nucleophilic than the selenium analogues, the benzyl methyl ethers were suitable substrates in the reaction, due the highly (Lewis) acidic and oxophilic character of iron(III), which probably support the nucleophilic substitution step allowing the cyclization. In general, larger reaction times (up to 24 h) were required to give the respective isochromene-fused selenophenes 174i–n in 41%–83% yields. The protocol was compatible to diyne 173o, containing the heteroaryl group 3-thienyl, giving the corresponding product 173o in 62% yield after 8 h. No reaction was observed when an alkyl group was introduced at the carbon–carbon triple bond of the benzyl methyl selenoether 173p (R1 = C4H9), as well as when the benzyl methyl thioether 173q (XR = SMe) were substrates. In the case of thioether, the respective selenophene 174q was not formed, probably because the sulfur atom is not sufficiently nucleophilic to promote the cyclization step.
The proposed mechanism for the synthesis of the fused selenophenes 174 is similar to that described in Scheme 119, for the synthesis of 172. Firstly, FeCl3 reacts with dibutyl diselenide promoting a Se-Se bond heterolytic cleavage to form a butylselanyl cation and butylselenolate anion. Then, the electrophilic portion of dibutyl diselenide adds to the C-C triple bond of the substrate 173, generating the three-membered cyclic seleniranium cation CXXXV. Subsequently, an intramolecular attack by the selenium atom from the benzylic position occurs, affording the isoselenochromene intermediate CXXXVI, through a 6-endo-dig cyclization and releasing BuSe- to the reaction medium. In the sequence, the butyl group bonded to the selenium cation is removed by the reaction with butylselenolate, giving the 4-(butylselanyl)-isoselenochromene CXXXVII. Finally, this intermediary reacts with iron(III) chloride and dibutyl diselenide again, in a second cyclization reaction, now via a 5-endo-dig mode, to give the product 174 (Scheme 119).
This year (2020), an alternative approach for the synthesis of selenophene-fused chromene derivatives 176 was described [187]. The method involves the one-pot formation of C-C, C-Se, Se-C and C-Se bonds, using FeCl3/Bu2Se2 to promote the cyclization reaction of 1,3-diynyl propargyl aryl ethers 175, in DCM, at room temperature under argon atmosphere (Scheme 120). The extent and limitations of the protocol was firstly investigated by evaluating the influence of the substituents directly bonded to the alkyne triple bond. The electron-rich 1,3-diynyl propargyl aryl ether 175b (R = 4-MeO-C6H4) reacted under the optimal conditions to afford the respective fused selenophene 176b in 61% yield after 45 min, while the electron-poor 175c (R = 4-Cl-C6H4) was less reactive, giving 176c in 40% yield after 1 h. An equally good result was obtained using the sterically hindered alkyne 175d (R = 2-naphthyl), and the selenophene derivative 176d was isolated in 66% yield after 1 h of reaction. According to the authors, the expected products from 1,3-diynyl propargyl aryl ethers with para-fluoro, meta-methoxy and ortho-amino phenyl groups and 3-pyridyl could not be isolated, due to their decomposition during the purification process. The tentative reactions starting from alkyl, propargyl alcohol, and terminal 1,3-diynes hydrogen failed once the starting materials were unstable under the reaction conditions. Regarding the phenyl group directly bonded to the oxygen atom of the substrate 175, the influence of the presence of ortho- and para-substituents was evaluated, and the respective cyclized products 176e-l were obtained in moderate to good yields. The regioselectivity of the nucleophilic attack of the aromatic ring to the triple bond was studied by the reaction of the meta-chloro-substituted 1,3-diyne 175m. In this case, it was observed the formation of a 3:1 mixture (determined by GC/MS) of 176m and 176m’ in 70% overall yield. The moderate regioselectivity in favor of 176m may be influenced by the steric hindrance caused by the chlorine atom in the 176m’ regioisomer. The reaction was explored in the cyclization of other chalcogenoethers (S, Se and Te), and only the thioether derivative was a suitable substrate, affording the respective thiochromene derivative 176n in only 33% yield. This outcome indicates a strong influence of the identity of the chalcogen atom in the reactivity of the whole molecule.
The first step of the reaction mechanism is the formation of a butylselenium iron complex, which activates the triple of the substrate 175, forming the intermediate CXXXVIII. Then, an attack of the electron cloud from the aromatic ring at the activated intermediate CXXXVIII occurs, giving the cyclized species CXXXIX, which rapidly releases butylselenolate and iron salt to the reaction medium, to form the intermediate CXL. The removal of a proton from intermediate CXL by a chloride anion, restores the aromaticity, giving the 3-(butylselanyl)-2H-chromene CXLI. In the sequence, the organoselenium iron complex promotes the activation of the remaining triple bond of intermediate CXLI, and an intramolecular nucleophilic attack by the selenium generates the selenonium CXLII. The removal of the butyl group bonded to the selenium atom by the chloride ion via an SN2 displacement affords the selenophene chromene derivative 176 and butyl chloride (detected by GC/MS) (Scheme 121).
The optimal conditions were successfully extended to the cyclization of propargyl anilines 177, leading to the formation of 1-(butylselanyl)-selenophene-fused quinolines 178. The 1,3-Diynes containing both electron-donor or electron-withdrawing groups at the pendant aromatic rings were suitable substrates for the reaction, and the respective products 178a–e were obtained in 49%–78% yield after 1 h (Scheme 122).
The reactivity of the new fused-selenophenes 176a and 178a was explored in the Pd-catalyzed Suzuki cross-coupling reaction with arylboronic acids (Scheme 123). The selenophene-fused chromene 176a reacted with phenyl and p-tolylboronic acids under Suzuki conditions to produce the corresponding products 179a and 179b in 82% and 72% yields, respectively. Similarly, the selenophene-fused quinoline 178a coupled with 4-methoxyphenylboronic acid to give 179c in 43% yield.
In 2019, some of us [188] reported the synthesis of 5-H-selenopheno[3,2-c]isochromen-5-ones 181 through a double intramolecular cyclization of methyl 2-(organyl-1,3-diynyl)benzoate 180 promoted by electrophilic species of selenium generated in situ by the reaction of dialkyl diselenides with Oxone®, under the reflux of EtOH, in an open flask (Scheme 124). This protocol was suitable for different dialkyl diselenides (R2 = Bu, Et, Oct), which reacted with the diyne 180 to give the expected isochromenones 181a-c in 77–86% yields, while no reaction was observed starting from dibenzyl- and bis(2-naphthylmethyl)diselenide. Regarding the 1,3-diyne counterpart, both electron-releasing and electron-withdrawing groups at the para-position of the pendant phenyl ring (R1) of 180 were tolerated in the reaction with dibutyl diselenide (181f–h). Similar results were obtained when naphthyl and butyl groups are directly bonded to the alkyne. An interesting result was obtained when ortho-methoxy-1,3-diyne 180g (R1 = 2-MeOC6H4) was used as substrate, with the desired fused selenophene 181g being obtained in only 40% yield (Scheme 125). This result can be explained by the two competing intramolecular Se-cyclization and the O-cyclization reactions in the intermediate 184c, formed in the first step of the reaction (see Scheme 126, for the reaction mechanism), producing the benzofuran derivative 181g’ in 50% yield as a coproduct of the reaction. However, when the dimer dimethyl 2,2′-(buta-1,3-diyne-1,4-diyl)dibenzoate was submitted the optimal conditions in the reaction with Bu2Se2, the double intramolecular O-cyclization product was not formed, and the fused isochromenones selenophene 181k was obtained in 80% yield after 1.5 h. The presence of a fluor-group at the pendant ring of the ester group in 180l (R = F) did not alter the reactivity, affording the respective selenophene 181l in 84% yield after 2.5 h (Scheme 124).
To better understand the mechanism for this cyclization reaction, a series of control experiments were carried out by the authors. For the identification of the active electrophilic selenium species involved in the reaction, 77Se NMR spectra were collected from the reaction mixture between Oxone® and dibutyl diselenide in an NMR tube, using MeOD-d4 as the solvent. This was observed as a signal at 984 ppm, which was attributed to methyl butane-1-seleninate 182, formed from the reaction between butylseleninic acid 183 and MeOD-d4. The formation of butylseleninic acid 182 is a result of the overoxidation of dibutyl diselenide by Oxone®, which could pass by the intermediates BuSeOSO3- (CXLIV) and BuSeOH (CXLV). The reaction between the diyne 180a and Bu2Se2/Oxone® was monitored by GC/MS. Aliquots of the reaction mixture were collected, pretreated by a “micro work-up” and injected into the GC/MS spectrometer. This experiment was carried out under the optimal conditions, and aliquots were collected at 15, 30, 60 and 120 min. From these experiments, the intermediate 184 was identified (formed by O-cyclization reactions). With these elements in hand, a mechanism was proposed, which firstly involves the reaction between dibutyl diselenide and Oxone® to form the intermediates CXLIV and CXLV (Scheme 126, path a). The species CXLV can react with H+ from the reaction medium, leading to BuSeOH2+ (CXLVIII). After, diyne 180a reacts with CXLIV and CXLVIII, leading to the cyclic intermediate CXLIX, releasing HSO4- and H2O to the medium. The displacement of the methyl group from CXLIX by a nucleophile (HSO4- from previous step and/or SO42-, from Oxone®), affords the key intermediate 184. Following this, the remaining triple bond of 187 reacts in the same way with CXLIV and CXLVIII, giving the fused-selenophene cation intermediate CL. The displacement of the butyl group from the selenonium cation affords the expected 5H-selenopheno[3,2-c]isochromen-5-one 181a.
In 2019, the Fe(III)-promoted intramolecular cascade cyclization of 1,3-diynes 185 and 1,3,5-triynes 186 under reflux of DCM for the construction of selenophene-fused, quinoline- and acridine-based heteroacene scaffolds 187–190 was described (Scheme 127) [189]. In this protocol, the 1,3-diyne and 1,3,5-triyne were cyclized through diversified internal nucleophiles by using dialkyl diselenides, which play a dual role, as a cyclizing agent and in the insertion of one and/or two selenium atoms and one R2Se group in the final product. The synthesis of selenophene-fused thieno/selenophene[2,3-b]quinolines 187 and selenophene-fused thieno/furo[2,3-c]acridine 188 was evaluated in the electrophilic cyclization of ortho-functionalized 1,3-diynes with SMe/SeMe or 3-thionyl/3-furyl, respectively, using the FeCl3·6H2O/dialkyl diselenide system (2.5:2 equiv). Under these reaction conditions, several selenophene-fused quinoline/acridine 187 and 188 were obtained in 70–88% after 5–8 h. The cascade cyclization of 1,3,5-triynes 186 was achieved in the presence of Fe(III) (3 equiv) and dibutyl diselenide (2.5 equiv) conditions, and several diselenophene-fused thieno/selenophene-[2,3-b]quinolines 188, and diselenophene-fused thieno/furo[2,3-c]acridine 190 were obtained in 68%–84% yields after 8 h.
