
Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pre-Docking Preparation
2.2. Molecular Docking Simulation of the Fragment
2.3. Fragment Growing
2.4. Molecular Docking Simulation of the Ligand
2.5. ADME-Tox Analysis
3. Results
3.1. Initial Toxicological Screening
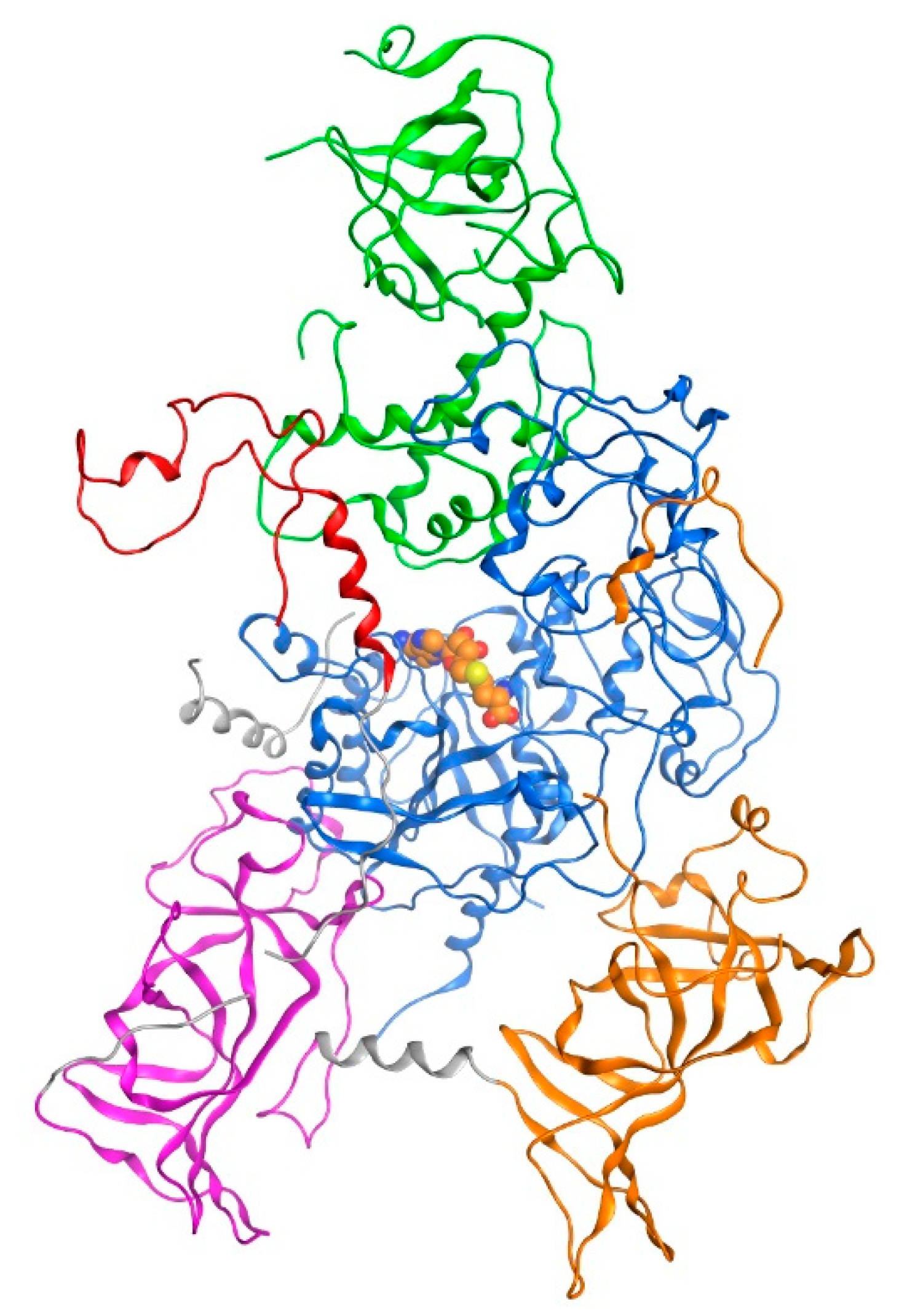
3.2. Molecular Docking Simulation of the Fragment
