
Secoiridoid Glucosides and Anti-Inflammatory Constituents from the Stem Bark of Fraxinus chinensis

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Results and Discussion
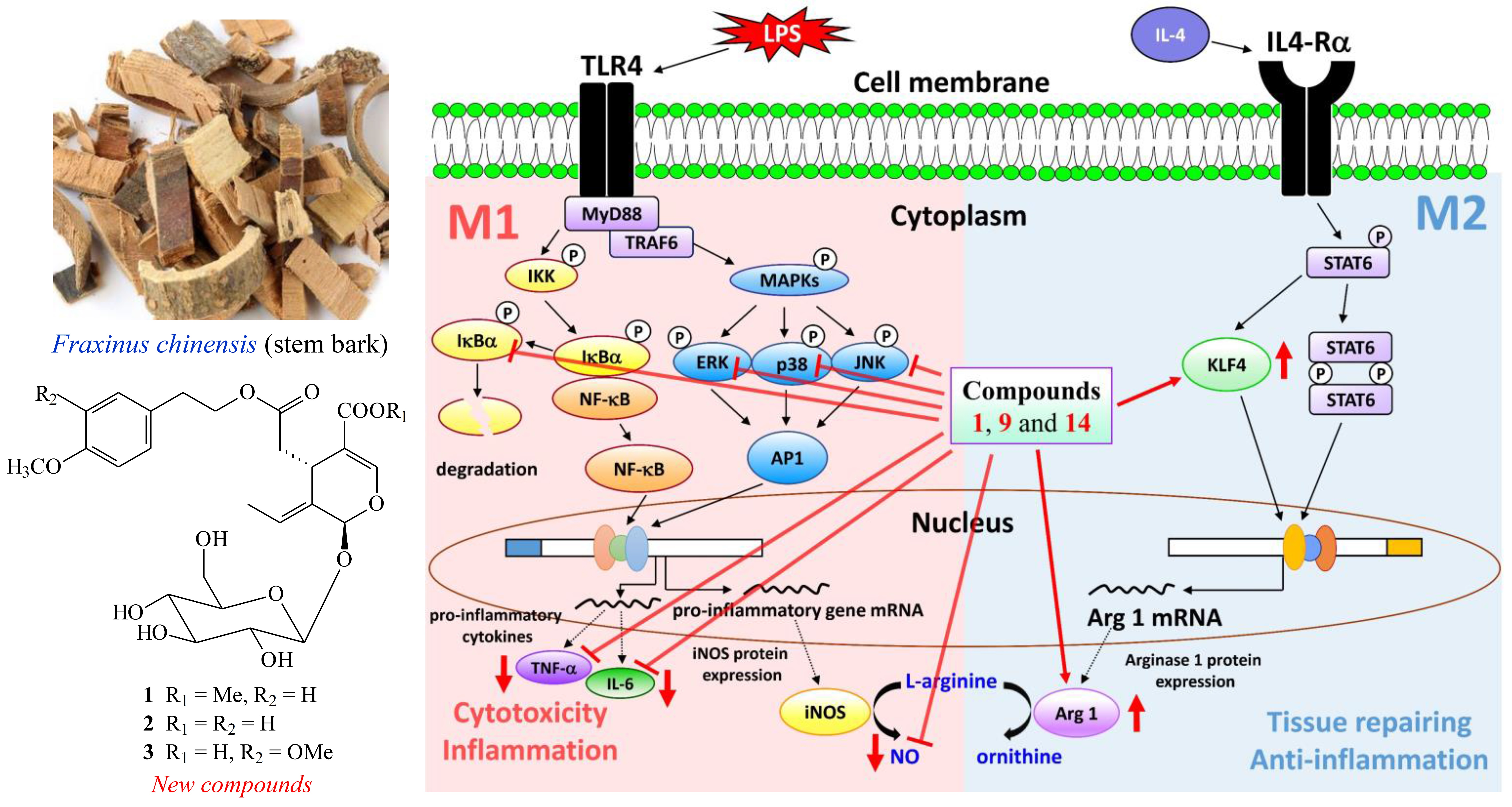
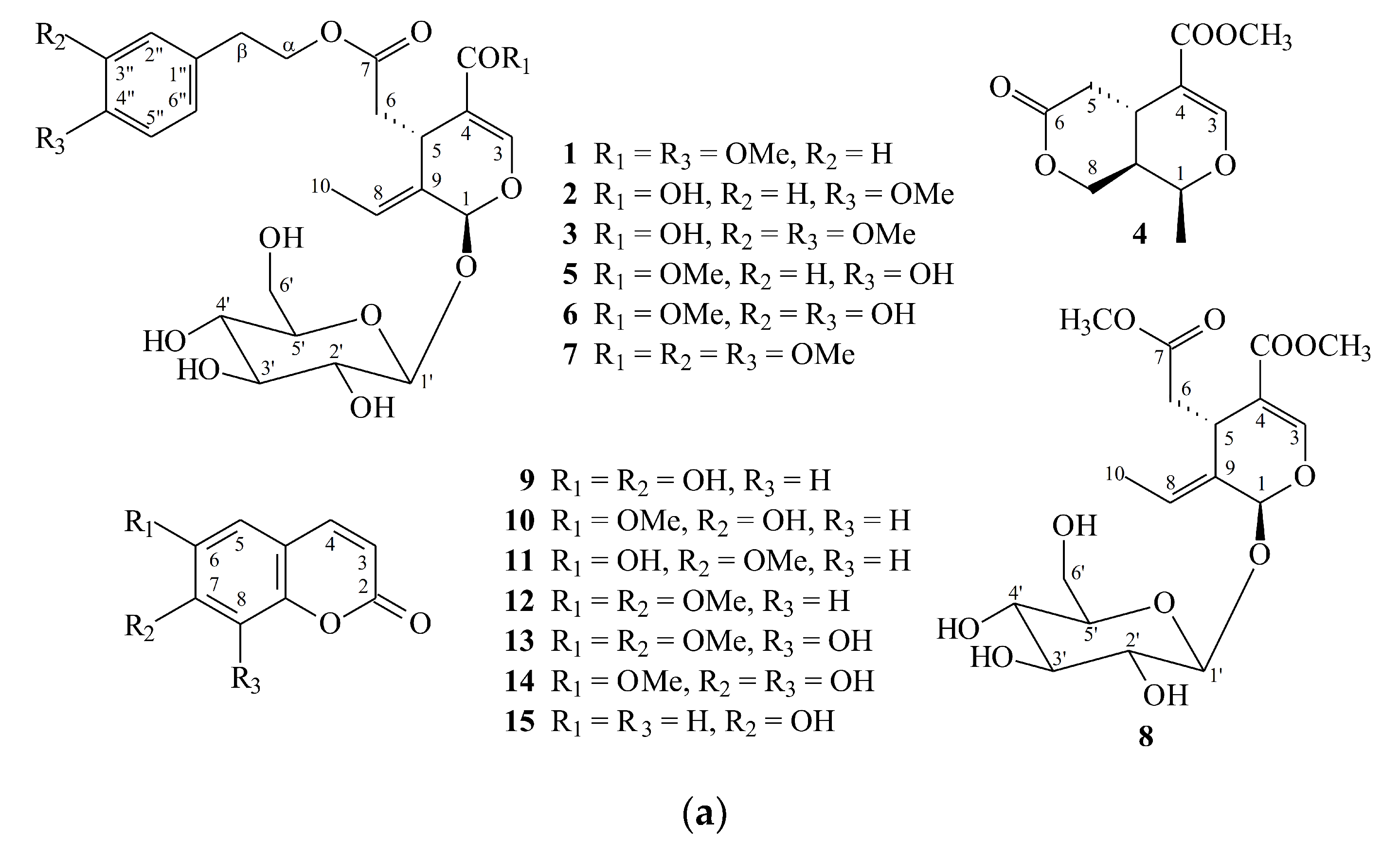
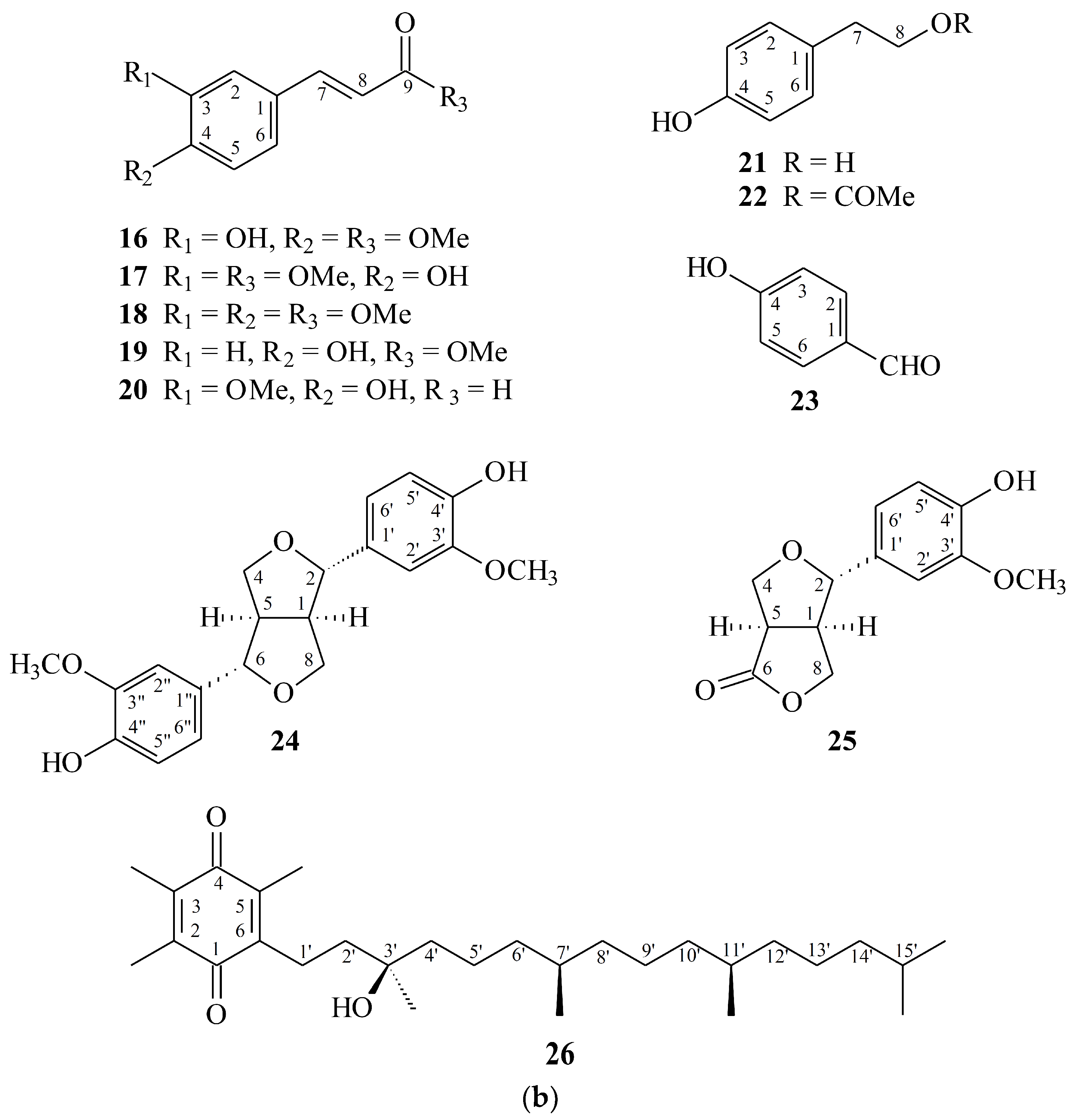
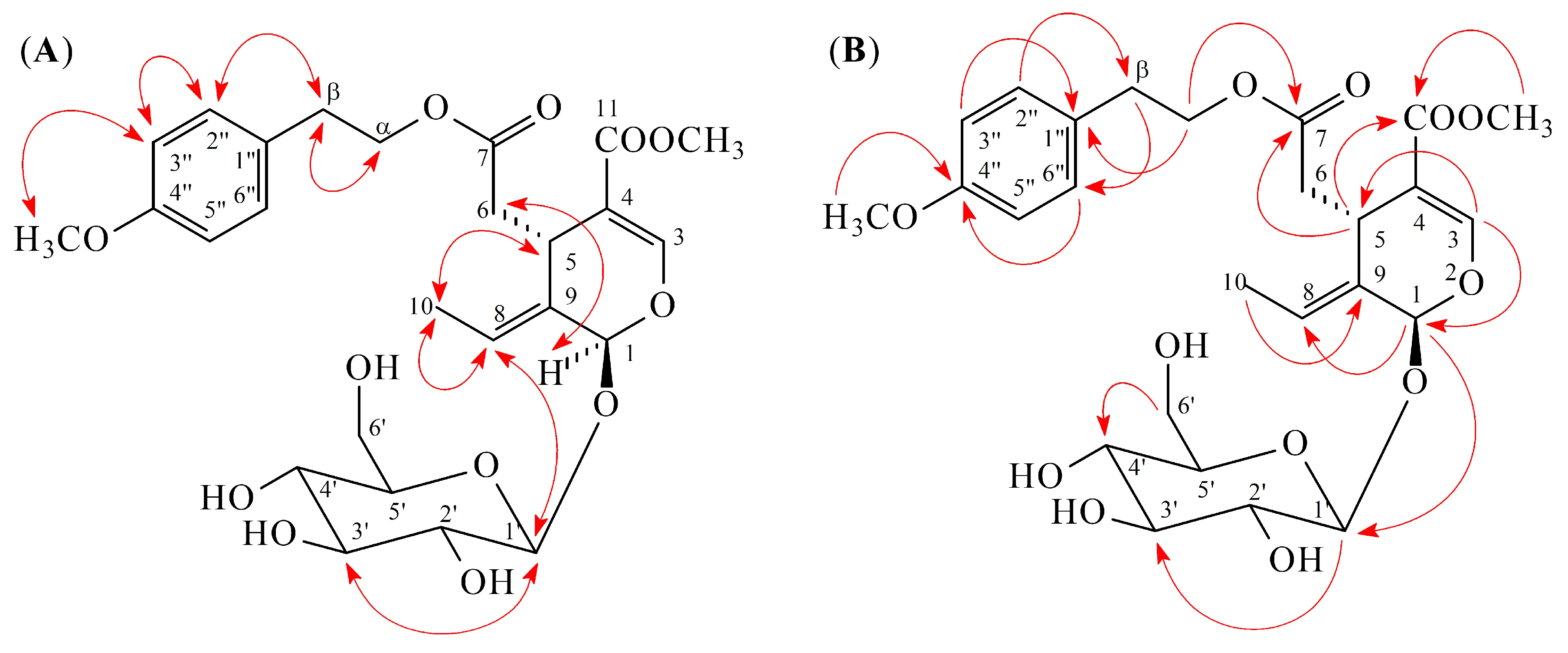
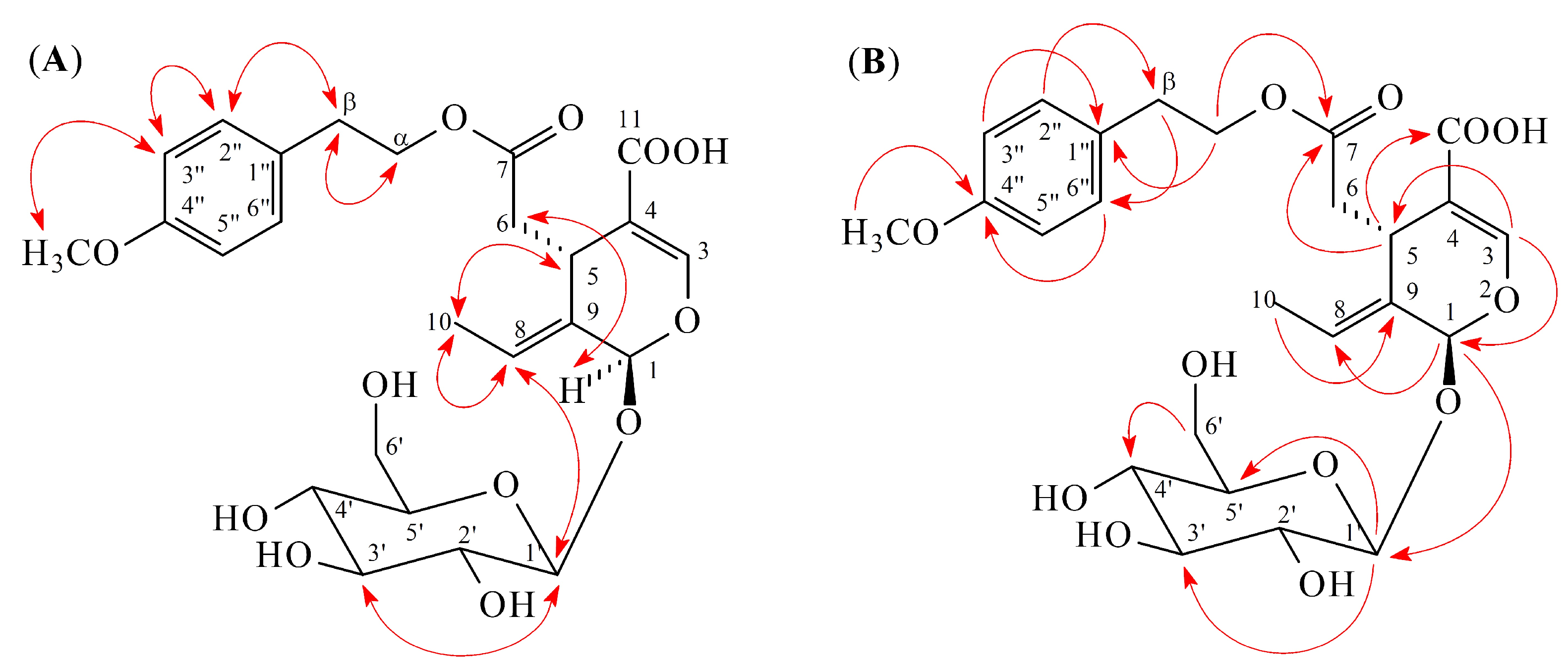
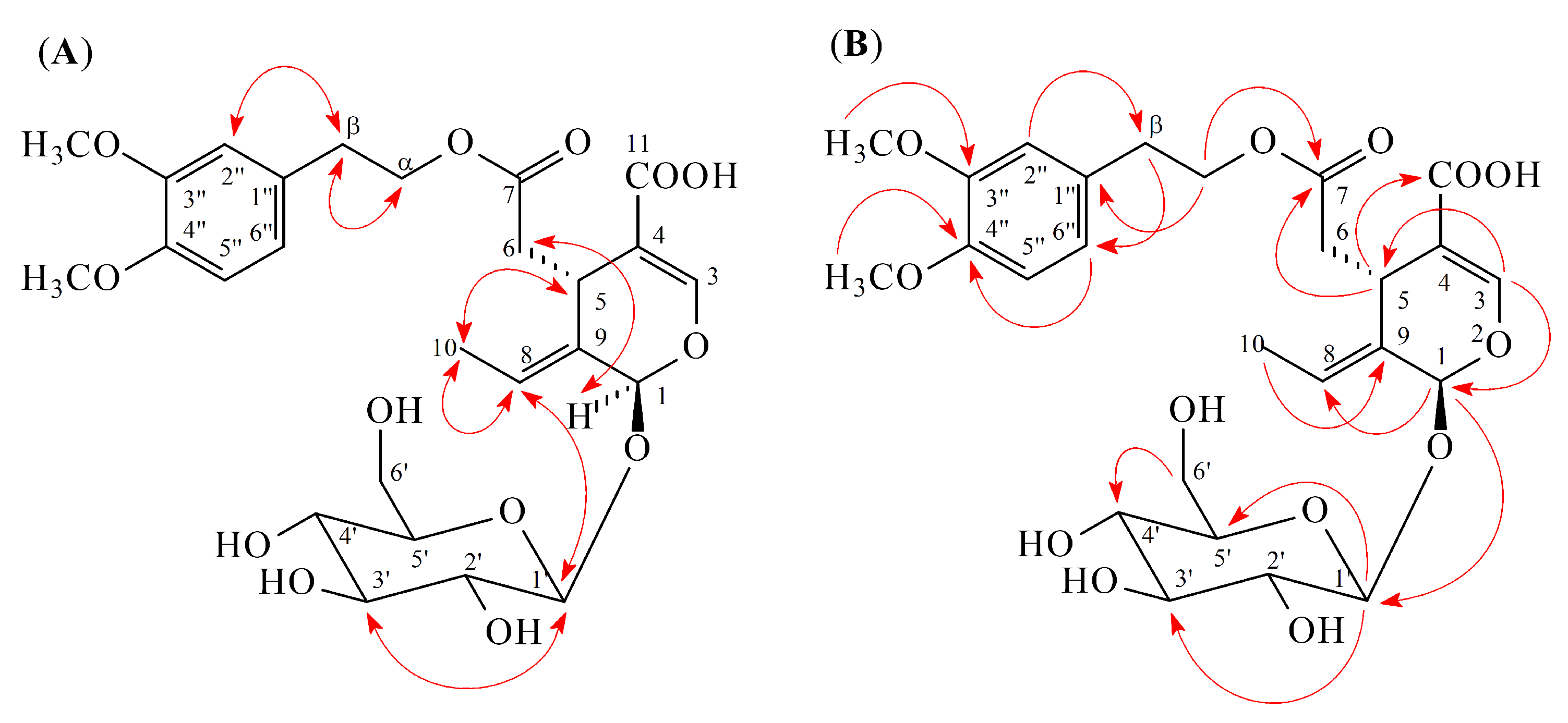
2.1. Isolation and Structural Elucidation
2.2. Structure Identification of the Known Isolates
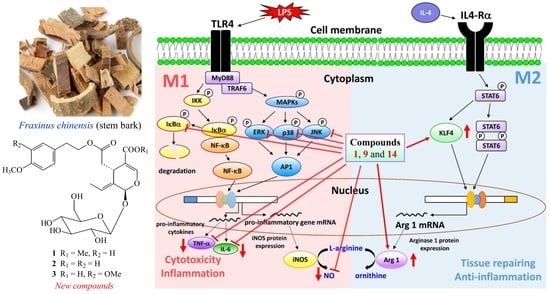
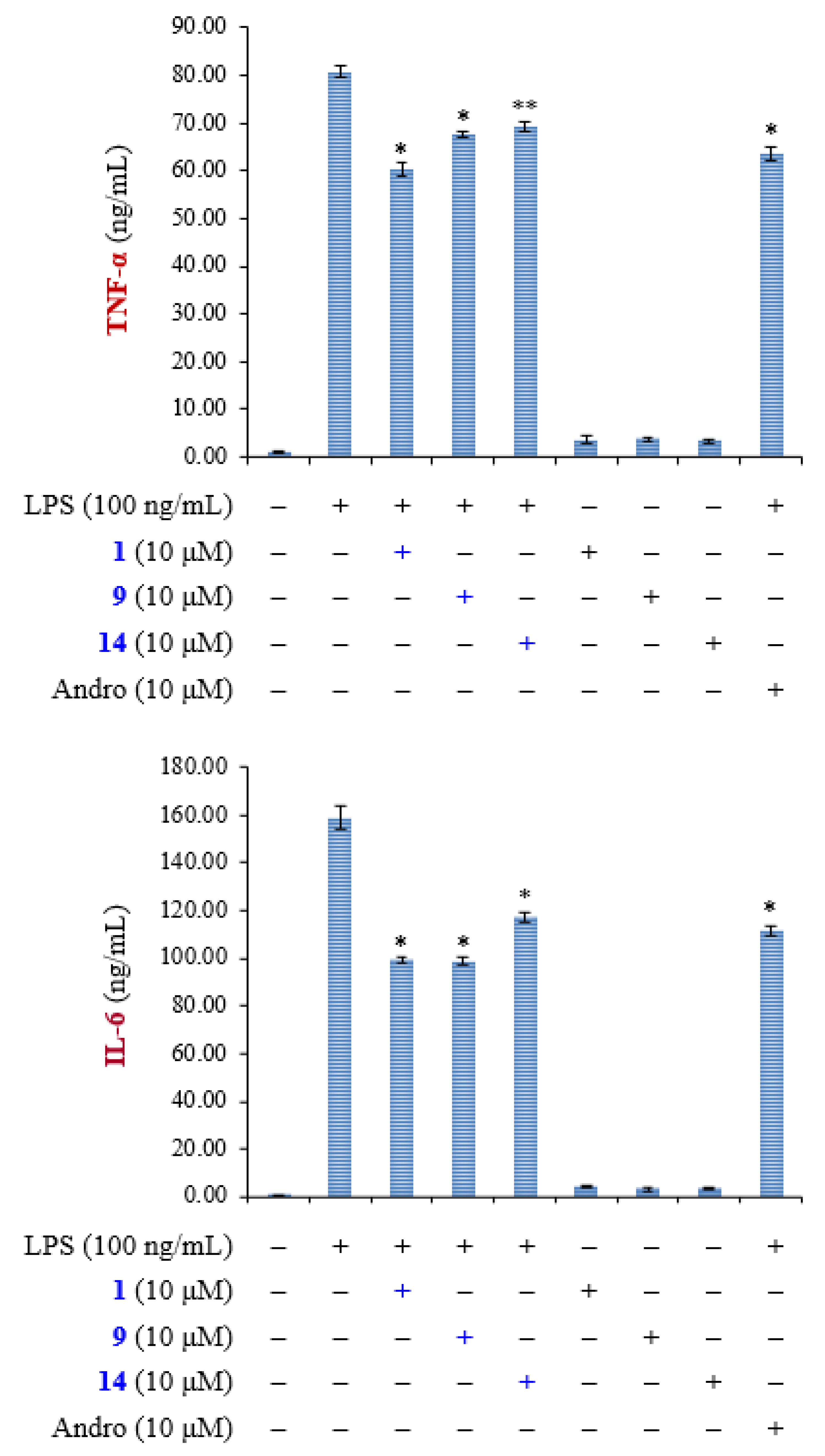
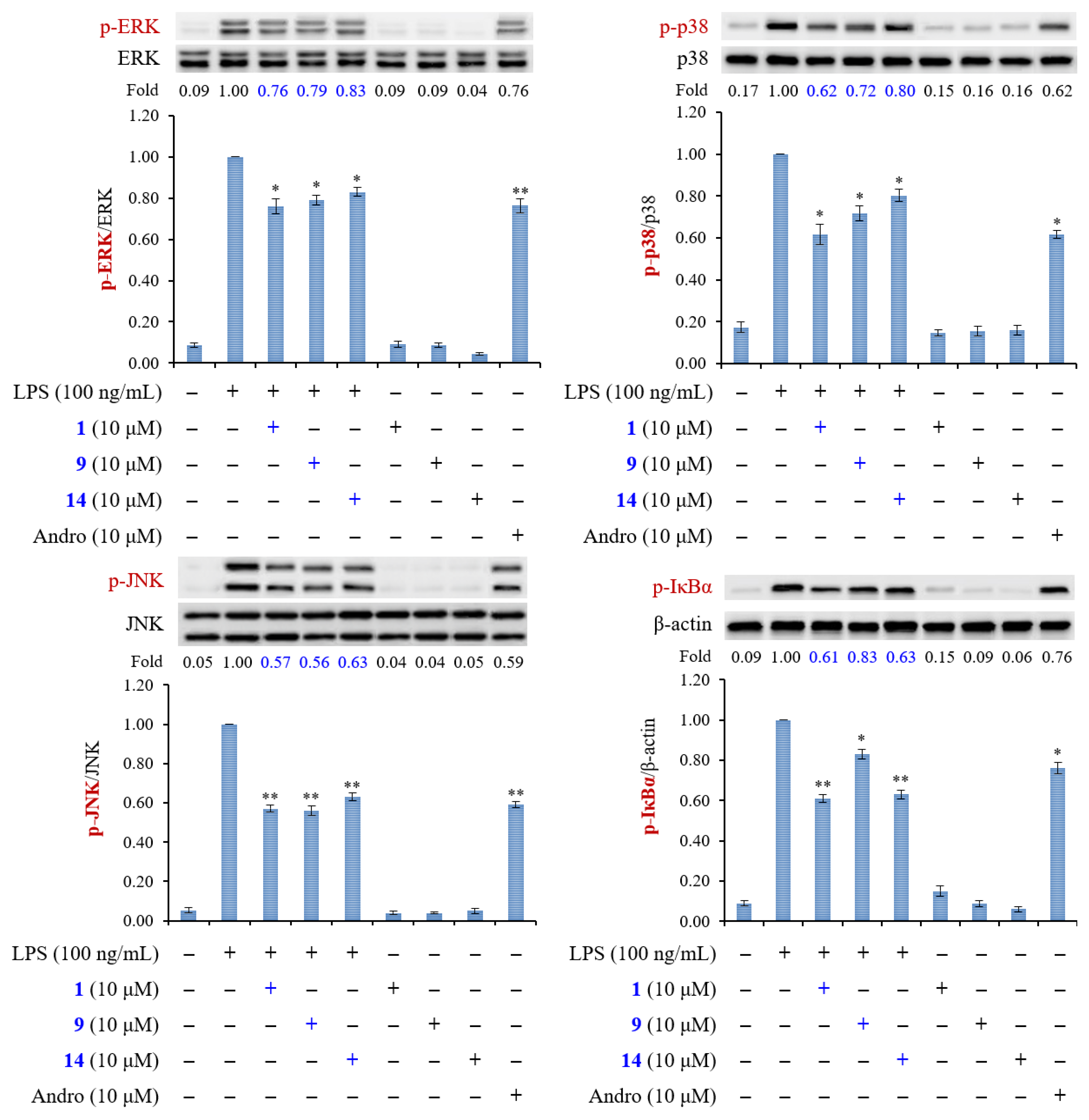
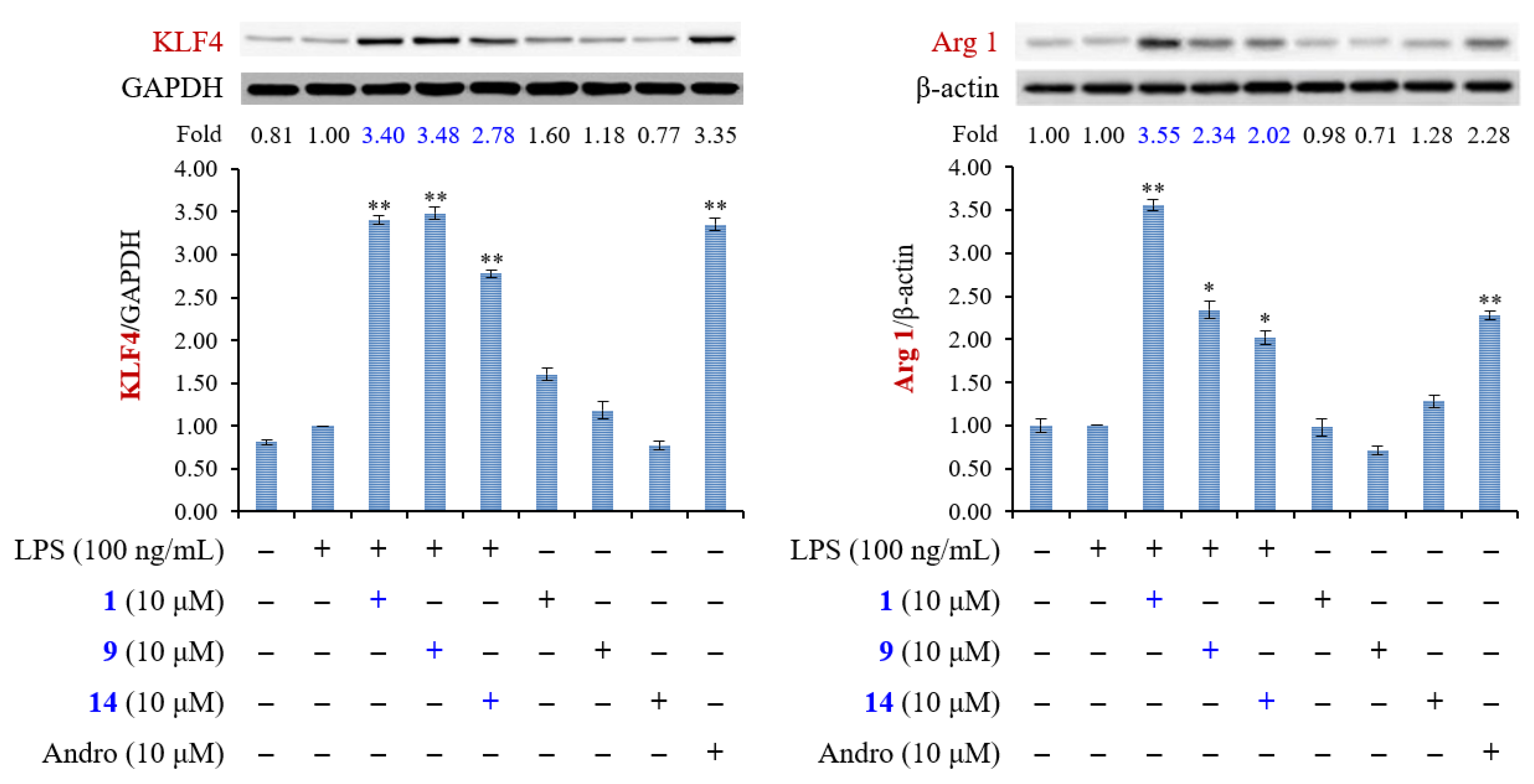
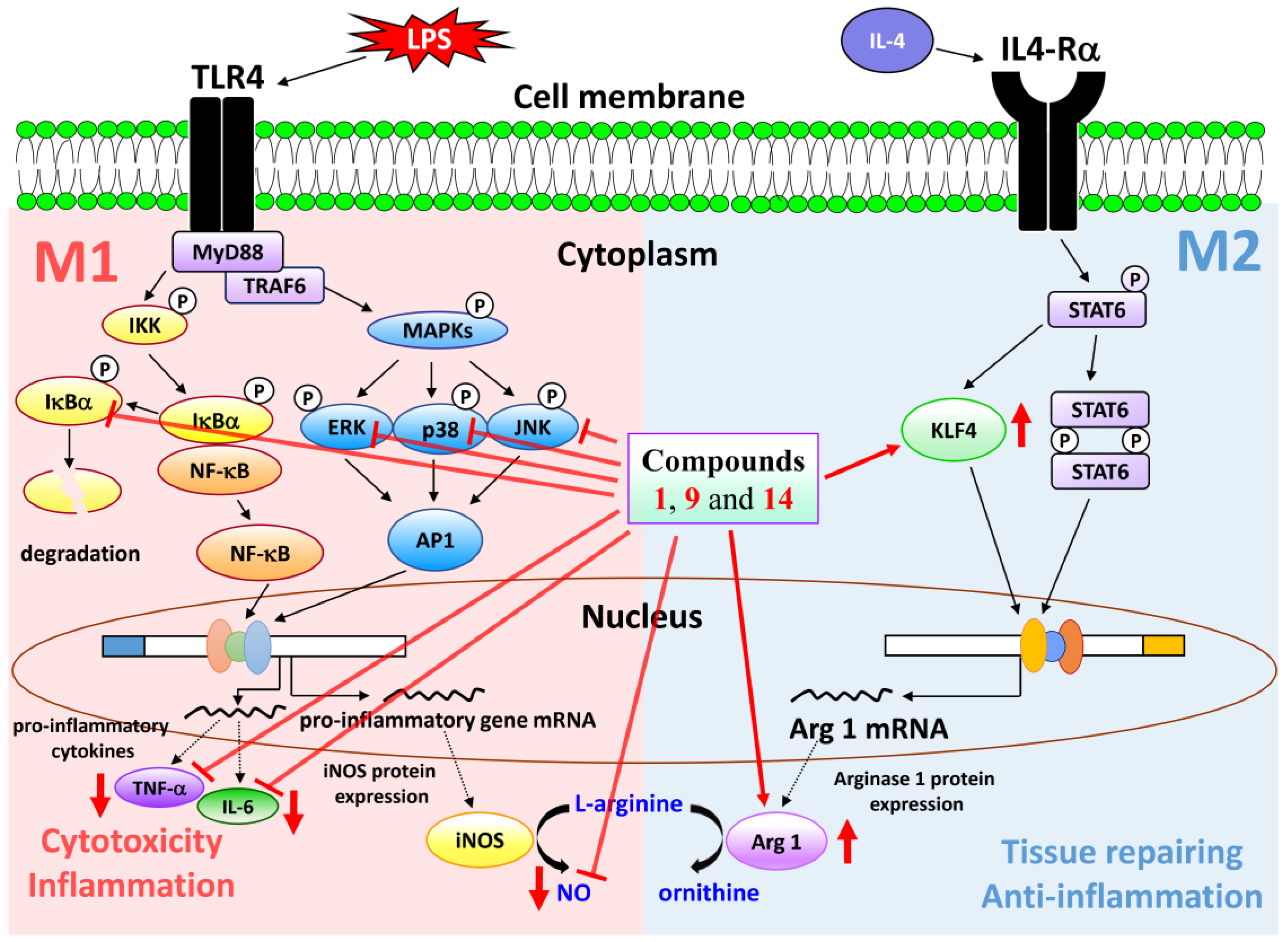
2.3. Biological Studies
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Biological Assay
3.4.1. Mensuration of Human Neutrophils
3.4.2. Mensuration of Superoxide Anion (O2•−) Generation
3.4.3. Measurement of Elastase Release
3.4.4. Determination of NO Production
3.4.5. Cell Viability Assay
3.4.6. Enzyme-Linked Immunosorbent Assay
3.4.7. Western Blot
3.4.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wei, Z.; Green, S.P. ‘Fraxinus chinensis’ in ‘Flora of China’; Science Press: Beijing, China, 1996; Volume 15, p. 277. [Google Scholar]
- Huang, Y.L.; Oppong, M.B.; Guo, Y.; Wang, L.Z.; Fang, S.M.; Deng, Y.R.; Gao, X.M. The Oleaceae family: A source of secoiridoids with multiple biological activities. Fitoterapia 2019, 136, 104155. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, Y.; Tanahashi, T.; Shintaku, M.; Sakai, T.; Nagakura, N.; Parida. Secoiridoid glucosides from Fraxinus americana. Phytochemistry 2000, 55, 275–284. [Google Scholar] [CrossRef]
- Xiao, K.; Song, Q.H.; Zhang, S.W.; Xuan, L.J. Water-soluble constituents of the root barks of Fraxinus rhynchophylla (Chinese drug Qinpi). J. Asian Nat. Prod. Res. 2008, 10, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Wnag, L.L.; Li, J.; Hu, L.H. Two new coumarins from Fraxinus chinensis Roxb. J. Integr. Plant Biol. 2007, 31, 1277–1280. [Google Scholar]