In this reaction, when 3-furan and 3-thiophene were used as internal nucleophiles, the nucleophilic attack takes place from the ortho-position of furan and thiophene, which interestingly resulted in six-membered acridine-core heterocycles 188 and 190. On the other hand, when sulfur and selenium were used instead, five-membered heterocycles 188 and 189 were obtained (Scheme 127). Based on these results, a mechanism for the synthesis of diselenophene-fused thiene[2,3-c]acridine 190 was proposed, which involves, in the first step, the reaction of iron salt with dibutyl diselenide to promote the heterolytic cleavage of the Se-Se bond, producing the butylselanyl cation and butylselenolate anion (Scheme 128). After the Fe(III) coordinates with one selenium atom from dibutyl diselenide, it favors the addition to a triple bond and affording the seleniranium intermediate CLI. In the sequence, the intermediate CLI undergoes an anti-attack by the thiophene nucleophile, resulting in the cyclic intermediate CLII. The rearomatization of intermediate CLIII is achieved via the SN2 displacement by the butylselenolate anion, giving the thieno[2,3-c]acridine CLIV. The second cyclization step proceeds with the BuSe nucleophilic moiety in CLV, which resulted in the intermediate selenophene-fused thieno[2,3-c]acridine CLVI. On continuation, the cascade cyclization proceeds for the remaining triple bond in CLVI, which finally affords the product diselenophene-fused thieno[2,3-c]acridine 190.
The same strategy for the selenocyclization of properly substituted alkynes was used this year for the synthesis of 3-(aryl/alkylselanyl)-2-arylselenophene[2,3-b]quinoxaline 193 and 6-aryl-7-(phenylselanyl)selenopheno[2,3-b]pyrazine 194 [190]. The authors developed a Fe(III)-promoted electrophilic cascade cyclization of 2-(methylselanyl)-3-(arylethynyl)quinoxaline 191 and 2-(methylselanyl)-3-(arylethynyl)pyrazine 192, respectively, using diorganyl diselenides and diorganyl disulfides as chalcogen sources, in DCM at 45 °C (Scheme 129). Differently substituted 2-(methylselanyl)-3-(arylethynyl)quinoxaline 191 bearing neutral (R = H), electron-donor (R = 4-Me and 4-MeO) and electron-withdrawing (R = 4-F, 3-F and 2-F) groups were suitable substrates in the reaction using FeCl3/PhSeSePh, affording the corresponding 3-(phenylselanyl)-2-arylselenopheno[2,3-b]quinoxaline 193a-f in moderate to good yields (61–92%). The presence of substituents in the phenyl ring reduced the reactivity of the substrate (193a vs. 193b-f), and this decrease was less pronounced in the case of the electron-deficient alkynes. When alkyl diselenides (dibutyl and dimethyl) were employed instead of diphenyl diselenide, the respective selenophenes 193g and 193h were obtained in 66% and 80% yields after 22 and 5 h, respectively. Diorganyl disulfides were suitable dichalcogenides in the reaction, despite the low yields compared to the diselenides analogues. The reaction was more efficient using dimethyl disulfide in comparation to diphenyl disulfide, and the sulfides 193i and 193j were obtained in 57% and 5% yields, after 21 and 37 h, respectively. The developed strategy was suitable for the cyclization of differently substituted 2-(methylselanyl)-3-(arylethynyl)pyrazines 192 by diphenyl diselenide, giving the respective 6-aryl-7-(phenylselanyl)selenopheno[2,3-b]pyrazine derivatives 194a-e in 47%–70% yields after 3–4 h, and there does not seem to be a clear electronic effect from the aromatic ring. The proposed mechanism for the novel seleno-cascade cyclization is similar to that described in Scheme 128.
In the same work [190], the authors described the synthesis of 2-arylselenopheno[2,3-b]quinoxaline derivatives 196 through a SNAr reaction of 2-chloro-3-(arylethynyl)quinoxaline derivatives 195 using NaHSe as a nucleophile (Scheme 130). NaHSe was prepared in situ by the reaction of elemental selenium (1 equiv) with sodium borohydride (2 equiv) in a 1:2 EtOH/H2O mixture at 0 °C for 15 min. Once the color of the reaction mixture changed from black to colorless, indicating the formation of NaHSe, a solution of 2-chloro-3-(arylethynyl)quinoxaline 195 (1 equiv) in ethanol was added, and the resulting mixture was stirred under reflux. After 1 h of reaction, the corresponding 2-arylselenopheno[2,3-b]quinoxaline derivatives 196a-f were obtained in 69–95% yield. The lower reactivity was observed starting from 3-fluoro-substituted phenyl derivative 195e (R = 3-F) while the best substrate for the reaction was 195f, substituted with a fluorine atom at the ortho-position (R = 2-F).
Recently, the one-step polyelectrophilic cyclization of polyynes 197 using the ambiphilic reagent MeSeCl for the synthesis of polyselenophenes fused to benzothiophenes 198 in DCM/DCE as solvent under N2 atmosphere for 1 h was reported (Scheme 131) [191]. Under these reaction conditions, the fused selenophenes 198a-c were obtained in moderate to excellent yields (67–100%). In the tetracyclization sequence to form the polyselenophene 198d, the fully annulated product was obtained together with a complex mixture of other byproducts (suspected MeSe/Cl addition products). This drawback was circumvented by heating the reaction mixture in refluxing DCE for 48 h, giving semi-pure 198d in 50% yield (by 1H NMR). Chromatographic separation of 198d from these byproducts proved to be difficult, and recrystallization afforded pure 198d in only 10% yield. The ambiphilic reagent MeSeCl was prepared by the reaction of dimethyl diselenide with sulfuryl chloride (SO2Cl2) in dichloromethane at 0 °C under a nitrogen atmosphere, with a loss of SO2(g).
The proposed mechanism of the polyelectrophilic cyclization of polyynes 197 with MeSeCl for the synthesis of selenophenes fused to benzothiophenes 198, is similar to those previously described selenocyclizations. Firstly, there is the activation of the triple bond by the MeSeCl, forming the complex CLVII, which could exist as the equilibrium between CLVIII and CLVIIIa. Then, the seleniranion CLVIIIa undergoes an intramolecular nucleophilic attack by the sulfur atom, producing the benzothiophene cation CLIX through a 5-endo-dig cyclization (Scheme 132). The removal of the methyl group bonded to the sulfur atom via SN2 displacement by the chloride ion affords the intermediate CLX. Finally, a second cyclization process occurs with intermediate CLX to give the final product 198.
The protocol depicted in Scheme 131 was extended to the bidirectional polyelectrophilic cyclization (biPEC) of tetraynes 199 and 201 using 4.5 equiv of MeSeCl. The diverging biPEC of 199 afforded the fused selenophene 200 in only 30% yield, probably due to the poor solubility of the product. In contrast, the converging biPEC of tetrayne 201 produced the dibenzo[b]selenopheno[2,3-d]thiophene 202 in 82% yield (Scheme 133).
The synthesis of dibenzo[b]selenopheno[2,3-d]thiophene 202 from tetrayne 201 with MeSeCl occurs by a “seesawing” mechanism, involving alternating shifts in electron density through the polyyne chain (Scheme 134). The reaction initiates by the cyclization of the symmetrical tetrayne 201 with MeSeCl to give the monocyclized product CLXIII. Then, the resultant electron-rich 3-(methylseleno)-benzothiophenyl group (it is more electron-rich than the phenyl one) drives the electron density to distal alkynyl carbon (indicated by δ−), favoring the cyclization via the stabilized cation intermediate CLXIV, giving the diyne CLXV. In the sequence, the monocylization of symmetrical diyne CLXV, via the intermediate CLXVI, gives the unsymmetrical alkyne CLXVII, that is finally cyclized after attack by MeSeCl to the remote carbon, favoring the cyclization through the more stable cation CLXVIII, to give the product 202.
5. Conclusions
This review highlights the protocols that were developed in recent years for the synthesis of selenophenes and their derivatives, as well as the mechanistic insights of the different reactions. Most of the synthetic approaches covered in this review are conducted under relatively mild conditions and tolerate a wide variety of functional groups. However, there is still room to improve existing synthetic methods, as well as to develop new ones. We believe that, in the coming years, there will be a further development of protocols to access the selenophene core, especially its fused derivatives. In addition, we see good prospects in the use of these derivatives as building blocks in the formation of new carbon–carbon bonds, in pharmacological enhancement, in addition to the emergence of new materials based on selenophene for optoelectronic applications. In this sense, we hope that this review can stimulate the research in preparative methods to access this peculiar class of organoselenium compounds, thus providing new alternatives for accessing the selenophene family, through more efficient, robust, and greener methods.
Author Contributions
Conceptualization, G.P. and E.J.L.; methodology, G.P.; writing—original draft preparation, P.S.H., T.J.P. and F.P.; writing—review and editing, F.P., L.B., G.P., E.J.L.; supervision, G.P. and E.J.L.; All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001; FAPERGS, grant number 17/2551-0000964-9; CNPq and FINEP.
Acknowledgments
We would like to thank all the authors cited in this review, without whom it would not be possible to write it. This work has been undertaken under the umbrella of the Selenium Sulfur Redox and Catalysis Network (SeSRedCat).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Godoi, M.; Paixão, M.W.; Braga, A.L. Chiral Organoselenium-transition-metal Catalysts in Asymmetric Transformations. Dalton Trans. 2011, 40, 11347–11355. [Google Scholar] [CrossRef] [PubMed]
- Lenardão, E.J.; Santi, C.; Sancineto, L. Organoselenium Compounds as Reagents and Catalysts to Develop New Green Protocols; Springer: Cham, Switzerland, 2018; pp. 1–98. [Google Scholar]
- Singh, F.V.; Wirth, T. Selenium Reagents as Catalysts. Catal. Sci. Technol. 2019, 9, 1073–1091. [Google Scholar] [CrossRef]
- Shao, L.; Li, Y.; Lu, J.; Jiang, X. Recent Progress in Selenium-catalyzed Organic Reactions. Org. Chem. Front. 2019, 6, 2999–3041. [Google Scholar] [CrossRef]
- Stein, A.L.; Bilheri, F.N.; Zeni, G. Application of Organoselenides in the Suzuki, Negishi, Sonogashira and Kumada Cross-coupling Reactions. Chem. Commun. 2015, 51, 15522–15525. [Google Scholar] [CrossRef] [PubMed]
- Grélaud, S.; Desvergnes, V.; Landais, Y. Stereocontrolled (Me3Si)3SiH-Mediated Radical and Ionic Hydride Transfer in Synthesis of 2,3,5-Trisubstituted THF. Org. Lett. 2016, 18, 1542–1545. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, K.; Wang, L.; Yu, Z. Iron-Promoted Difunctionalization of Alkenes by Phenylselenylation/1,2-Aryl Migration. Org. Lett. 2017, 19, 5450–5453. [Google Scholar] [CrossRef] [PubMed]
- Temperini, A.; Piazzolla, F.; Minuti, L.; Curini, M.; Siciliano, C. General, Mild, and Metal-Free Synthesis of Phenyl Selenoesters from Anhydrides and Their Use in Peptide Synthesis. J. Org. Chem. 2017, 82, 4588–4603. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, D.Y. Electrochemical Radical Selenylation/1,2-Carbon Migration and Dowd–Beckwith-Type Ring-Expansion Sequences of Alkenylcyclobutanols. Org. Lett. 2019, 21, 1021–1025. [Google Scholar] [CrossRef]