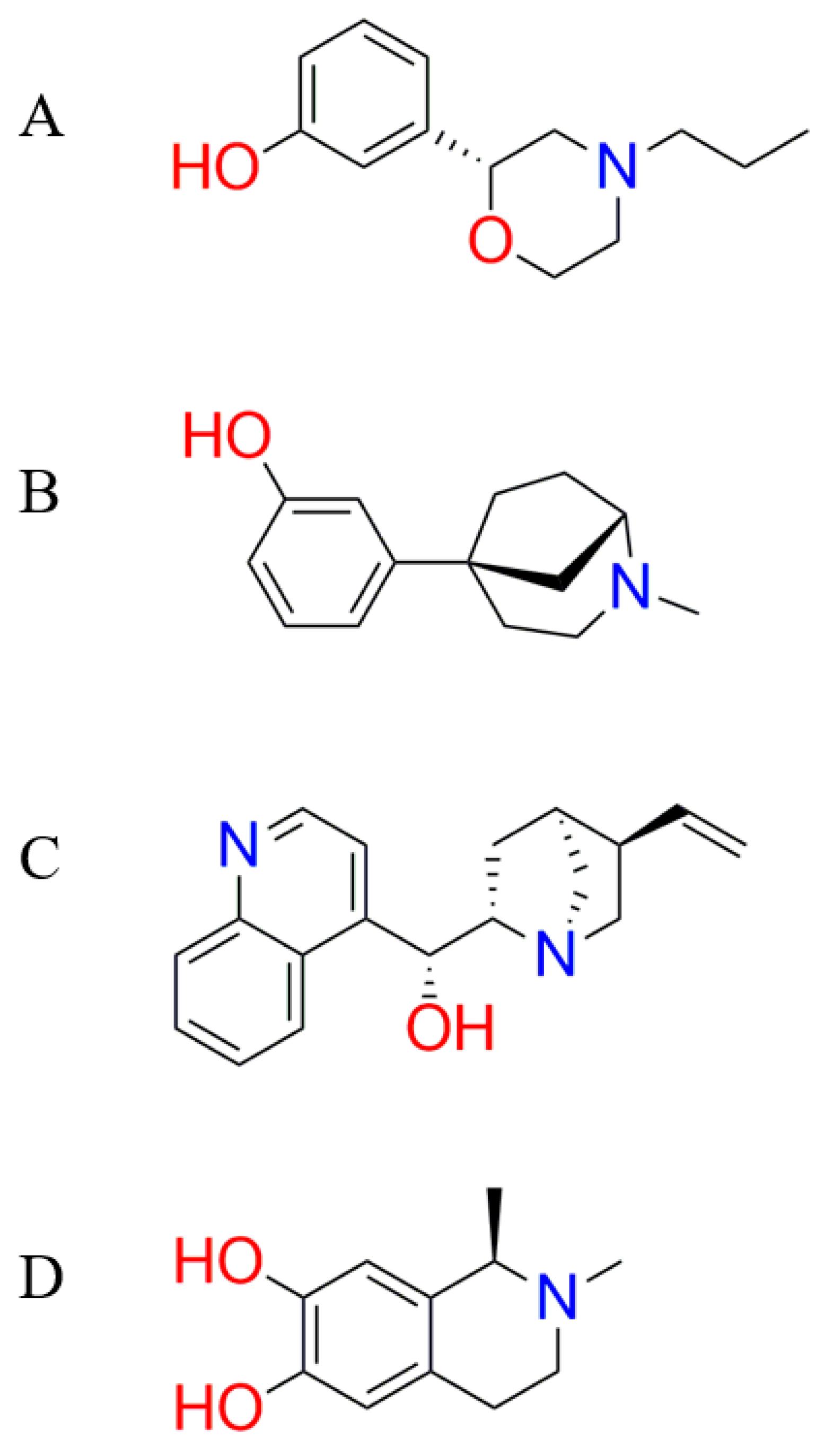
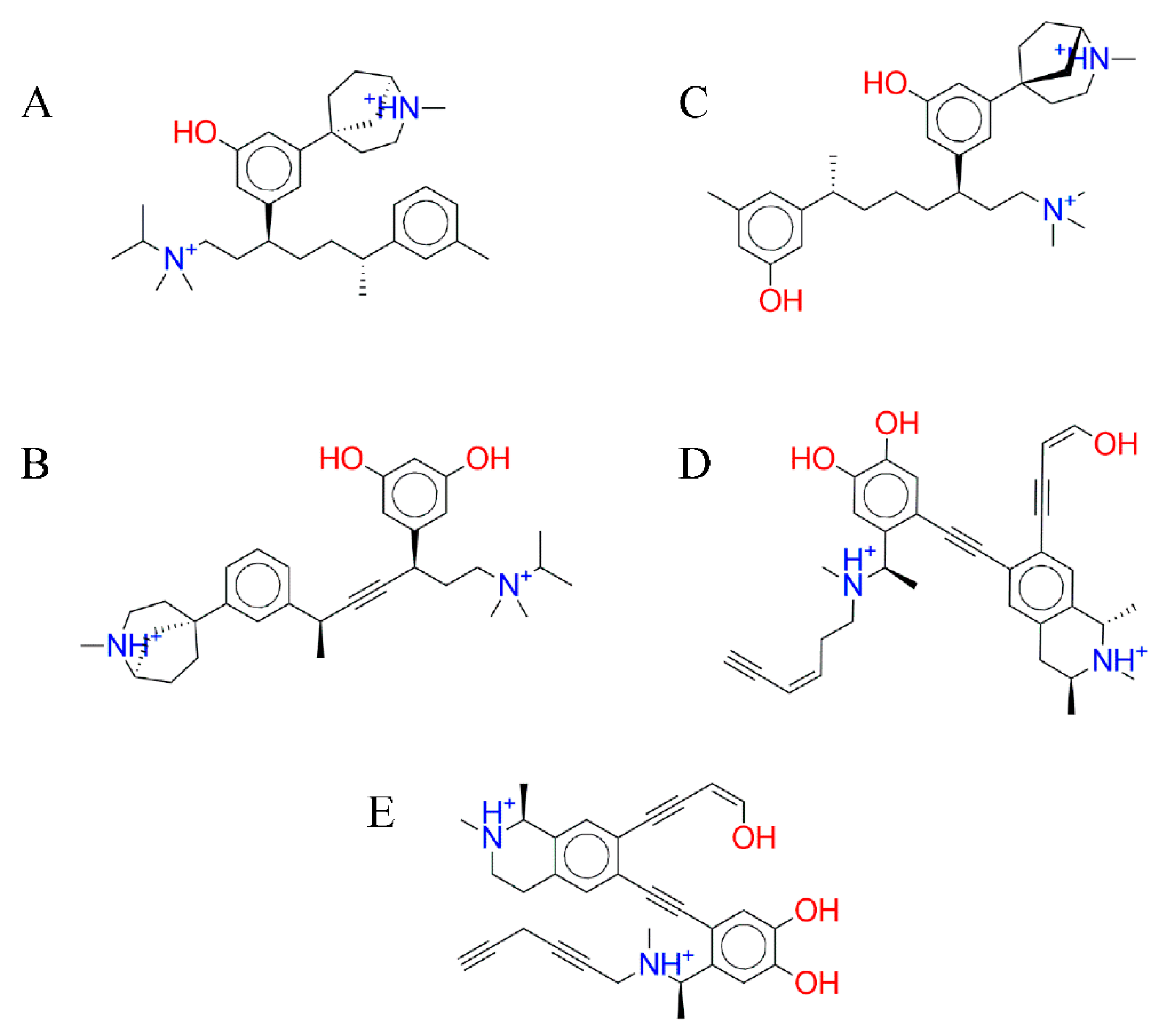
3.3. Fragment Growing
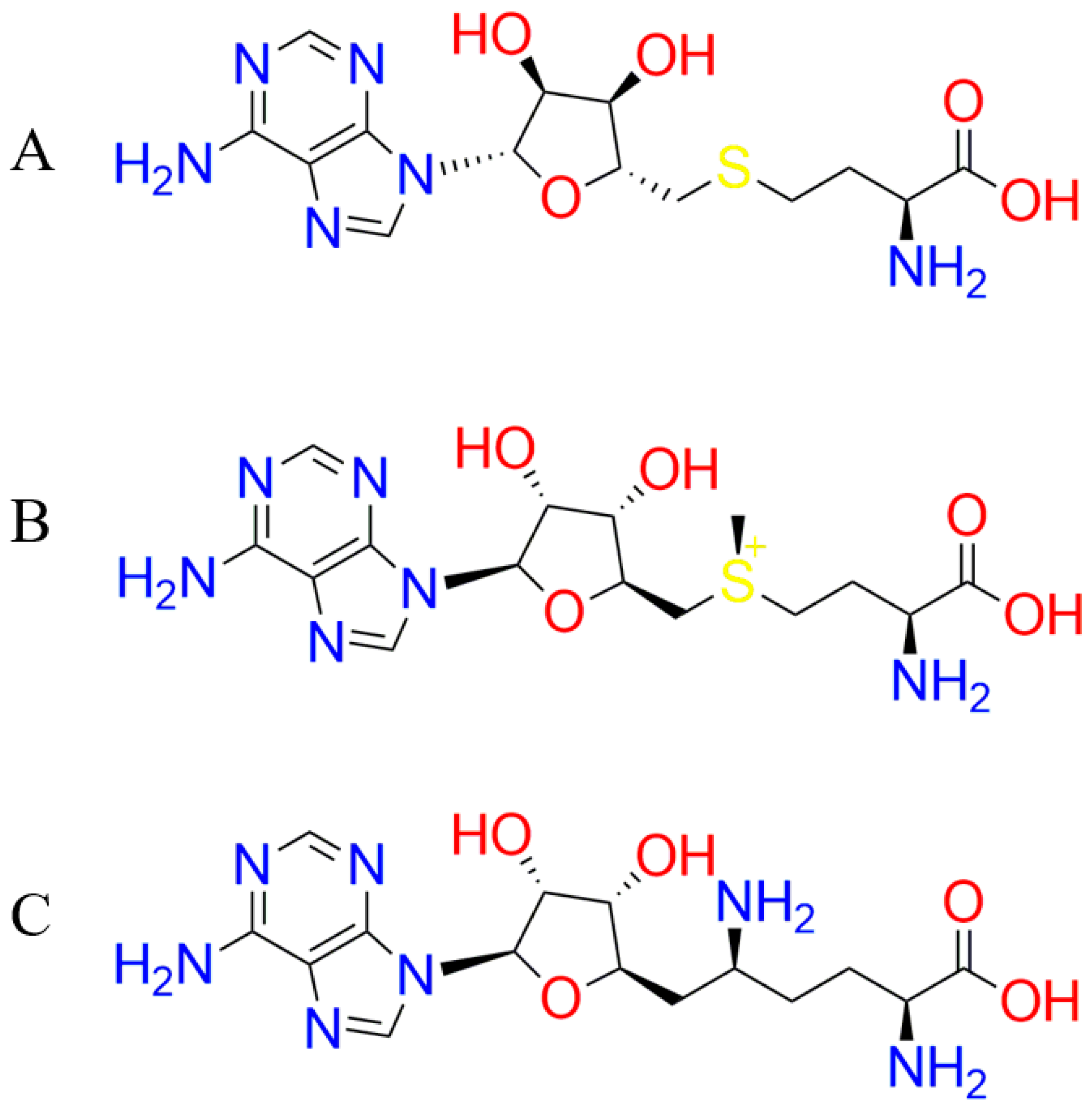