- Wang, L.J.; Sun, F.; Zhang, X.Y.; Ma, Z.J.; Cheng, L. A secoiridoid with quinone reductase inducing activity from Cortex fraxini. Fitoterapia 2010, 81, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.G.; Choi, J.S.; Yeon, S.W.; Cui, E.J.; Park, H.J.; Yoo, J.S.; Chung, I.S.; Baek, N.L. Secoiridoid glycoside from the flowers of Osmanthus fragrans var. aurantiacus Makino inhibited the activity of β-secretase. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 371–374. [Google Scholar] [CrossRef]
- Darmak, N.; Allouche, N.; Hamdi, B.; Litaudon, M.; Darmak, M. New secoiridoid from olive mill wastewater. Nat. Prod. Res. 2012, 26, 125–131. [Google Scholar] [CrossRef]
- Tanahashi, T.; Takenaka, Y.; Nagakura, N. Two dimeric secoiridoid glycosides from Jasminum polyanthum. Phytochemistry 1996, 41, 1341–1345. [Google Scholar] [CrossRef]
- He, Z.D.; But, P.H.; Chan, T.W.; Dong, H.; Xu, H.X.; Lau, C.P.; Sun, H.D. Antioxdative glucosides from the fruits of Ligustrum lucidium. Chem. Pharm. Bull. 2001, 49, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.C.; Lin, S.L.; Hsieh, P.W.; Chein, C.C. Secoiridoid glycosides from Jasminum polyanthum. J. Chin. Chem. Soc. 1996, 43, 171–176. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Hisada, S.; Nishibe, S.; Rouxt, D.G.; Rourke, J.P. Coumarin from Olea Africana and Olea capensis. Phytochemistry 1984, 23, 699–700. [Google Scholar] [CrossRef]
- Wu, T.S.; Shi, L.S.; Wang, J.J.; Iou, S.C.; Chang, H.C.; Chen, Y.P.; Kuo, Y.H.; Chang, Y.L.; Teng, C.M. Cytotoxic and antiplatelet aggregation principles of Ruta graveolens. J. Chin. Chem. Soc. 2003, 50, 171–178. [Google Scholar] [CrossRef]
- Lee, C.K.; Lee, P.H.; Kuo, Y.H. The chemical constituents from the aril of Cassia fistula L. J. Chin. Chem. Soc. 2001, 48, 1053–1058. [Google Scholar] [CrossRef]
- Yu, H.J.; Chen, C.C.; Shieh, B.J. The constituents from the leaves of Magnolia coco. J. Nat. Prod. 1998, 61, 1017–1019. [Google Scholar] [CrossRef]