- Perin, G.; Lenardão, E.J.; Jacob, R.G.; Panatieri, R.B. Synthesis of Vinyl Selenides. Chem. Rev. 2009, 109, 1277–1301. [Google Scholar] [CrossRef]
- Wirth, T. Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Santoro, S.; Azeredo, J.B.; Nascimento, V.; Sancineto, L.; Braga, A.L.; Santi, C. The Green Side of the Moon: Ecofriendly Aspects of Organoselenium Chemistry. RSC Adv. 2014, 4, 31521–31535. [Google Scholar] [CrossRef]
- Perin, G.; Alves, D.; Jacob, R.G.; Barcellos, A.M.; Soares, L.K.; Lenardão, E.J. Synthesis of Organochalcogen Compounds using Non-Conventional Reaction Media. ChemistrySelect 2016, 2, 205–258. [Google Scholar] [CrossRef]
- Palomba, M.; Bagnoli, L.; Marini, F.; Santi, C.; Sancineto, L. Recent advances in the chemistry of vinylchalcogenides. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 235–244. [Google Scholar] [CrossRef]
- Santi, C.; Jacob, R.G.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardão, E.J. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules 2016, 21, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos, A.M.; Abenante, L.; Sarro, M.T.; Leo, I.D.; Lenardão, E.J.; Perin, G.; Santi, C. New Prospective for Redox Modulation Mediated by Organo selenium and Organotellurium Compounds. Curr. Org. Chem. 2017, 21, 2044–2061. [Google Scholar] [CrossRef]
- Jain, V.K.; Priyadarsini, K.I. Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; The Royal Society of Chemistry: Croydon, UK, 2017. [Google Scholar]
- Sudati, J.H.; Nogara, P.A.; Saraiva, R.A.; Wagner, C.; Alberto, E.E.; Braga, A.L.; Fachinetto, R.; Piquini, P.C.; Rocha, J.B.T. Diselenoamino acid Derivatives as GPx Mimics and as Substrates of TrxR: In vitro and in silico Studies. Org. Biomol. Chem. 2018, 16, 3777–3783. [Google Scholar] [CrossRef]
- Leo, I.D.; Messina, F.; Nascimento, V.; Nacca, F.G.; Pietrella, D.; Lenardão, E.J.; Perin, G.; Sancineto, L. Synthetic Approaches to Organoselenium Derivatives with Antimicrobial and Anti -Biofilm Activity. Mini-Rev. Org. Chem. 2019, 16, 589–601. [Google Scholar] [CrossRef]
- Ninomiya, M.; Garud, D.R.; Koketsu, M. Biologically Significant Selenium-containing Heterocycles. Chem. Rev. 2011, 255, 2968–2990. [Google Scholar] [CrossRef]
- Ruberte, A.C.; Sanmartin, C.; Aydillo, C.; Sharma, A.K.; Plano, D. Development and Therapeutic Potential of Selenazo Compounds. J. Med. Chem. 2020, 63, 1473–1489. [Google Scholar] [CrossRef]
- Abdelwahab Mahmoud, A.B.; Kirsch, G.; Peagle, E. Biologically Active Selenophenes and Benzo[b]selenophenes. Curr. Org. Synth. 2017, 14, 1091–1101. [Google Scholar] [CrossRef]
- Gay, B.M.; Prigol, M.; Stein, A.L.; Nogueira, C.W. Antidepressant-like Pharmacological Profile of 3-(4-Fluorophenylselenyl)-2,5-diphenylselenophene: Involvement of Serotonergic System. Neuropharmacology 2010, 59, 172–179. [Google Scholar] [CrossRef]
- Gai, B.M.; Stein, A.L.; Roehrs, J.A.; Bilheri, F.N.; Nogueira, C.W.; Zeni, G. Synthesis and Antidepressant-like Activity of Selenophenes Obtained via Iron(III)–PhSeSePh-mediated Cyclization of Z-Selenoenynes. Org. Biomol. Chem. 2012, 10, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Gai, B.M.; Bortolatto, C.F.; Heck, S.O.; Stein, A.L.; Duarte, M.M.M.F.; Zeni, G.; Nogueira, C.W. An Organoselenium Compound Improves Behavioral, Endocrinal and Neurochemical Changes Induced by Corticosterone in Mice. Psychopharmacology 2014, 231, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Gai, B.M.; Bortolatto, C.F.; Brüning, C.A.; Zborowski, V.A.; Stein, A.L.; Zeni, G.; Nogueira, C.W. Depression-Related Behavior and Mechanical Allodynia are Blocked by 3-(4-Fluorophenylselenyl)-2,5-diphenylselenophene in a Mouse Model of Neuropathic Pain Induced by Partial Sciatic Nerve Ligation. Neuropharmacology 2014, 79, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Gai, B.M.; Sanna, M.D.; Stein, A.L.; Zeni, G.; Galeotti, N.; Nogueira, C.W. ERK1/2 Phosphorylation is Involved in the Antidepressant-like Action of 2,5-Diphenyl-3-(4-fluorophenylseleno)-selenophene in Mice. Eur. J. Pharmacol. 2014, 736, 44–54. [Google Scholar] [CrossRef]
- Velasquez, D.; Quines, C.; Pistóia, R.; Zeni, G.; Nogueira, C.W. Selective inhibition of MAO-A Activity Results in an Antidepressant-like Action of 2-Benzoyl 4-iodoselenophene in Mice. Physiol. Behav. 2017, 170, 100–105. [Google Scholar] [CrossRef]
- Schumacher, R.F.; Rosário, A.R.; Souza, A.C.G.; Acker, C.I.; Nogueira, C.W.; Zeni, G. The Potential Antioxidant Activity of 2,3-Dihydroselenophene, a Prototype Drug of 4-Aryl-2,3-dihydroselenophenes. Bioorg. Med. Chem. 2011, 19, 1418–1425. [Google Scholar] [CrossRef] [Green Version]
- Tanini, D.; Panzella, L.; Amorati, R.; Capperucci, A.; Pizzo, E.; Napolitano, A.; Menichetti, S.; d’Ischia, M. Resveratrol-based Benzoselenophenes with an Enhanced Antioxidant and Chain Breaking Capacity. Org. Biomol. Chem. 2015, 13, 5757–5764. [Google Scholar] [CrossRef] [Green Version]
- Paegle, E.; Domracheva, I.; Turovska, B.; Petrova, M.; Kanepe-Lapsa, I.; Gulbe, A.; Liepinsh, E.; Arsenyan, P. Natural-Antioxidant-Inspired Benzo[b]selenophenes: Synthesis, Redox Properties, and Antiproliferative Activity. Chem. Asian J. 2016, 11, 1929–1938. [Google Scholar] [CrossRef]
- Tavadyan, L.A.; Manukyan, Z.H.; Harutyunyan, L.H.; Musayelyan, M.V.; Sahakyan, A.D.; Tonikyan, H.G. Antioxidant Properties of Selenophene, Thiophene and Their Aminocarbonitrile Derivatives. Antioxidants 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Yan, J.; Poon, J.-F.; Gates, P.J.; Butcher, R.J.; Engman, L. Chain-Breaking Phenolic 2,3-Dihydrobenzo[b]selenophene Antioxidants: Proximity Effects and Regeneration Studies. Chem. Eur. J. 2017, 23, 15080–15088. [Google Scholar] [CrossRef] [Green Version]
- Manikova, D.; Sestakova, Z.; Rendekova, J.; Vlasakova, D.; Lukacova, P.; Paegle, E.; Arsenyan, P.; Chovanec, M. Resveratrol-Inspired Benzo[b]selenophenes Act as Anti-Oxidants in Yeast. Molecules 2018, 23, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsherbini, M.; Hamama, W.S.; Zoorob, H.H. An Easy Synthetic Approach to Construct Some Ebselen Analogues and Benzo[b]selenophene Derivatives: Their Antioxidant and Cytotoxic Assessment. J. Heterocycl. Chem. 2018, 55, 1645–1650. [Google Scholar] [CrossRef]
- Wilhelm, E.A.; Jesse, C.R.; Bortolatto, C.F.; Nogueira, C.W.; Savegnago, L. Anticonvulsant and Antioxidant Effects of 3-Alkynyl Selenophene in 21-Day-old Rats on Pilocarpine Model of Seizures. Brain Res. Bull. 2009, 79, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, E.A.; Gai, B.M.; Souza, A.C.G.; Bortolatto, C.F.; Roehrs, J.A.; Nogueira, C.W. Involvement of GABAergic and Glutamatergic Systems in the Anticonvulsant Activity of 3-Alkynyl Selenophene in 21 Day-old Rats. Mol. Cell. Biochem. 2012, 365, 175–180. [Google Scholar] [CrossRef]
- Wiles, J.A.; Phadke, A.S.; Bradbury, B.J.; Pucci, M.J.; Thanassi, J.A.; Deshpande, M. Selenophene-Containing Inhibitors of Type IIA Bacterial Topoisomerases. J. Med. Chem. 2011, 54, 3418–3425. [Google Scholar] [CrossRef]
- Shiah, H.-S.; Lee, W.-S.; Juang, S.-H.; Hong, P.-C.; Lung, C.-C.; Chang, C.-J.; Chou, K.-M.; Chang, J.-Y. Mitochondria-mediated and p53-Associated Apoptosis Induced in Human Cancer Cells by a Novel Selenophene Derivative, D-501036. Biochem. Pharmacol. 2007, 73, 610–619. [Google Scholar] [CrossRef]
- Chou, L.-C.; Huang, L.-J.; Hsu, M.-H.; Fang, M.-C.; Yang, J.-S.; Zhuang, S.-H.; Lin, H.-Y.; Lee, F.-Y.; Teng, C.-M.; Kuo, S.-C. Synthesis of 1-Benzyl-3-(5-hydroxymethyl-2-furyl)selenolo[3,2-c]pyrazole Derivatives as New Anticancer Agents. Eur. J. Med. Chem. 2010, 45, 1395–1402. [Google Scholar] [CrossRef]
- Arsenyan, P.; Paegle, E.; Domracheva, I.; Gulbe, A.; Kanepe-Lapsa, I.; Shestakova, I. Selenium Analogues of Raloxifene as Promising Antiproliferative Agents in Treatment of Breast Cancer. Eur. J. Med. Chem. 2014, 87, 471–483. [Google Scholar] [CrossRef]
- Doganay, K.; Kelestemur, U.; Balcioglu, S.; Ates, B.; Altundas, A. Synthesis, Reaction, and Evaluation of the Anticancer Activity of 6,7,8,9-Tetrahydro-5H-cyclohepta[4,5]selenopheno[2,3-d]pyrimidine Derivatives. Turk. J. Chem. 2016, 40, 631–640. [Google Scholar] [CrossRef]
- Luo, J.; Hu, Z.; Xiao, Y.; Yang, T.; Dong, C.; Huang, J.; Zhou, H.-B. Rational Design and Optimization of Selenophenes with Basic Side Chains as Novel Potent Selective Estrogen Receptor Modulators (SERMs) for Breast Cancer Therapy. Med. Chem. Commun. 2017, 8, 1485–1497. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z.; Hu, Z.; Li, C.; Tang, C.; Carlson, K.E.; Luo, J.; Dong, C.; Katzenellenbogen, J.A.; Huang, J.; et al. Selenophenes: Introducing a New Element into the Core of Non-Steroidal Estrogen Receptor Ligands. ChemMedChem 2017, 12, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liang, P.; Si, W.; Zhao, B.; Shao, J.; Huang, W.; Zhang, Y.; Zhang, Q.; Dong, X. A Selenophene Substituted Diketopyrrolopyrrole Nanotheranostic Agent for Highly Efficient Photoacoustic/infrared-thermal Imaging-guided phototherapy. Org. Chem. Front. 2018, 5, 98–105. [Google Scholar] [CrossRef]
- Karaman, O.; Almammadov, T.; Emre Gedik, M.; Gunaydin, G.; Kolemen, S.; Gunbas, G. Mitochondria-Targeting Selenophene-Modified BODIPY Based Photosensitizers for the Treatment of Hypoxic Cancer Cells. ChemMedChem 2019, 14, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Adly, M.E.; Gedawy, E.M.; El-Malah, A.A.; El-Telbany, F.A. Synthesis and Anticancer Activity of Certain Selenophene Derivatives. Russ. J. Org. Chem. 2019, 55, 1189–1196. [Google Scholar] [CrossRef]
- Wilhelm, E.A.; Jesse, C.R.; Bortolatto, C.F.; Nogueira, C.W.; Savegnago, L. Antinociceptive and Anti-allodynic Effects of 3-Alkynyl Selenophene on Different Models of Nociception in Mice. Pharmacol. Biochem. Behav. 2009, 93, 419–425. [Google Scholar] [CrossRef]
- Wilhelm, E.A.; Jesse, C.R.; Roman, S.S.; Nogueira, C.W.; Savegnago, L. Hepatoprotective Effect of 3-Alkynyl Selenophene on Acute Liver Injury Induced by d-Galactosamine and Lipopolysaccharide. Exp. Mol. Pathol. 2009, 87, 20–26. [Google Scholar] [CrossRef]
- Wilhelm, E.A.; Jesse, C.R.; Prigol, M.; Alves, D.; Schumacher, R.F.; Nogueira, C.W. 3-Alkynyl Selenophene Protects against Carbon-tetrachloride-induced and 2-Nitropropane-induced Hepatic Damage in Rats. Cell Biol. Toxicol. 2010, 26, 569–577. [Google Scholar] [CrossRef]
- Juang, S.H.; Lung, C.C.; Hsu, P.C.; Hsu, K.S.; Li, Y.C.; Hong, P.C.; Shiah, H.S.; Kuo, C.C.; Huang, C.W.; Wang, Y.C.; et al. D-501036, A Novel Selenophene-based Triheterocycle Derivative, Exhibits Potent in vitro and in vivo Antitumoral Activity which Involves DNA Damage and Ataxia telangiectasia–mutated Nuclear Protein Kinase Activation. Mol. Cancer. Ther. 2007, 6, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Xu, C.; Okamoto, T. Ladder π-Conjugated Materials with Main Group Elements. Pure Appl. Chem. 2006, 78, 721–730. [Google Scholar] [CrossRef]
- Mohanakrishnan, A.K.; Kumar, N.S.; Amaladass, P. Synthesis and Characterization of 9,9-Dialkylfluorene Capped Benzo[c]thiophene/benzo[c]selenophene Analogs as Potential OLEDs. Tetrahedron Lett. 2008, 49, 4792–4795. [Google Scholar] [CrossRef]
- Onk, I.; Hizalan, G.; Cevher, S.C.; Hacioglu, S.O.; Toppare, L.; Cirpan, A.J. Multipurpose Selenophene Containing Conjugated Polymers for Optoelectronic Applications. Macromol. Sci. Pure Appl. Chem. 2017, 54, 133–139. [Google Scholar] [CrossRef]