- Kaufmann, F.; Lam, J. Chemical constituents of the genus of Dahlia II. The isolation of two new aromatic compounds: Naringenin trimethylether and fraxedin. Acta Chem. Scand. 1967, 21, 311–313. [Google Scholar] [CrossRef] [Green Version]
- Aiyelaagbe, J.B.; Gloer, J.B. Japodic acid, a novel aliphatic acid from Jatropha podagrica Hook. Rec. Nat. Prod. 2008, 2, 100–106. [Google Scholar]
- Kumar, S.; Ray, A.B.; Konno, C.; Oshima, Y.; Hikino, H. Cleomiscosin D, a coumarinolignan from seeds of Cleome Viscosa. Phytochemistry 1988, 27, 636–638. [Google Scholar] [CrossRef]
- Weight, E.S.; Razdan, T.K.; Qadni, B.; Harkar, S. Chromones and coumarin from Skimmia larueola. Phytochemistry 1982, 26, 2063–2069. [Google Scholar] [CrossRef]
- Rose, B.R.; Kaustuv, B. Anullmann ether reaction involving an alkynyl substrate: A convergent route to a combretastatin intermediate. Synth. Commun. 1995, 25, 3187–3197. [Google Scholar]
- Lee, T.H.; Chiou, J.L.; Lee, C.K.; Kuo, Y.H. Separation and determination of chemical constituents in the root of Rhus javanica L. var. roxburghiana. J. Chin. Chem. Soc. 2005, 52, 833–841. [Google Scholar] [CrossRef]
- Bowden, B.F.; Camie, R.C.; Parnell, J.C. Constituents of the fruit of Pseudopanax arboreun (Araliaceae). Aust. J. Chem. 1975, 28, 91–107. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Subbarao, G.V.; Nakahara, K.; Yoshihashi, T.; Ito, O.; Maeda, I.; Ono, H.; Yoshida, M. Nitrification inhibitors from root tissues of Brachiaria humidicola, a tropical grass. J. Agric. Food. Chem. 2007, 55, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Barakat, H.H.; Nawwar, M.A.M.; Buddrust, J.; Linscheid, M. Niloticol, a phenolic glyceride and two phenolic aldehydes from the roots of Tamarix nilotica. Phytochemistry 1987, 26, 1837–1838. [Google Scholar] [CrossRef]