- Cho, W.; Sarada, G.; Lee, H.; Song, M.; Gal, Y.-S.; Lee, Y.; Jin, S.-H. Highly Efficient, Conventional and Flexible Deep-red Phosphorescent OLEDs using Ambipolar Thiophene/selenophene-phenylquinoline ligand-based Ir(III) complexes. Dyes Pigm. 2017, 136, 390–397. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, D.; Yang, X.; Jin, D.; Zhong, D.; Liu, B.; Zhou, G.; Ma, M.; Wu, Z. Asymmetric Heteroleptic Ir(III) Phosphorescent Complexes with Aromatic Selenide and Selenophene Groups: Synthesis and Photophysical, Electrochemical, and Electrophosphorescent Behaviors. Inorg. Chem. 2018, 57, 11027–11043. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deng, Y.; Zhu, X.; Wang, L.; Lv, L.; Wu, X.; Li, Z.; Shi, Q.; Peng, A.; Peng, Q.; et al. Toward Achieving Single-Molecule White Electroluminescence from Dual Emission of Fluorescence and Phosphorescence. Chem. Mater. 2020, 32, 4038–4044. [Google Scholar] [CrossRef]
- Petrenko, A.; Leitonas, K.; Volyniuk, D.; Baryshnikov, G.V.; Belyakov, S.; Minaev, B.F.; Ågren, H.; Durgaryan, H.; Gražulevičius, J.V.; Arsenyan, P. Benzoselenophenylpyridine Platinum Complexes: Green versus Red Phosphorescence towards Hybrid OLEDs. Dalton Trans. 2020, 49, 3393–3397. [Google Scholar] [CrossRef]
- Takimiya, K.; Kunugi, Y.; Konda, Y.; Niihara, N.; Otsubo, T. 2,6-Diphenylbenzo[1,2-b:4,5-b’]dichalcogenophenes: A New Class of High-Performance Semiconductors for Organic Field-Effect Transistors. J. Am. Chem. Soc. 2004, 126, 5084–5085. [Google Scholar] [CrossRef]
- Takimiya, K.; Kunugi, Y.; Konda, Y.; Ebata, H.; Toyoshima, Y.; Otsubo, T. 2,7-Diphenyl[1]benzoselenopheno[3,2-b][1]benzoselenophene as a Stable Organic Semiconductor for a High-Performance Field-Effect Transistor. J. Am. Chem. Soc. 2006, 128, 3044–3050. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takimiya, K. Facile Synthesis of Highly ð-Extended Heteroarenes, Dinaphtho[2,3-b:2′,3′-f]chalcogenopheno[3,2-b]chalcogenophenes, and Their Application to Field-Effect Transistors. J. Am. Chem. Soc. 2007, 129, 2224–2225. [Google Scholar] [CrossRef]
- Otsubo, T.; Takimiya, K. Selenophenes as Hetero-analogues of Thiophene-based Materials. In Handbook of Thiophene-Based Materials: Applications. In Organic Electronics and Photonics; Perepichka, I.F., Perepichka, D.F., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 321–340. [Google Scholar]
- Nakano, M.; Mori, H.; Shinamura, S.; Takimiya, K. Naphtho[2,3-b:6,7-b′]dichalcogenophenes: Syntheses, Characterizations, and Chalcogene Atom Effects on Organic Field-Effect Transistor and Organic Photovoltaic Devices. Chem. Mater. 2012, 24, 190–198. [Google Scholar] [CrossRef]
- Hwang, Y.-J.; Murari, N.M.; Jenekhe, S.A. New n-Type Polymer Semiconductors Based on Naphthalene Diimide and Selenophene Derivatives for Organic Field-effect Transistors. Polym. Chem. 2013, 4, 3187–3195. [Google Scholar] [CrossRef]
- Hollinger, J.; Gao, D.; Seferos, D.S. Selenophene Electronics. Isr. J. Chem. 2014, 54, 440–453. [Google Scholar] [CrossRef]
- Shan, Z.; Shi, J.; Xu, W.; Li, C.; Wang, H. Organic Field-effect Transistors Based on Biselenophene Derivatives as Active Layers. Dyes Pigm. 2019, 171, 107675. [Google Scholar] [CrossRef]
- Jang, S.-Y.; Kim, I.-B.; Kang, M.; Fei, Z.; Jung, E.; McCarthy-Ward, T.; Shaw, J.; Lim, D.-H.; Kim, Y.-J.; Mathur, S.; et al. Diseleno[3,2-b:2′,3′-d]selenophene-Containing High-Mobility Conjugated Polymer for Organic Field-Effect Transistors. Adv. Sci. 2019, 6, 1900245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, B.; Dellago, B.; Thamm, A.-K.; Mathis, T.; Stoeger, B.; Horkel, E.; Hametner, C.; Batlogg, B.; Froehlich, J.; Lumpi, D. Symmetric Mixed Sulfur–Selenium Fused Ring Systems as Potential Materials for Organic Field-Effect Transistors. Chem. Eur. J. 2020, 26, 2869–2882. [Google Scholar] [CrossRef]
- Petrenko, A.; Bezvikonnyi, O.; Volyniuk, D.; Danyliv, Y.; Simokaitiene, J.; Belyakov, S.; Grazulevicius, J.V.; Arsenyan, P. Synthesis of Fused Chalcogenophenocarbazoles: Towards Dual Emission Resulting from Hybridized Local and Charge-transfer States. New J. Chem. 2020, 44, 3903–3911. [Google Scholar] [CrossRef]
- Gao, D.; Hollinger, J.; Seferos, D.S. Selenophene-Thiophene Block Copolymer Solar Cells with Thermostable Nanostructures. ACS Nano 2012, 6, 7114–7121. [Google Scholar] [CrossRef]
- Ashraf, R.S.; Meager, I.; Nikolka, M.; Kirkus, M.; Planells, M.; Schroeder, B.C.; Holliday, S.; Hurhangee, M.; Nielsen, C.B.; Sirringhaus, H.; et al. Chalcogenophene Comonomer Comparison in Small Band Gap Diketopyrrolopyrrole-Based Conjugated Polymers for High-Performing Field-Effect Transistors and Organic Solar Cells. J. Am. Chem. Soc. 2015, 137, 1314–1321. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhang, Q.; Wan, X.; Li, M.; Zhang, H.; Li, C.; Chen, Y. A Small Molecule with Selenophene as the Central Block for High Performance Solution-processed Organic Solar Cells. Org. Electron. 2015, 19, 98–104. [Google Scholar] [CrossRef]
- Meng, D.; Sun, D.; Zhong, C.; Liu, T.; Fan, B.; Huo, L.; Li, Y.; Jiang, W.; Choi, H.; Kim, T.; et al. High-Performance Solution-Processed Non-Fullerene Organic Solar Cells Based on Selenophene-Containing Perylene Bisimide Acceptor. J. Am. Chem. Soc. 2016, 138, 375–380. [Google Scholar] [CrossRef]
- Cao, F.-Y.; Tseng, C.-C.; Lin, F.-Y.; Chen, Y.; Yan, H.; Cheng, Y.-J. Selenophene-Incorporated Quaterchalcogenophene-Based Donor-Acceptor Copolymers to Achieve Efficient Solar Cells with Jsc Exceeding 20 mA/cm2. Chem. Mater. 2017, 29, 10045–10052. [Google Scholar] [CrossRef]
- Yin, Y.; Song, J.; Guo, F.; Sun, Y.; Zhao, L.; Zhang, Y. Asymmetrical vs. Symmetrical Selenophene-Annulated Fused Perylenediimide Acceptors for Efficient Non-Fullerene Polymer Solar Cells. ACS Appl. Energy Mater. 2018, 1, 6577–6585. [Google Scholar] [CrossRef]
- Li, C.; Xia, T.; Song, J.; Fu, H.; Ryu, H.S.; Weng, K.; Ye, L.; Woo, H.Y.; Sun, Y. Asymmetric Selenophene-based Non-fullerene Acceptors for High-performance Organic Solar Cells. J. Mater. Chem. A 2019, 7, 1435–1441. [Google Scholar] [CrossRef]
- Liao, X.; Shi, X.; Zhang, M.; Gao, K.; Zuo, L.; Liu, F.; Chen, Y.; Jen, A.K.-Y. Fused Selenophene-thieno[3,2-b]thiophene-selenophene (ST)-Based Narrow-bandgap Electron Acceptor for Efficient Organic Solar Cells with Small Voltage Loss. Chem. Commun. 2019, 55, 8258–8261. [Google Scholar] [CrossRef] [PubMed]
- Ramki, K.; Venkatesh, N.; Sathiyan, G.; Thangamuthu, R.; Sakthivel, P. A Comprehensive Review on the Reasons Behind Low Power Conversion Efficiency of Dibenzo Derivatives Based Donors in Bulk Heterojunction Organic Solar Cells. Org. Electron. 2019, 73, 182–204. [Google Scholar] [CrossRef]
- Wang, H.; Nakagawa, T.; Zhang, M.-M.; Ogumi, K.; Yang, S.; Matsuo, Y. High-yielding Pd2(dba)3·C6H6-based Four-fold Sonogashira Coupling with Selenophene-conjugated Magnesium Tetraethynylporphyrin for Organic Solar Cells. RSC Adv. 2019, 9, 32562–32572. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, V.; Vartanian, M.; Malhotra, P.; Biswas, S.; de la Cruz, P.; Sharma, G.D.; Langa, F. Increase in Efficiency on Using Selenophene Instead of Thiophene in π-Bridges for D-π-DPP-π-D Organic Solar Cells. J. Mater. Chem. A 2019, 7, 11886–11894. [Google Scholar] [CrossRef]
- Lee, J.-W.; Sung, M.J.; Kim, D.; Lee, S.; You, H.; Kim, F.S.; Kim, Y.-H.; Kim, B.J.; Kwon, S.-K. Naphthalene Diimide-Based Terpolymers with Controlled Crystalline Properties for Producing High Electron Mobility and Optimal Blend Morphology in All-Polymer Solar Cells. Chem. Mater. 2020, 32, 2572–2582. [Google Scholar] [CrossRef]
- Pan, J.; Wang, L.; Chen, W.; Sang, S.; Sun, H.; Wu, B.; Hang, X.-C.; Sun, Z.; Huang, W. Non-fullerene Small Molecule Acceptors with Three-dimensional Thiophene/selenophene-annulated Perylene Diimides for Efficient Organic Solar Cells. J. Mater. Chem. C 2020, 8, 6749–6755. [Google Scholar] [CrossRef]
- Kong, H.; Chung, D.S.; Kang, I.-N.; Park, J.-H.; Park, M.-J.; Jung, I.H.; Park, C.E.; Shim, H.-K. New Selenophene-based Semiconducting Copolymers for High Performance Organic Thin-Film Transistors. J. Mater. Chem. 2009, 19, 3490–3499. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.; Jung, Y.K.; Cho, N.S.; Kang, I.-N.; Park, J.-H.; Cho, S.; Shim, H.-K. New Semiconducting Polymers Containing 3,6-Dimethyl(thieno[3,2-b]-thiophene or Selenopheno[3,2-b]selenophene) for Organic Thin-Film Transistors. Chem. Mater. 2009, 21, 2650–2660. [Google Scholar] [CrossRef]
- Cho, M.J.; Shin, J.; Yoon, S.H.; Lee, T.W.; Kaur, M.; Choi, D.H. A High-mobility Terselenophene and Diketopyrrolopyrrole Containing Copolymer in Solution-processed Thin Film Transistors. Chem. Commun. 2013, 49, 7132–7134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, G.E.; Choi, S.; Lee, D.H.; Um, H.A.; Shin, J.; Cho, M.J.; Choi, D.H. Effect of the Thiophene and Selenophene Moiety in Regular Terpolymers on the Performance of Thin Film Transistors and Polymer Solar Cells. Polymer 2016, 94, 43–52. [Google Scholar] [CrossRef]
- Liu, L.; Ren, Z.; Xiao, C.; Dong, H.; Yan, S.; Hu, W.; Wang, Z. Surface-induced Highly Oriented Perylo[1,12-b,c,d]selenophene Thin Films for High Performance Organic Field-effect Transistors. Org. Electron. 2016, 35, 186–192. [Google Scholar] [CrossRef]
- Fei, Z.; Han, Y.; Gann, E.; Hodsden, T.; Chesman, A.S.R.; McNeill, C.R.; Anthopoulos, T.D.; Heeney, M. Alkylated Selenophene-Based Ladder-Type Monomers via a Facile Route for High-Performance Thin-Film Transistor Applications. J. Am. Chem. Soc. 2017, 139, 8552–8561. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Tang, L.; Guo, H.; Uddin, M.A.; Wang, H.; Yang, K.; Liu, B.; Wang, Y.; Sun, H.; Woo, H.Y.; et al. Bichalcogenophene Imide-Based Homopolymers: Chalcogen-Atom Effects on the Optoelectronic Property and Device Performance in Organic Thin-Film Transistors. Macromolecules 2019, 52, 7301–7312. [Google Scholar] [CrossRef]
- Park, K.; Shin, E.-Y.; Jiao, X.; McNeill, C.R.; Kim, Y.-H.; Kwon, S.-K.; Noh, Y.-Y. Effect of Backbone Sequence of a Naphthalene Diimide-Based Copolymer on Performance in n-Type Organic Thin-Film Transistors. ACS Appl. Mater. Interfaces 2019, 11, 35185–35192. [Google Scholar] [CrossRef]
- Jones, R.L.; Elder, M.J.; Ewen, J.A. Metallocenes with Cp Ring-Fused to Chalcogen Heterocycles: Synthesis and Polymerization Results. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 827–843. [Google Scholar] [CrossRef]
- Buccella, D.; Janak, K.E.; Parkin, G. Reactivity of Mo(PMe3)6 towards Benzothiophene and Selenophenes: New Pathways Relevant to Hydrodesulfurization. J. Am. Chem. Soc. 2008, 130, 16187–16189. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Renili, F.; Zacchini, S.; Zanotti, V. Addition of Alkynes to Zwitterionic μ-Vinyliminium Diiron Complexes: New Selenophene (Thiophene) and Vinyl Chalcogenide Functionalized Bridging Ligands. Organometallics 2010, 29, 1797–1805. [Google Scholar] [CrossRef]
- Ahmad, S.; Yadav, K.K.; Bhattacharya, S.; Chauhan, P.; Chauhan, S.M.S. Synthesis of 21,23-Selenium- and Tellurium-Substituted 5-Porphomethenes, 5,10-Porphodimethenes, 5,15-Porphodimethenes, and Porphotrimethenes and Their Interactions with Mercury. J. Org. Chem. 2015, 80, 3880–3890. [Google Scholar] [CrossRef]
- Hua, C.; D’Alessandro, D.M. Systematic Tuning of Zn(II) Frameworks with Furan, Thiophene, and Selenophene Dipyridyl and Dicarboxylate Ligands. Cryst. Growth Des. 2017, 17, 6262–6272. [Google Scholar] [CrossRef]