- Xu, Q.M.; Liu, Y.L.; Li, X.R.; Feng, Y.L.; Yang, S.L. Two new phenylglycol derivatives isolated from Syringa reticulata var. mandshurica and their antifungal activities. Chem. Pharm. Bull. 2009, 57, 863–866. [Google Scholar] [CrossRef] [Green Version]
- Assia, I.; Bouaziz, M.; Ghamgui, H.; Kamoun, A.; Miled, N.; Sayadi, S.; Gargouri, Y. Optimization of lipase-catalyzed synthesis of acetylated tyrosol by response surface methodology. J. Agric. Food Chem. 2007, 55, 10298–10305. [Google Scholar] [CrossRef]
- Chiang, C.Y.; Leu, Y.L.; Chan, Y.Y.; Wu, T.S. Sodium aristolochates from the flowers and fruits of Aristolochia zollingeriana. J. Chin. Chem. Soc. 1998, 45, 93–97. [Google Scholar] [CrossRef]
- El-Hassan, A.; El-Sayed, M.; Hamed, A.I.; Rhee, I.K.; Ahmed, A.A.; Zeller, K.P.; Verpoorte, R. Bioactive constituents of Leptadenia arborea. Fitoterapia 2003, 74, 184–187. [Google Scholar] [CrossRef]
- Marchand, P.A.; Zajick, J.; Lewis, N.G. Oxygen insertion in Sesamum indicum furanofuran lignans. Diastereoselective syntheses of enzyme substrate analogues. Can. J. Chem. 1997, 75, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.H.; Wang, G.J.; Kuo, Y.H.; Lee, C.K. The low polar constituents from Bidens pilosa L. var. minor (Blume) Sherff. J. Chin. Chem. Soc. 2000, 47, 1131–1136. [Google Scholar] [CrossRef]
- Borregaard, N. The human neutrophil. Function and dysfunction. Eur. J. Haematol. 1998, 41, 401–413. [Google Scholar] [CrossRef]
- Roos, D.; van Bruggen, R.; Meischl, C. Oxidative killing of microbes by neutrophils. Microbes Infect. 2003, 5, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Rieu, P.; Descamps-Latscha, B.; Lesavre, P.; Halbwachs-Mecarelli, L. Neutrophils: Molecules, functions and pathophysiological aspects. Lab. Investig. 2000, 80, 617–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. Res. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauleau, A.L.; Rutschman, R.; Lang, R.; Pernis, A.; Watowich, S.S.; Murray, P.J. Enhancer-mediated control of macrophage-specific arginase I expression. J. Immunol. 