- Barros, O.S.R.; Silva, F.R.; Nunes, V.L. Copper(I) Selenophene-2-carboxylate (CuSC) Promoted C-S Cross-coupling Reaction of Thiols with Aryl Iodides. J. Sulfur. Chem. 2019, 40, 9–17. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sawada, J.; Murahashi, T. Thiophene and Selenophene Binding at a Pd3 Cluster Site. Chem. Eur. J. 2020, 26, 8388–8392. [Google Scholar] [CrossRef] [PubMed]
- Barros, O.S.R.; Favero, A.; Nogueira, C.W.; Menezes, P.H.; Zeni, G. Palladium-catalyzed Cross-coupling of 2-Haloselenophene with Terminal Alkynes in the Absence of Additive. Tetrahedron Lett. 2006, 47, 2179–2182. [Google Scholar] [CrossRef]
- Prediger, P.; Moro, A.V.; Nogueira, C.W.; Savegnago, L.; Rocha, J.B.T.; Zeni, G. Palladium-Catalyzed Suzuki Cross-Coupling of 2-Haloselenophenes: Synthesis of 2-Arylselenophenes, 2,5-Diarylselenophenes, and 2-Arylselenophenyl Ketones. J. Org. Chem. 2006, 71, 3786–3792. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.; Luchese, C.; Nogueira, C.W.; Zeni, G. Electrophilic Cyclization of (Z)-Selenoenynes: Synthesis and Reactivity of 3-Iodoselenophenes. J. Org. Chem. 2007, 72, 6726–6734. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, R.F.; Alves, D.; Brandão, R.; Nogueira, C.W.; Zeni, G. 3-Iodoselenophene Derivatives: A Versatile Substrate for Negishi Cross-coupling Reaction. Tetrahedron Lett. 2008, 49, 538–542. [Google Scholar] [CrossRef]
- Alves, D.; Reis, J.S.; Luchese, C.; Nogueira, C.W.; Zeni, G. Synthesis of 3-Alkynylselenophene Derivatives by a Copper-Free Sonogashira Cross-Coupling Reaction. Eur. J. Org. Chem. 2008, 2008, 377–382. [Google Scholar] [CrossRef]
- Tùng, D.T.; Villinger, A.; Langer, P. Efficient Synthesis of Substituted Selenophenes Based on the First Palladium(0)-Catalyzed Cross-Coupling Reactions of Tetrabromoselenophene. Adv. Synth. Catal. 2008, 350, 2109–2117. [Google Scholar] [CrossRef]
- Ehlers, P.; Dang, T.T.; Patonay, T.; Villinger, A.; Langer, P. Synthesis of Alkynylated Selenophenes by Site-Selective Sonogashira Reactions of Tetrabromoselenophene. Eur. J. Org. Chem. 2013, 10, 2000–2007. [Google Scholar] [CrossRef]
- Skhiri, A.; Salem, R.B.; Soulé, J.-F.; Doucet, H. Reactivity of Bromoselenophenes in Palladium-catalyzed Direct Arylations. Beilstein J. Org. Chem. 2017, 13, 2862–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skhiri, A.; Salem, R.B.; Soul, J.-F.; Doucet, H. Access to (Hetero)arylated Selenophenes via Palladium catalysed Stille, Negishi or Suzuki Couplings or C-H Bond Functionalization Reaction. ChemCatChem 2017, 9, 2895–2913. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-Y.; Pao, Y.-C.; Sahoo, S.K.; Huang, W.-C.; Lai, Y.-Y.; Cheng, Y.-J. Synthesis of Unsymmetrical Benzotrichalcogenophenes by N-Heterocyclic Carbene-palladium-catalyzed Intramolecular Direct C3-Arylation of Chalcogenophenes. Chem. Commun. 2018, 54, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Barros, O.S.R.; Nogueira, C.W.; Stangherlin, E.C.; Menezes, P.H.; Zeni, G. Copper-Promoted Carbon-Nitrogen Bond Formation with 2-Iodo-selenophene and Amides. J. Org. Chem. 2006, 71, 1552–1557. [Google Scholar] [CrossRef]
- Prediger, P.; Brandão, R.; Nogueira, C.W.; Zeni, G. Palladium-Catalyzed Carbonylation of 2-Haloselenophenes: Synthesis of Selenophene-2-carboxamides, Selenophene-2,5-dicarboxamides and N,N-Bridged Selenophene-2-carboxamides. Eur. J. Org. Chem. 2007, 32, 5422–5428. [Google Scholar] [CrossRef]
- Wang, P.-Y.; Li, L.-C.; Yang, C.; Li, Y.-R. Theoretical Investigation on the Mechanism of the CuI-Catalyzed Carbon-Nitrogen Coupling Reaction of 2-Iodo-selenophene with Benzamide. J. Phys. Chem. A 2008, 112, 435–440. [Google Scholar] [CrossRef]
- Zeni, G. Carbon-sulfur Bond Formation from 2-Halochalcogenophenes via Copper Catalyzed Thiol Cross-coupling. Tetrahedron Lett. 2005, 46, 2647–2651. [Google Scholar] [CrossRef]
- Tamba, S.; Fujii, R.; Mori, A.; Hara, K.; Koumura, N. Synthesis and Properties of Seleno-analog MK-organic Dye for Photovoltaic Cells Prepared by CH Functionalization Reactions of Selenophene Derivatives. Chem. Lett. 2011, 40, 922–924. [Google Scholar] [CrossRef] [Green Version]
- Rampon, D.S.; Wessjohann, L.A.; Schneider, P.H. Palladium-Catalyzed Direct Arylation of Selenophene. J. Org. Chem. 2014, 79, 5987–5992. [Google Scholar] [CrossRef] [Green Version]
- Skhiri, A.; Salem, R.B.; Soule, J.-F.; Doucet, H. Unprecedented Access to ß-Arylated Selenophenes via Palladium-Catalysed Direct Arylation. Chem. Eur. J. 2017, 23, 2788–2791. [Google Scholar] [CrossRef]
- Shi, X.; Mao, S.; Roisnel, T.; Doucet, H.; Soule, J.-F. Palladium-catalyzed Successive C–H bond Arylations and Annulations toward the π-Extension of Selenophene-containing Aromatic Skeletons. Org. Chem. Front. 2019, 6, 2398–2403. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Sahoo, S.K.; Huang, C.-L.; Chan, T.-H.; Cheng, Y.-J. Pd(II)-Catalyzed Direct Dehydrogenative Mono- and Diolefination of Selenophenes. Org. Lett. 2020, 22, 2318–2322. [Google Scholar] [CrossRef] [PubMed]
- Sommen, G.L. Synthesis of Selenophenes. Mini-Rev. Org. Chem. 2005, 2, 375–388. [Google Scholar] [CrossRef]
- Młochowski, J.; Giurg, M. New trends in chemistry and application of aromatic and related selenaheterocycles. In Aromaticity in Heterocyclic Compounds; Krygowski, T.M., Cyrański, M.K., Eds.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2009; Volume 19, pp. 287–340. [Google Scholar]
- Rhoden, C.R.B.; Zeni, G. New Development of Synthesis and Reactivity of Seleno- and Tellurophenes. Org. Biomol. Chem. 2011, 9, 1301–1313. [Google Scholar] [CrossRef]
- Vasiljeva, E.; Arsenyan, P. Synthesis of Selenophenes Condensed with Six-Membered Nitrogen Heterocycles. Chem. Heterocycl. Comp. 2012, 48, 981–992. [Google Scholar] [CrossRef]
- Biehl, E.R. Five-membered ring systems: Thiophene and Se/Te derivatives. In Progress in Heterocycle Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 27, pp. 117–157. [Google Scholar]
- Wu, B.; Yoshikai, N. Recent Developments in Synthetic Methods for Benzo[b]heteroles. Org. Biomol. Chem. 2016, 14, 5402–5416. [Google Scholar] [CrossRef] [Green Version]
- Diakov, M.Y.; Prodanov, M.F.; Vashchenko, V.V. Recent Progress in Selenophenes Synthesis from Inorganic Se-Precursors. Curr. Org. Chem. 2017, 14, 683–690. [Google Scholar] [CrossRef]
- Ranu, B.; Ghosh, T.; Adak, L.; Panja, S. Synthesis and reactivity of selenophene and their benzo- and other carbocyclic-fused derivatives. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Paal, C. Ueber die Einwirkung von Phosphorpentaselenid auf des Acetonylaceton. Ber. Dtsch. Chem. Ges. 1885, 18, 2255–2256. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, H.V.A.; Peel, J.B. The Preparation and Properties of Selenophen and Certain Halogen Derivatives of Selenophen. J. Chem. Soc. 1928, 1741–1747. [Google Scholar] [CrossRef]
- McMahon, F.A.; Pearson, T.G.; Robinson, P.L. An Improved Preparation of Selenophen and Attempts to prepare Tellurophen. J. Chem. Soc. 1933, 1643–1644. [Google Scholar] [CrossRef]
- Korchevin, N.A.; Ostroukhova, L.A.; Sukhomazova, E.N.; Zhnikin, A.R.; Deryagina, E.N.; Voronkov, M.G. Gas phase synthesis of selenophene from dimethyl selenides and acetylene. Chem. Heterocycl. Compds. 1987, 23, 237. [Google Scholar] [CrossRef]
- Tsuboi, S.; Watanabe, K.; Mimura, S.; Takeda, A. A novel synthesis of 2,5-disubstituted furans and selenophenes via the oxidation of 2,4-alkadienoic esters with SeO2. Tetrahedron Lett. 1986, 27, 2643–2644. [Google Scholar] [CrossRef]
- Maity, P.; Kundu, D.; Roy, R.; Ranu, B.C. A Direct Synthesis of Selenophenes by Cu-Catalyzed One-Pot Addition of a Selenium Moiety to (E,E)-1,3-Dienyl Bromides and Subsequent Nucleophilic Cyclization. Org. Lett. 2014, 16, 4122–4125. [Google Scholar] [CrossRef] [PubMed]
- Roehrs, J.A.; Pistoia, R.P.; Back, D.F.; Zeni, G. Diorganyl Dichalcogenides-Promoted Nucleophilic Closure of 1,4-Diyn-3-ols: Synthesis of 2-Benzoyl Chalcogenophenes. J. Org. Chem. 2015, 80, 12470–12481. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Bilheri, F.N.; Stein, A.L.; Zeni, G. Synthesis of Chalcogenophenes via Cyclization of 1,3-Diynes Promoted by Iron(III) Chloride and Dialkyl Dichalcogenides. Adv. Synth. Catal. 2015, 357, 1221–1228. [Google Scholar] [CrossRef]
- Maity, P.; Ranu, B.C. Iodine-Catalyzed Synthesis of Chalcogenophenes by the Reaction of 1,3-Dienyl Bromides and Potassium Selenocyanate/Potassium Sulfide (KSeCN/K2S). Adv. Synth. Catal. 2017, 359, 4369–4378. [Google Scholar] [CrossRef]
- Jin, S.; Kuang, Z.; Song, Q. Precise Construction of SCF2H or SeCF2H Groups on Heteroarenes Generated in Situ from CF3-Containing 1,3-Enynes. Org. Lett. 2020, 22, 615–619. [Google Scholar] [CrossRef]
- Barancelli, D.A.; Schumacher, R.F.; Leite, M.R.; Zeni, G. Copper(II)-Mediated Intramolecular Cyclization of (Z)-Chalcogenoenynes: Synthesis of 3-Halochalcogenophene Derivatives. Eur. J. Org. Chem. 2011, 2011, 6713–6718. [Google Scholar] [CrossRef]
- Barancelli, D.A.; Acker, C.I.; Menezes, P.H.; Zeni, G. Selective Base-promoted Synthesis of Substituted Selenophenes by Carbocyclization of (Z)-Benzylselenoenynes. Org. Biomol. Chem. 2011, 9, 1529–1537. [Google Scholar] [CrossRef]
- Schumacher, R.F.; Rosário, A.R.; Leite, M.R.; Zeni, G. Cyclization of Homopropargyl Chalcogenides by Copper(II) Salts: Selective Synthesis of 2,3-Dihydroselenophenes, 3-Arylselenophenes, and 3-Haloselenophenes/thiophenes. Chem. Eur. J. 2013, 19, 13059–13064. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, T.; Giraudy, K.A.; Morgan, J.L.; Kornman, C.; Olaitan, A.D. Green Synthesis of Halogenated Thiophenes, Selenophenes and Benzo[b]selenophenes using Sodium Halides as a Source of Electrophilic Halogens. Tetrahedron Lett. 2017, 58, 638–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoi, B.; Schumacher, R.F.; Zeni, G. Synthesis of Heterocycles via Electrophilic Cyclization of Alkynes Containing Heteroatom. Chem. Rev. 2011, 111, 2937–2980. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, R.P.; Roehrs, J.A.; Back, D.F.; Zeni, G. Iodine-mediated Regioselective 5-endo-dig Electrophilic Cyclization Reaction of Selenoenynes: Synthesis of Selenophene Derivatives. Org. Chem. Front. 2017, 4, 277–282. [Google Scholar] [CrossRef]
- Casola, K.K.; Gomes, M.R.; Back, D.F.; Zeni, G. Electrophilic Cyclization Involving Carbon-Selenium/Carbon-Halide Bond Formation: Synthesis of 3-Substituted Selenophenes. J. Org. Chem. 2018, 83, 6706–6718. [Google Scholar] [CrossRef]
- Soares, L.K.; Barcellos, A.M.; Neto, J.S.S.; Alves, D.; Lenardão, E.J.; Rosati, O.; Santi, C.; Perin, G. Dichalcogenides/Oxone®-Mediated Cyclization of (Z)-Chalcogenoenynes under Ultrasound Irradiation. ChemistrySelect 2020, 5, 9813–9819. [Google Scholar] [CrossRef]
- Kesharwani, T.; Worlikar, S.A.; Larock, R.C. Synthesis of 2,3-Disubstituted Benzo[b]selenophenes via Electrophilic Cyclization. J. Org. Chem. 2006, 71, 2307–2312. [Google Scholar] [CrossRef]
- Mehta, S.; Waldo, J.P.; Larock, R.C. Competition Studies in Alkyne Electrophilic Cyclization Reactions. J. Org. Chem. 2009, 74, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Nakamura, I.; Terada, M. Platinum-Catalyzed Multisubstituted Benzo[b]selenophene Synthesis. Eur. J. Org. Chem. 2009, 32, 5509–5512. [Google Scholar] [CrossRef]
- Krzyzanowski, A.; Saleeb, M.; Elofsson, M. Synthesis of Indole-, Benzo[b]thiophene-, and Benzo[b]selenophene-Based Analogues of the Resveratrol Dimers Viniferifuran and (±)-Dehydroampelopsin B. Org. Lett. 2018, 20, 6650–6654. [Google Scholar] [CrossRef]