2004, 172, 7565–7573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Ming, X.F. Functions of arginase isoforms in macrophage inflammatory responses: Impact on cardiovascular diseases and metabolic disorders. Front. Immunol. 2014, 5, 533. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Boyum, A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand. J. Clin. Lab. Investig. 1968, 97, 77–89. [Google Scholar]
- Jauregui, H.O.; Hayner, N.T.; Driscoll, J.L.; Williams-Holland, R.; Lipsky, M.H.; Galletti, P.M. Trypan blue dye uptake and lactate dehydrogenase in adult rat hepatocytes-freshly isolated cells, cell suspensions, and primary monolayer cultures. In Vitro 1981, 17, 1100–1110. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Hwang, T.L.; Leu, Y.L.; Kao, S.H.; Tang, M.C.; Chang, H.L. Viscolin, a new chalcone from Viscum coloratum, inhibits human neutrophil superoxide anion and elastase release via a cAMP-dependent pathway. Free Radic. Biol. Med. 2006, 41, 1433–1441. [Google Scholar] [CrossRef]
- Chen, J.J.; Ting, C.W.; Wu, Y.C.; Hwang, T.L.; Cheng, M.J.; Sung, P.J.; Wang, T.C.; Chen, J.F. New labdane-type diterpenoids and anti-inflammatory constituents from Hedychium Coronarium. Int. J. Mol. Sci. 2013, 14, 13063–13077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.; Köpcke, B.; Anke, H.; Sterner, O. Biologically active secondary metabolites from the ascomycete A111-95. 2. Structure elucidation. J. Antibiot. 2002, 55, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lai, J.L.; Liu, Y.H.; Liu, C.; Qi, M.P.; Liu, R.N.; Zhu, X.F.; Zhou, Q.G.; Chen, Y.Y.; Guo, A.Z.; Hu, C.M. Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways. Inflammation 2017, 40, 1–12. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | 3 a |
|---|---|---|---|
| 1 | 5.92 br s | 5.87 br s | 5.86 br s |
| 3 | 7.51 s | 7.39 s | 7.38 s |
| 5 | 3.95 dd (9.5, 5.0) | 4.01 dd (9.5, 5.0) | 4.00 dd (9.5, 4.5) |
| 6 | 2.44 dd (14.0, 9.5) | 2.41 dd (14.0, 9.5) | 2.41 dd (14.0, 9.5) |