- Pistoia, R.P.; Roehrs, J.A.; Back, D.F.; Zeni, G. Synthesis of 2-Acylselenophenes via Iodine-Promoted Nucleophilic Cyclization of [2-(Butylselanyl)phenyl]-propynols. Adv. Synth. Catal. 2015, 357, 3655–3665. [Google Scholar] [CrossRef]
- Perin, G.; Roehrs, J.A.; Hellwig, P.S.; Stach, G.; Barcellos, T.; Lenardão, E.J.; Jacob, R.G.; Luz, E.Q. Synthesis of 2-Organylchalcogenyl-benzo[b]selenophenes: 1-(2,2-Dibromovinyl)-2-butylselenanylbenzenes as Precursors to Access Alkynes Susceptible to Cyclization. ChemistrySelect 2017, 2, 4561–4566. [Google Scholar] [CrossRef]
- Bilheri, F.N.; Pistoia, R.P.; Back, D.F.; Zeni, G. Copper/Palladium-Catalyzed Cyclization/Cross-Coupling Cascade Reaction of 2-gem-Dibromovinyl Aryl Selenides: Synthesis of 2-Substituted Benzo[b]selenophenes. Adv. Synth. Catal. 2017, 359, 4208–4216. [Google Scholar] [CrossRef]
- Stach, G.; Peglow, T.J.; Roehrs, J.A.; Penteado, F.; Barcellos, T.; Jacob, R.G.; Lenardão, E.J.; Perin, G. Synthesis of 2-(Arylselanyl)benzo[b]chalcogenophenes via Intramolecular Cyclization of Vinyl Selenides. Synthesis 2019, 51, 578–586. [Google Scholar]
- Perin, G.; Soares, L.K.; Hellwig, P.S.; Silva, M.S.; Neto, J.S.S.; Roehrs, J.A.; Barcellos, T.; Lenardão, E.J. Synthesis of 2,3-Bis-organochalcogenylbenzo[b]chalcogenophenes Promoted by Oxone®. New J. Chem. 2019, 43, 6323–6331. [Google Scholar] [CrossRef]
- Xu, J.; Yu, X.; Yan, J.; Song, Q. Synthesis of 3-(Arylsulfonyl)benzothiophenes and Benzoselenophenes via TBHP-Initiated Radical Cyclization of 2-Alkynylthioanisoles or -selenoanisoles with Sulfinic Acids. Org. Lett. 2017, 19, 6292–6295. [Google Scholar] [CrossRef]
- Yan, J.; Xu, J.; Zhou, Y.; Chen, J.; Song, Q. Photoredox-catalyzed Cascade Annulation of Methyl(2-(phenylethynyl)phenyl)sulfanes and Methyl(2-(phenylethynyl)phenyl)selanes with Sulfonyl Chlorides: Synthesis of Benzothiophenes and Benzoselenophenes. Org. Chem. Front. 2018, 5, 1483–1487. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Busto, E.; Herrera, F.; Lázaro-Milla, C.; Luna, A. Photopromoted Entry to Benzothiophenes, Benzoselenophenes, 3H-Indoles, Isocoumarins, Benzosultams, and (Thio)flavones by Gold-Catalyzed Arylative Heterocyclization of Alkynes. Adv. Synth. Catal. 2017, 359, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Liu, J.; Zhang, H.; Wang, X.; Feng, J.; Shen, R.; Gao, Y. Ag-Mediated Radical Cyclization of 2-Alkynylthio(seleno)anisoles: Direct Synthesis of 3-Phosphinoylbenzothio(seleno)phenes. Org. Lett. 2019, 21, 4605–4608. [Google Scholar] [CrossRef]
- Lu, S.-C.; Wu, B.; Zhang, S.-P.; Gong, Y.-L.; Xu, S. K2S2O8-Mediated Radical Cyclisation of 2-Alkynylthioanisoles or -selenoanisoles: A Green and Regioselective Route to 3-Nitrobenzothiophenes and Benzoselenophenes. RSC Adv. 2020, 10, 19083–19087. [Google Scholar] [CrossRef]
- Zang, H.; Sun, J.-G.; Dong, X.; Li, P.; Zhang, B. Preparation of Benzothiophenes and Benzoselenophenes from Arylamines and Alkynes via Radical Cascade Reactions. Adv. Synth. Catal. 2016, 358, 1746–1752. [Google Scholar] [CrossRef]
- Klayman, D.L.; Griffin, T.S. Reaction of Selenium with Sodium Borohydride in Protic Solvents. A Facile Method for the Introduction of Selenium into Organic Molecules. J. Am. Chem. Soc. 1973, 95, 197–199. [Google Scholar] [CrossRef]
- Hoenig, S.M.; Yan, Y.; Dougherty, E.A.; Hudson, R.; Petovic, S.; Lee, C.K.; Hu, Y.; Gomez, L.S.; Katz, J.L. Convergent Strategy for the Synthesis of Oxa-, Thia-, and Selena[5]helicenes by Acetylene-Activated SNAr Reactions. J. Org. Chem. 2020, 85, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Kaname, M.; Sashida, H. Tandem Addition-Cyclization of o-Ethynylphenyllithiums and Isoselenocyanates: A Convenient Preparation of Functionalized Benzo[c]selenophenes. Tetrahedron Lett. 2011, 52, 3279–3282. [Google Scholar] [CrossRef]
- Lyapunova, A.G.; Androsov, D.A.; Petrov, M.L. A Convenient Synthesis of Benzo[b]chalcogenophenes from 4-(2-Chloro-5-nitrophenyl)-1,2,3-chalcogenadiazoles. Tetrahedron Lett. 2013, 54, 3427–3430. [Google Scholar] [CrossRef]
- Lyapunova, A.G.; Petrov, M.L.; Androsov, D.A. A Novel Synthesis of Benzo[b]selenophenes via Regioselective Intramolecular Transformation of 4-(3-Nitroaryl)-1,2,3-selenadiazoles. Org. Lett. 2013, 15, 1744–1747. [Google Scholar] [CrossRef]
- Paegle, E.; Belyakov, S.; Arsenyan, P. An Approach to the Selenobromination of Aryl(thienyl)alkynes: Access to 3-Bromobenzo[b]selenophenes and Selenophenothiophenes. Eur. J. Org. Chem. 2014, 2014, 3831–3840. [Google Scholar] [CrossRef]
- Paegle, E.; Belyakov, S.; Petrova, M.; Liepinsh, E.; Arsenyan, P. Cyclization of Diaryl(hetaryl)alkynes under Selenobromination Conditions: Regioselectivity and Mechanistic Studies. Eur. J. Org. Chem. 2015, 2015, 4389–4399. [Google Scholar] [CrossRef]
- Wu, B.; Yoshikai, N. Versatile Synthesis of Benzothiophenes and Benzoselenophenes by Rapid Assembly of Arylzinc Reagents, Alkynes, and Elemental Chalcogens. Angew. Chem. Int. Ed. 2013, 52, 10496–10499. [Google Scholar] [CrossRef]
- Matsumura, M.; Sakata, Y.; Iwase, A.; Kawahata, M.; Kitamura, Y.; Murata, Y.; Kakusawa, N.; Yamaguchi, K.; Yasuike, S. Copper-catalyzed Tandem Cyclization of 2-(2-Iodophenyl)imidazo[1,2-a]pyridine Derivatives with Selenium: Synthesis of Benzo[b]selenophene Fused Imidazo[1,2-a]pyridines. Tetrahedron Lett. 2016, 57, 5484–5488. [Google Scholar] [CrossRef]
- Sun, P.; Jiang, M.; Wei, W.; Min, Y.; Zhang, W.; Li, W.; Yang, D.; Wang, H. Copper-Catalyzed Selenylation of Imidazo[1,2-a]pyridines with Selenium Powder via a Radical Pathway. J. Org. Chem. 2017, 82, 2906–2913. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, J.; Wang, J.; Xu, S.; Lin, A.; Yao, H.; Jiang, S.; Xu, J. Cobalt-catalyzed Carbon-Sulfur/Selenium Bond Formation: Synthesis of Benzo[b]thio/selenophene-fused Imidazo[1,2-a]pyridines. Org. Biomol. Chem. 2018, 16, 3721–3725. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wu, G.; Yang, H.; Liu, M.; Gao, W.; Huang, X.; Chen, J.; Wu, H. Copper-Catalyzed Three-Component Reaction for Regioselective Aryl- and Heteroarylselenation of Indoles using Selenium Powder. J. Org. Chem. 2016, 81, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Tanini, D.; Viglianisi, C.; Panzella, L.; Capperucci, A.; Menichetti, S.; Supuran, C.T. Evaluation of Selenide, Diselenide and Selenoheterocycle Derivatives as Carbonic Anhydrase I, II, IV, VII and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 2518–2523. [Google Scholar] [CrossRef]
- Wang, M.; Fan, Q.; Jiang, X. Transition-Metal-Free Diarylannulated Sulfide and Selenide Construction via Radical/Anion-Mediated Sulfur-Iodine and Selenium-Iodine Exchange. Org. Lett. 2016, 18, 5756–5759. [Google Scholar] [CrossRef]
- Maity, P.; Paroi, B.; Ranu, B.C. Transition-Metal-Free Iodine Catalyzed Selenocayanation of Styrenyl Bromides and an Easy Access to Benzoselenophenes via Intermediacy of Styrenyl Selenocyanate. Org. Lett. 2017, 19, 5748–5751. [Google Scholar] [CrossRef]
- Hu, Y.; Ma, J.; Li, L.; Hu, Q.; Lv, S.; Liu, B.; Wang, S. Fused Multifunctionalized Dibenzoselenophenes from Tetraynes. Chem. Commun. 2017, 53, 1542–1545. [Google Scholar] [CrossRef]
- Ni, P.; Tan, J.; Zhao, W.; Huang, H.; Xiao, F.; Deng, G.-J. A Three-Component Strategy for Benzoselenophene Synthesis under Metal-Free Conditions Using Selenium Powder. Org. Lett. 2019, 21, 3518–3522. [Google Scholar] [CrossRef]
- Sperança, A.; Godoi, B.; Costa, M.D.; Menezes, P.H.; Zeni, G. Electrophilic Cyclization of 3-Alkynyl-4-chalcogen-2-H-chromenes: Synthesis of 3-Halo-chalcogenophene[3,2-c]chromene Derivatives. Tetrahedron Lett. 2011, 52, 388–391. [Google Scholar] [CrossRef] [Green Version]
- Stein, A.L.; Bilheri, F.N.; Rosário, A.R.; Zeni, G. FeCl3-Diorganyl Dichalcogenides Promoted Cyclization of 2-Organochalcogen-3-alkynylthiophenes: Synthesis of Chalcogenophene[2,3-b]thiophenes. Org. Biomol. Chem. 2013, 11, 2972–2978. [Google Scholar] [CrossRef]
- Racharlawar, S.S.; Shankar, D.; Karkhelikar, M.V.; Sridhar, B.; Likhar, P.R. Intramolecular Heterocyclization and Cyclopalladation of Selenoanisole Substituted Propargyl Imines: Synthesis and Reactivity of P-C Bond Towards Alkynes. J. Organomet. Chem. 2014, 757, 14–20. [Google Scholar] [CrossRef]
- Sonawane, A.D.; Garud, D.R.; Udagawa, T.; Koketsu, M. Synthesis of thieno[2,3-b]quinoline and Selenopheno[2,3-b]quinoline Derivatives via Iodocyclization Reaction and a DFT Mechanistic Study. Org. Biomol. Chem. 2018, 16, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Leonel, G.; Back, D.F.; Zeni, G. Synthesis of 3-Substituted Chalcogenophene-Fused Indoles from 2-Alkynylindoles. Adv. Synth. Catal. 2020, 362, 585–593. [Google Scholar] [CrossRef]
- Peglow, T.J.; Bartz, R.H.; Martins, C.C.; Belladona, A.L.; Luchese, C.; Wilhelm, E.A.; Schumacher, R.F.; Perin, G. Synthesis of 2-Organylchalcogenopheno[2,3-b]pyridines from Elemental Chalcogen and NaBH4/PEG-400 as Reducing System: Antioxidant and Antinociceptive Properties. ChemMedChem 2020, 15, 1741–1751. [Google Scholar] [CrossRef]
- Perin, G.; Peglow, T.J.; Bartz, R.H.; Cargnelutti, R.; Lenardão, E.J.; Schumacher, R.F. Synthesis of 2-Organylselenopheno[2,3-b]pyridines and 1,6-Diazaselenanthrenes via Radical Cascade Reactions using tert-Butyl Nitrite. Arkivoc 2019, 2019, 50–64. [Google Scholar] [CrossRef]
- Arsenyan, P.; Vasiljeva, J.; Shestakova, I.; Domracheva, I.; Belyakov, S. The synthesis and Cytotoxic Properties of Selenopheno[3,2-c]- and Selenopheno[2,3-c]quinolones. Chem. Heterocycl. Comp. 2014, 49, 1674–1680. [Google Scholar] [CrossRef]
- Martins, G.M.; Back, D.F.; Kaufman, T.S.; Silveira, C.C. SeCl2-Mediated Approach toward Indole-Containing Polysubstituted Selenophenes. J. Org. Chem. 2018, 83, 3252–3264. [Google Scholar] [CrossRef]
- Neto, J.S.S.; Iglesias, B.A.; Back, D.F.; Zeni, G. Iron-Promoted Tandem Cyclization of 1,3-Diynyl Chalcogen Derivatives with Diorganyl Dichalcogenides for the Synthesis of Benzo[b]furan-Fused Selenophenes. Adv. Synth. Catal. 2016, 358, 3572–3585. [Google Scholar] [CrossRef]
- Pinheiro, R.C.; Back, D.F.; Zeni, G. Iron(III) Chloride/Dialkyl Diselenides-Promoted Cascade Cyclization of ortho-Diynyl Benzyl Chalcogenides. Adv. Synth. Catal. 2019, 361, 1866–1873. [Google Scholar] [CrossRef]
- Lutz, G.; Back, D.F.; Zeni, G. Iron-Mediated Cyclization of 1,3-Diynyl Propargyl Aryl Ethers with Dibutyl Diselenide: Synthesis of Selenophene-Fused Chromenes. Adv. Synth. Catal. 2020, 362, 1096–1105. [Google Scholar] [CrossRef]
- Goulart, H.A.; Neto, J.S.S.; Barcellos, A.M.; Barcellos, T.; Silva, M.S.; Alves, D.; Jacob, R.; Lenardão, E.J.; Perin, G. Synthesis of 5H-Selenopheno[3,2-c]isochromen-5-ones Promoted by Dialkyl Diselenides and Oxone®. Adv. Synth. Catal. 2019, 361, 3403–3411. [Google Scholar] [CrossRef]
- Sonawane, A.D.; Kubota, Y.; Koketsu, M. Iron-Promoted Intramolecular Cascade Cyclization for the Synthesis of Selenophene-Fused, Quinoline-Based Heteroacenes. J. Org. Chem. 2019, 84, 8602–8614. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.D.; Shimozuma, A.; Udagawa, T.; Ninomiya, M.; Koketsu, M. Synthesis and Photophysical Properties of Selenopheno[2,3-b]quinoxaline and Selenopheno[2,3-b]pyrazine heteroacenes. Org. Biomol. Chem. 2020, 18, 4063–4070. [Google Scholar] [CrossRef] [PubMed]
- Dillon, A.S.; Flynn, B.L. Polyynes to Polycycles: Domino Reactions Forming Polyfused Chalcogenophenes. Org. Lett. 2020, 22, 2987–2990. [Google Scholar] [CrossRef]
Figure 1.
Selected examples of biologically active and technologically interesting selenophenes.