| 2.69 dd (14.0, 5.0) | 2.79 dd (14.0, 4.5) | 2.80 dd (14.0, 4.5) | |
| 8 | 6.06 br q (7.0) | 6.05 br q (7.0) | 6.06 br q (7.0) |
| 10 | 1.62 dd (7.0, 1.0) | 1.66 dd (7.0, 1.0) | 1.66 dd (7.0, 1.0) |
| α | 4.12 dt (10.5, 7.0) | 4.11 dt (10.5, 7.0) | 4.15 dt (10.5, 7.0) |
| 4.24 dt (10.5, 7.0) | 4.23 dt (10.5, 7.0) | 4.27 dt (10.5, 7.0) | |
| β | 2.85 t (7.0) | 2.85 t (7.0) | 2.86 t (7.0) |
| 1′ | 4.80 d (7.5) | 4.80 d (8.0) | 4.80 d (8.0) |
| 2′ | 3.28–3.36 m | 3.29–3.35 m | 3.28–3.36 m |
| 3′ | 3.42 dd (8.5, 8.5) | 3.41 dd (9.0, 8.5) | 3.41 dd (8.5, 8.5) |
| 4′ | 3.28–3.36 m | 3.29–3.35 m | 3.28–3.36 m |
| 5′ | 3.28–3.36 m | 3.29–3.35 m | 3.28–3.36 m |
| 6′ | 3.66 dd (12.0, 5.5) | 3.67 dd (12.0, 5.5) | 3.66 dd (12.0, 5.5) |
| 3.88 dd (12.0, 1.5) | 3.88 dd (12.0, 1.5) | 3.88 dd (12.0, 1.5) | |
| 2′′ | 7.15 d (9.0) | 7.15 d (9.0) | 6.86 d (2.0) |
| 3′′ | 6.85 d (9.0) | 6.85 d (9.0) | |
| 5′′ | 6.85 d (9.0) | 6.85 d (9.0) | 6.88 d (8.5) |
| 6′′ | 7.15 d (9.0) | 7.15 d (9.0) | 6.79 dd (8.5, 2.0) |
| OMe-11 | 3.71 s | ||
| OMe-3′′ | 3.82 s | ||
| OMe-4′′ | 3.76 s | 3.76 s | 3.80 s |
| Position | 1 a | 2 a | 3 a |
|---|---|---|---|
| 1 | 95.3 | 95.0 | 95.3 |
| 3 | 155.3 | 152.8 | 155.3 |
| 4 | 109.5 | 110.2 | 109.5 |
| 5 | 32.0 | 31.9 | 32.0 |
| 6 | 41.3 | 41.3 | 41.4 |
| 7 | 173.4 | 173.3 | 173.4 |
| 8 | 125.1 | 124.6 | 125.0 |
| 9 | 130.5 | 130.7 | 130.6 |
| 10 | 13.7 | 13.6 | 13.7 |
| 11 | 168.9 | 171.0 | 168.8 |
| α | 67.0 | 66.9 | 66.9 |
| β | 35.2 | 35.2 | 35.7 |
| 1′ | 101.0 | 101.0 | 101.0 |
| 2′ | 74.9 | 74.9 | 74.9 |
| 3′ | 78.1 | 78.1 | 78.1 |
| 4′ | 71.7 | 71.6 | 71.6 |
| 5′ | 78.6 | 78.6 | 78.6 |
| 6′ | 62.9 | 62.9 | 62.9 |
| 1′′ | 131.5 | 131.5 | 132.4 |
| 2′′ | 131.2 | 131.2 | 114.1 |
| 3′′ | 115.1 | 115.1 | 150.5 |
| 4′′ | 160.0 | 160.0 | 149.3 |
| 5′′ | 115.1 | 115.1 | 113.4 |
| 6′′ | 131.2 | 131.2 | 122.6 |
| OMe-11 | 52.1 | ||
| OMe-3′′ | 56.7 | ||
| OMe-4′′ | 55.9 | 55.9 | 56.7 |
| Compounds | Superoxide Anion | Elastase |