Scheme 1.
Two common protocols to the synthesis of selenophenes.

Scheme 2.
Common protocols used to prepare benzoselenophenes.

Scheme 3.
Scope of Cu-catalyzed one-pot synthesis of selenophenes 2 via nucleophilic cyclization.

Scheme 4.
Scope of Cu-catalyzed one-pot synthesis of selanyl selenophenes 4 via nucleophilic cyclization.
Scheme 4.
Scope of Cu-catalyzed one-pot synthesis of selanyl selenophenes 4 via nucleophilic cyclization.

Scheme 5.
Synthesis of selenophenes via cyclization of diynols using Bu2Se2 and a halogen source.

Scheme 6.
Proposed mechanism of the synthesis of 3-iodoselenophene 6.

Scheme 7.
Key intermediates in the synthesis of 4-iodoselenophenes 6 and 4-butylselanylselenophenes 7.
Scheme 7.
Key intermediates in the synthesis of 4-iodoselenophenes 6 and 4-butylselanylselenophenes 7.

Scheme 8.
Proposed mechanism of the synthesis of 4-butylselanylselenophenes 7.

Scheme 9.
Cross-coupling reactions of 3-iodoselenophene 6a.

Scheme 10.
Reaction scope of the synthesis of 3,4-bis(butylselanyl)selenophenes 12.

Scheme 11.
Key intermediates in the cyclization of unsymmetrical 1,3-diynes.

Scheme 12.
Scope of the iodine-catalyzed reaction of 1,3-dienyl bromides with KSeCN.

Scheme 13.
Proposed reaction pathway to prepare the selenophene 14.

Scheme 14.
Scope of the synthesis of selenophenes from CF3-containing 1,3-enynes.

Scheme 15.
Cu-promoted cyclization of chalcogenoenynes to form 3-halo-chalcogenophenes.

Scheme 16.
Mechanism of the Cu(II)-mediated intramolecular cyclization of (Z)-selenoenynes.

Scheme 17.
Synthesis of 2,3,5-triarylselenophenes by Pd-catalyzed Suzuki cross-coupling reaction.

Scheme 18.
Synthesis of 3-benzyl-2,5-diarylselenophenes 21 via carbocyclization of (Z)-benzylselenoenynes 20.
Scheme 18.
Synthesis of 3-benzyl-2,5-diarylselenophenes 21 via carbocyclization of (Z)-benzylselenoenynes 20.

Scheme 19.
Palladium-catalyzed Suzuki cross-coupling reaction of selenophene 22.

Scheme 20.
Mechanism of the carbocyclization of the (Z)-benzylselenoenynes 20.

Scheme 21.
Scope of the synthesis of 3-organochalcogen selenophenes.

Scheme 22.
Reactions of 3-lithio-selenophene with aldehydes.

Scheme 23.
Scope of the Cu(II)/halide-mediated cyclization of homopropargyl selenides.

Scheme 24.
Mechanism of the Cu(II) halide-mediated cyclization of homopropargyl selenides 27.

Scheme 25.
Reaction scope of the synthesis of halogenated selenophenes 32 and benzo[b]selenophenes 34.
Scheme 25.
Reaction scope of the synthesis of halogenated selenophenes 32 and benzo[b]selenophenes 34.

Scheme 26.
Proposed mechanism of the synthesis of chloroselenophene 32a.

Scheme 27.
Scope and limitations of the synthesis of 3-iodoselenophenes 36.

Scheme 28.
Scope of synthesis of 3-organoselanyl-selenophenes 37.

Scheme 29.
Proposed mechanism of the synthesis of 3-iodoselenophenes 36 from 35.

Scheme 30.
Scope of the synthesis of 3-iodoselenophenes 39.

Scheme 31.
Scope of the synthesis of 3-bromo-selenophenes 40.

Scheme 32.
Scope of the synthesis of 3-(phenylselanyl)selenophenes 41.

Scheme 33.
Mechanism of the synthesis of 3-iodoselenophenes 39.

Scheme 34.
Cross-coupling reactions using 3-iodoselenophene 39a.

Scheme 35.
Scope and limitations of the synthesis of 3-selanylselenophenes 46.

Scheme 36.
Proposed mechanism for the synthesis of 3-selanylselenophenes 46.

Scheme 37.
Electrophilic (a) and radical (b) cyclization processes starting from type C precursors.

Scheme 38.
Larock’s (a) and Nakamura’s (b) protocols to prepare benzoselenophenes.

Scheme 39.
Use of type C precursor in the synthesis of viniferifuran and (±)-dehydroampelopsin B Se-analogues.
Scheme 39.
Use of type C precursor in the synthesis of viniferifuran and (±)-dehydroampelopsin B Se-analogues.

Scheme 40.
Scope of the synthesis of 2-acylbenzo[b]selenophenes 58.

Scheme 41.
Proposed mechanism for the synthesis of 2-acylbenzo[b]selenophenes 58.

Scheme 42.
1,1-Dibromoalkenes as precursors of benzo[b]selenophenes.

Scheme 43.
Protocols for obtaining type C precursors and benzo[b]selenophenes 67.

Scheme 44.
Synthesis of 2-substituted benzo[b]selenophenes.

Scheme 45.
Proposed mechanism of the synthesis of 2-substituted benzo[b]selenophenes 68 and 69.

Scheme 46.
Synthesis of 2-arylselanyl-benzo[b]selenophenes 70.

Scheme 47.
Mechanism of the synthesis of 2-arylselanyl-benzo[b]selenophenes 70.

Scheme 48.
Scope of the synthesis of 2,3-bis-organoselanyl-benzo[b]selenophenes 71 using Oxone®.

Scheme 49.
The reaction mechanism of the synthesis of 71.

Scheme 50.
Scope of the formation of 3-(arylsulfonyl)benzoselenophenes 74 using sulfinic acid.

Scheme 51.
Proposed Mechanism of the formation of 74 using TBHP.

Scheme 52.
Scope of the synthesis of benzoselenophenes 74 using sulfuryl chloride.

Scheme 53.
The reaction mechanism of the photocatalyzed synthesis of 74.

Scheme 54.
Gold-photoredox catalysis in the synthesis of 2,3-diarylbenzo[b]selenophenes.

Scheme 55.
Gold-photoredox catalysis mechanism in the synthesis of selenophene 79.

Scheme 56.
Scope of the synthesis of 3-phosphinoylbenzo[b]selenophenes 81.

Scheme 57.
Scope of the synthesis of 3-nitrobenzo[b]selenophenes 82.

Scheme 58.
Synthesis of 2-substituted benzo[b]selenophenes and AT1 receptors.

Scheme 59.
Proposed mechanism of the radical cascade reaction to prepare 85.

Scheme 60.
General protocol for cyclization of type D precursors using NaBH4.

Scheme 61.
Synthesis of mono- and diselena[5]helicenes 89 and 91.

Scheme 62.
Synthesis of 3-methylidenebenzo[c]selenophenes 94.

Scheme 63.
Proposed mechanism of the synthesis of benzo[c]selenophenes 94.

Scheme 64.
General protocol to obtaining selenoketenes 96 from 95.

Scheme 65.
Synthesis of benzo[b]selenophenes 97.

Scheme 66.
Control in the formation of 97 and 98 (a) and possible pathways of the cyclization of selenoketene 96 (b).
Scheme 66.
Control in the formation of 97 and 98 (a) and possible pathways of the cyclization of selenoketene 96 (b).

Scheme 67.
Regioselective synthesis of benzo[b]selenophenes 97 and 98.

Scheme 68.
Formation of σH- and σCl-adducts from selenoketene 96.

Scheme 69.
Synthesis of 3-bromo-benzo[b]selenophenes 103.

Scheme 70.
Synthesis of 3-substituted-benzo[b]selenophenes 105.

Scheme 71.
Synthesis of 2-aryl-3-bromo-benzo[b]selenophenes 107.

Scheme 72.
Reaction pathways to prepare benzoselenophenes 107.

Scheme 73.
Retrosynthetic strategy for the preparation of 2-aryl 109 and 3-aryl-benzo[b]selenophenes 110.
Scheme 73.
Retrosynthetic strategy for the preparation of 2-aryl 109 and 3-aryl-benzo[b]selenophenes 110.

Scheme 74.
Scope of the synthesis of benzo[b]selenophenes through copper-catalyzed C-Se coupling/cyclization.
Scheme 74.
Scope of the synthesis of benzo[b]selenophenes through copper-catalyzed C-Se coupling/cyclization.

Scheme 75.
Scope of the synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 116.

Scheme 76.
Mechanism of the synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 116.

Scheme 77.
Scope of the synthesis of benzo[b]selenophenes fused to imidazo[1,2-a]-pyridines 118.

Scheme 78.
Mechanism of the synthesis of the fused benzo[b]selenophene 118.

Scheme 79.
Cobalt-catalyzed synthesis of benzo[b]selenophene-fused imidazo[1,2-a]pyridines 118.

Scheme 80.
Proposed mechanism of the cobalt-catalyzed synthesis of fused benzo[b]selenophene 118.

Scheme 81.
Scope of the synthesis of the benzoselenopheno[3,2-b]indoles 120.

Scheme 82.
Synthesis of the benzoselenophenes from Resveratrol.

Scheme 83.
Scope of the synthesis of diaryl[b,d]selenophenes 123.

Scheme 84.
Scope of the synthesis of benzo[b]selenophenes 125 from styryl bromides.

Scheme 85.
Proposed mechanism of the iodine-catalyzed synthesis of benzo[b]selenophene 125.

Scheme 86.
One-pot formation of fused dibenzoselenophenes 128.

Scheme 87.
Plausible mechanism of the HDDA annulation to prepare dibenzoselenophenes 128.

Scheme 88.
Synthesis of 3-benzo[b]selenophenes-2-aryl-1H-indoles 131.

Scheme 89.
The reaction mechanism of the synthesis of selenophene 131a.

Scheme 90.
Scope of the synthesis of selenophene[3,2-c]chromene 3-substituted 134.

Scheme 91.
Reactivity of 3-iodo-selenophene[3,2-c]chromene 134a toward Sonogashira and Suzuki cross-couplings.
Scheme 91.
Reactivity of 3-iodo-selenophene[3,2-c]chromene 134a toward Sonogashira and Suzuki cross-couplings.

Scheme 92.
Scope of the synthesis of 3-organochalcogen-selenophen[2,3-b]thiophenes 138.

Scheme 93.
Suzuki and Li-Se exchange reactions of fused selenophenes 138f and 138j.

Scheme 94.
Scope and mechanism of the synthesis of benzoselenophene-based palladacycles 142.

Scheme 95.
Synthesis of benzoselenophene[2,3-c]pyridinones 144.

Scheme 96.
Mechanism of the synthesis of benzoselenophene[2,3-c]pyridinone 144.

Scheme 97.
Scope of the synthesis of selenopheno[2,3-b]quinolines 146.

Scheme 98.
Proposed mechanism for the synthesis of selenopheno[2,3-b]quinoline 146.

Scheme 99.
Scope of the synthesis of 3-iodo-4H-selenopheno[3,2-b]indoles 148.

Scheme 100.
Mechanism of the synthesis of 3-iodo-4H-selenopheno[3,2-b]indoles 148.

Scheme 101.
Scope of the synthesis of 3-(alkylselanyl)-4H-selenopheno[3,2-b]indoles 149.

Scheme 102.
Scope of the synthesis of selenophenes fused to pyridines.

Scheme 103.
Proposed mechanism of the synthesis of 151.

Scheme 104.
Scope of the synthesis of 2-organylselenopheno[2,3-b]pyridines 151.

Scheme 105.
Mechanism of the synthesis of 2-organylselenopheno[2,3-b]pyridines 151.

Scheme 106.
Synthetic applications for compound 151a.

Scheme 107.
Scope of the synthesis of selenopheno[3,2-c]- and selenopheno[2,3-c]quinolones.

Scheme 108.
Proposed mechanism of the synthesis of selenopheno[3,2-c]quinolones.

Scheme 109.
Scope of the synthesis of selenopheno[3,2-b]- and selenopheno[2,3-b]thiophenes 162.

Scheme 110.
Scope of the synthesis of the selenophenothiophenes 164.

Scheme 111.
Scope of the synthesis of polysubstituted selenophenes 166.

Scheme 112.
Proposed mechanism of the synthesis of polysubstituted selenophenes 166.

Scheme 113.
Scope and mechanism of the synthesis of 2,3-diaryl-benzo[b]selenopheno[3,2-d]selenophenes 168.
Scheme 113.
Scope and mechanism of the synthesis of 2,3-diaryl-benzo[b]selenopheno[3,2-d]selenophenes 168.

Scheme 114.
Other common strategies to access fused selenophenes.

Scheme 115.
Scope of the synthesis of benzo[b]selenopheno[2,3-d]furans/thiophene/selenophenes 172.

Scheme 116.
Competition between selenium, sulfur and oxygen as nucleophiles in the cyclization reaction.
Scheme 116.
Competition between selenium, sulfur and oxygen as nucleophiles in the cyclization reaction.

Scheme 117.
Proposed mechanism of the synthesis of 172.

Scheme 118.
Scope of the synthesis of chalcogenochromene-fused selenophenes 174.

Scheme 119.
Mechanism of the synthesis of 3-(butylselanyl)-5H-selenopheno[3,2-c]isoselenochromene 174.
Scheme 119.
Mechanism of the synthesis of 3-(butylselanyl)-5H-selenopheno[3,2-c]isoselenochromene 174.

Scheme 120.
Scope of the synthesis of 1-(butylselanyl)-selenophene-fused chromenes 176.

Scheme 121.
Proposed mechanism of the synthesis of 4H-selenopheno[2,3-c]chromene 176.

Scheme 122.
Scope of the synthesis of selenophene-fused quinolines 178.

Scheme 123.
Suzuki cross-coupling reactions of 176a and 178a.

Scheme 124.
Scope of the synthesis of isochromenones fused to selenophenes 181.

Scheme 125.
Seleno- and oxo-cyclization competing reactions.

Scheme 126.
Mechanism of the synthesis of isochromenones fused to selenophene 181a.

Scheme 127.
Scope of the synthesis of selenophene and diselenophene-fused compounds.

Scheme 128.
Mechanism of the synthesis of diselenophene-fused thieno[2,3-c]acridine 190.

Scheme 129.
Scope for the synthesis of 3-(arylselanyl/sulfanyl)-2-arylselenopheno[2,3-b]quinoxaline 193 and 6-aryl-7-(phenylselanyl)selenopheno[2,3-b]pyrazine derivatives 194.
Scheme 129.
Scope for the synthesis of 3-(arylselanyl/sulfanyl)-2-arylselenopheno[2,3-b]quinoxaline 193 and 6-aryl-7-(phenylselanyl)selenopheno[2,3-b]pyrazine derivatives 194.

Scheme 130.
Scope of the synthesis of 2-arylselenopheno[2,3-b]quinoxaline derivatives 196.

Scheme 131.
Scope for the synthesis of polyselenophenes fused to benzothiophenes 198.

Scheme 132.
Proposed mechanism for the synthesis of selenophene-fused benzothiophene.

Scheme 133.
Synthesis of fused selenophenes 200 and 202 by biPEC.

Scheme 134.
Proposed mechanism for the synthesis of dibenzo[b]selenopheno[2,3-d]thiophene 202.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hellwig, P.S.; Peglow, T.J.; Penteado, F.; Bagnoli, L.; Perin, G.; Lenardão, E.J. Recent Advances in the Synthesis of Selenophenes and Their Derivatives. Molecules 2020, 25, 5907. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245907
AMA Style
Hellwig PS, Peglow TJ, Penteado F, Bagnoli L, Perin G, Lenardão EJ. Recent Advances in the Synthesis of Selenophenes and Their Derivatives. Molecules. 2020; 25(24):5907. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245907
Chicago/Turabian StyleHellwig, Paola S., Thiago J. Peglow, Filipe Penteado, Luana Bagnoli, Gelson Perin, and Eder J. Lenardão. 2020. "Recent Advances in the Synthesis of Selenophenes and Their Derivatives" Molecules 25, no. 24: 5907. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245907