|---|---|---|
| IC50 [µg/mL] b or (Inh %) c | ||
| (8E)-4′′-O-Methylligstroside (1) | 0.08 ± 0.01 *** | 2.57 ± 0.76 *** |
| (8E)-4′′-O-Methyldemethylligstroside (2) | 2.66 ± 0.33 *** | (42.92 ± 4.45) *** |
| 3′′,4′′-Di-O-methyldemethyloleuropein (3) | 5.22 ± 2.34 *** | (33.78 ± 1.64) *** |
| Olenoside A (4) | (8.67 ± 1.62) ** | (19.87 ± 2.94) ** |
| (8E)-Ligstroside (5) | (1.30 ± 1.88) | (26.58 ± 3.94) ** |
| Oleuropein (6) | 2.90 ± 0.46 | (23.76 ± 0.50) *** |
| (8E)-3′′,4′′-Di-O-methyloleuropein (7) | (11.34 ± 6.05) * | (32.73 ± 4.35) ** |
| Jaspolyside methyl ester (8) | (14.39 ± 3.28) * | (20.54 ± 2.24) *** |
| Esculetin (9) | 0.17 ± 0.03 | 2.41 ± 0.60 |
| Copoletin (10) | (−0.91 ± 1.16) | (8.98 ± 1.68) ** |
| Soscopoletin (11) | 5.20 ± 1.52 | 3.23 ± 0.68 |
| Esculetin dimethyl ester (12) | 7.65 ± 1.62 | (8.95 ± 2.94) * |
| Raxidin (13) | (8.95 ± 2.94) * | (12.14 ± 1.91) ** |
| Raxetin (14) | 0.19 ± 0.01 | 0.50 ± 0.10 |
| Umbelliferone (15) | (3.38 ± 1.99) | (27.92 ± 4.88) |
| Methyl isoferulate (16) | (9.03 ± 1.65) ** | (-2.76 ± 0.84) * |
| Methyl ferulate (17) | (23.02 ± 4.18) ** | (24.12 ± 4.58) ** |
| Methyl 3,4-dimethoxycinnamate (18) | (42.90 ± 3.97) *** | (7.05 ± 0.68) *** |
| Methyl (E)-p-coumarate (19) | (8.01 ± 0.66) *** | (20.30 ± 3.37) ** |
| (E)-Ferulaldehyde (20) | (31.40 ± 7.95) ** | (38.61 ± 3.64) *** |
| Tyrosol (21) | 4.93 ± 0.19 | 2.64 ± 0.22 |
| 4-Hydroxyphenethyl acetate (22) | 2.50 ± 0.35 | 3.03 ± 0.48 |
| p-Hydroxybenzaldehyde (23) | (16.16 ± 2.03) ** | (24.35 ± 4.45) ** |
| (+)-Pinoresinol (24) | 2.01 ± 0.38 | (42.37 ± 2.06) *** |
| (+)-Salicifoliol (25) | (3.70 ± 2.59) | (9.14 ± 1.58) ** |
| α-Tocopheryl quinone (26) | (17.88 ± 2.82) ** | (11.35 ± 4.41) |
| Diphenyleneiodonium (DPI) d | 0.52 ± 0.19 *** | - |
| Phenylmethylsulfonyl fluoride (PMSF) d | - | 34.4 ± 5.1 *** |
| Compounds | IC50 [μM] a |
|---|---|
| (8E)-4′′-O-Methylligstroside (1) | 12.38 ± 0.86 * |
| (8E)-4′′-O-Methyldemethylligstroside (2) | 24.72 ± 1.25 ** |
| 3′′,4′′-Di-O-methyldemethyloleuropein (3) | 37.14 ± 2.51 * |
| Olenoside A (4) | >100 |
| (8E)-Ligstroside (5) | 42.78 ± 3.23 * |
| Oleuropein (6) | 40.02 ± 2.69 * |
| (8E)-3′′,4′′-Di-O-methyloleuropein (7) | 53.44 ± 4.19 |
| Jaspolyside methyl ester (8) | 65.82 ± 5.64 |
| Esculetin (9) | 9.36 ± 0.25 ** |
| Copoletin (10) | 53.05 ± 3.63 * |
| Soscopoletin (11) | 15.36 ± 0.81 * |
| Esculetin dimethyl ester (12) | 31.80 ± 2.17 * |
| Raxidin (13) | 50.62 ± 3.08 * |
| Raxetin (14) | 10.11 ± 0.47 * |
| Umbelliferone (15) | 48.24 ± 3.22 |
| Methyl isoferulate (16) | >100 |
| Methyl ferulate (17) | >100 |
| Methyl 3,4-dimethoxycinnamate (18) | 75.84 ± 6.28 |
| Methyl (E)-p-coumarate (19) | >100 |
| (E)-Ferulaldehyde (20) | 67.38 ± 4.09 |
| Tyrosol (21) | 27.11 ± 1.87 * |
| 4-Hydroxyphenethyl acetate (22) | 35.36 ± 2.54 * |
| p-Hydroxybenzaldehyde (23) | 55.13 ± 4.25 |
| (+)-Pinoresinol (24) | 41.69 ± 3.02 * |
| (+)-Salicifoliol (25) | >100 |
| α-Tocopheryl quinone (26) | >100 |
| Quercetin b | 33.95 ± 2.34 * |
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.-C.; Wang, S.-W.; Chen, C.-Y.; Hwang, T.-L.; Cheng, M.-J.; Sung, P.-J.; Liao, K.-W.; Chen, J.-J. Secoiridoid Glucosides and Anti-Inflammatory Constituents from the Stem Bark of Fraxinus chinensis. Molecules 2020, 25, 5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245911
Chang H-C, Wang S-W, Chen C-Y, Hwang T-L, Cheng M-J, Sung P-J, Liao K-W, Chen J-J. Secoiridoid Glucosides and Anti-Inflammatory Constituents from the Stem Bark of Fraxinus chinensis. Molecules. 2020; 25(24):5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245911
Chicago/Turabian StyleChang, Hao-Chiun, Shih-Wei Wang, Chin-Yen Chen, Tsong-Long Hwang, Ming-Jen Cheng, Ping-Jyun Sung, Kuang-Wen Liao, and Jih-Jung Chen. 2020. "Secoiridoid Glucosides and Anti-Inflammatory Constituents from the Stem Bark of Fraxinus chinensis" Molecules 25, no. 24: 5911. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25245911