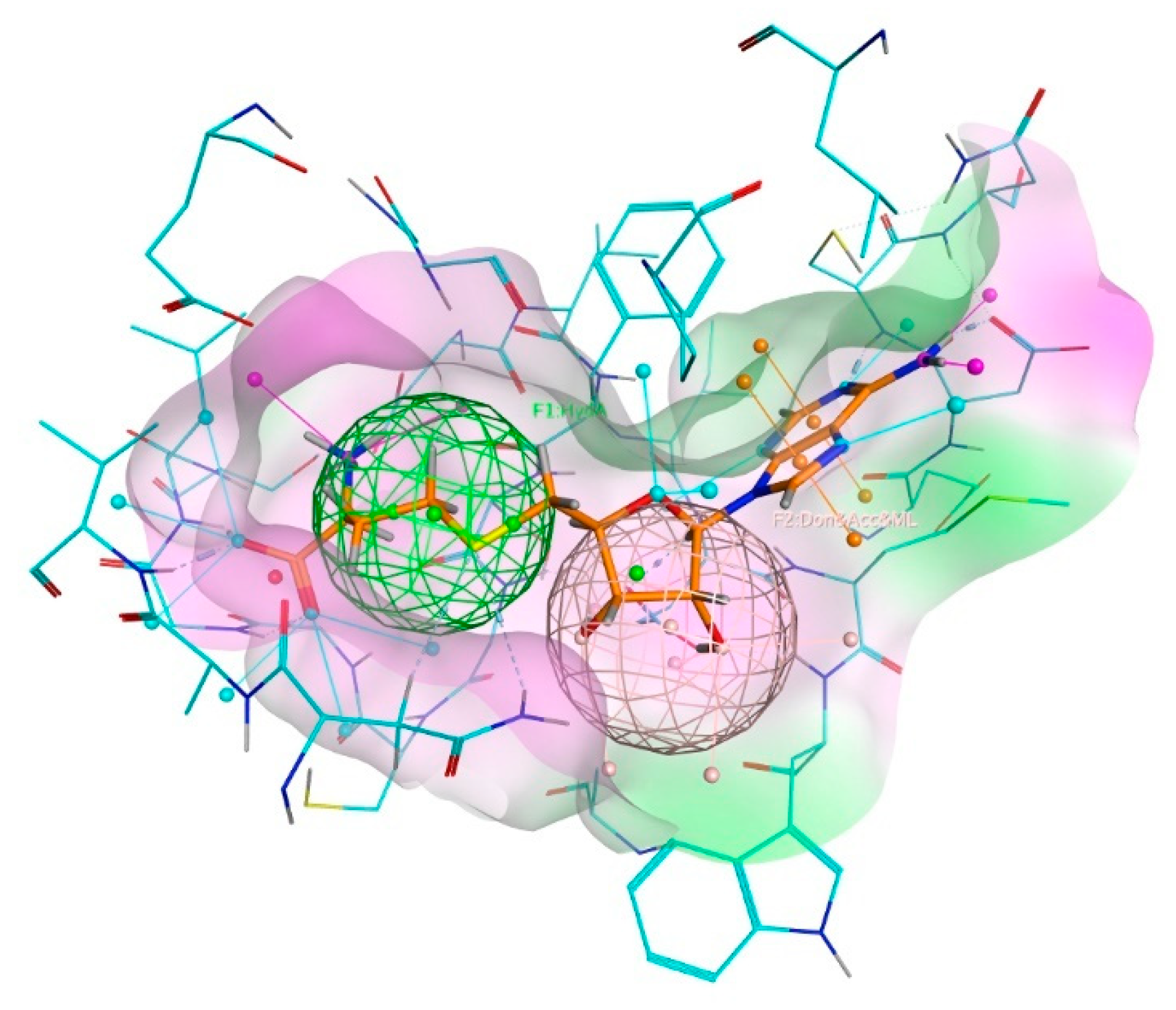
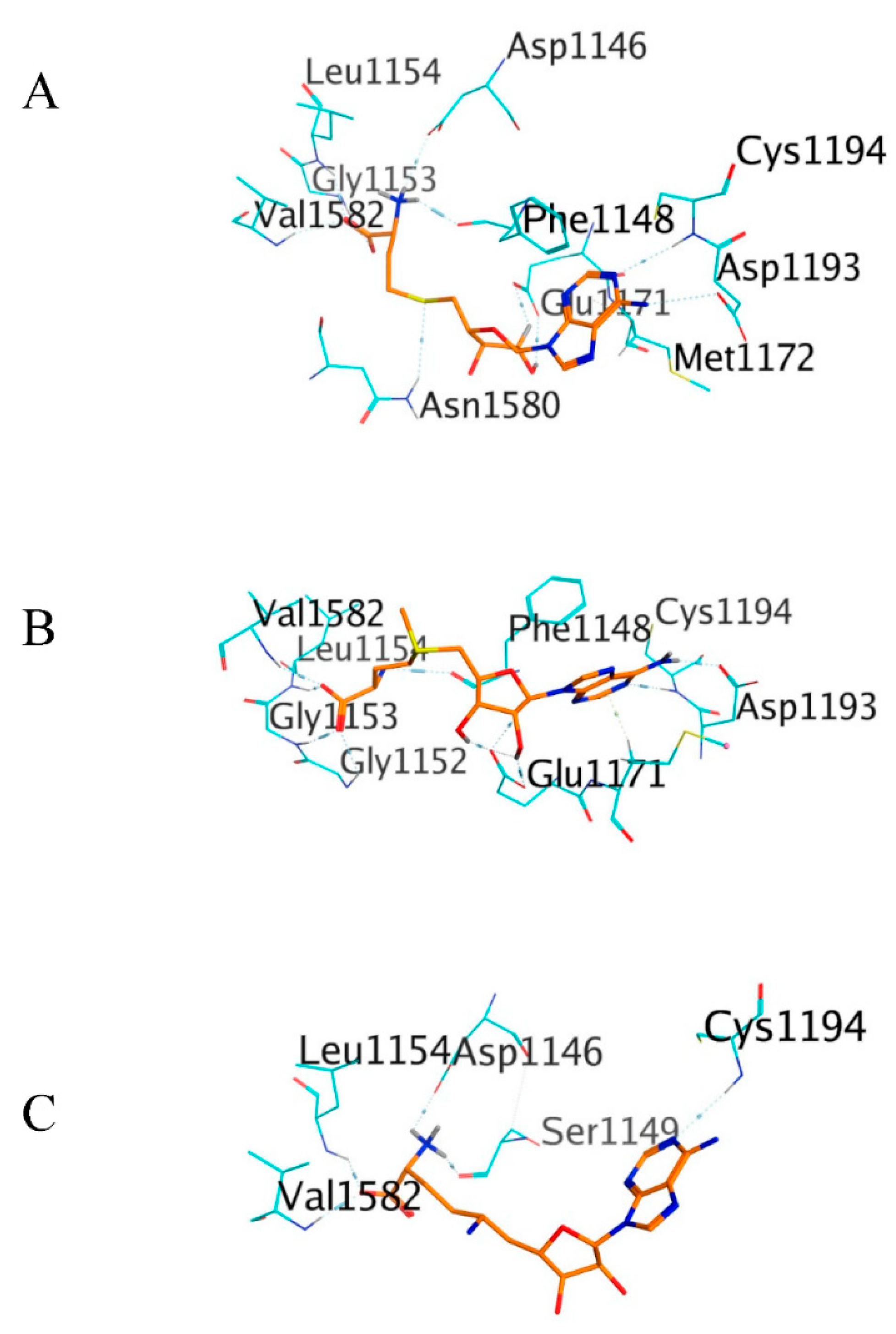
3.4. Molecular Docking Simulation of the Ligand
3.5. ADME-Tox Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Florin-Andrei Taran, A.; Schneeweiss, A.; Lux, M.P.; Janni, W.; Hartkopf, A.D.; Nabieva, N.; Overkamp, F.; Kolberg, H.-C.; Hadji, P.; Tesch, H.; et al. Update Breast Cancer 2018 (Part 1)—Primary Breast Cancer and Biomarkers. Geburtshilfe Frauenheilkd. 2018. [Google Scholar] [CrossRef] [Green Version]
- World Health Organisation. Latest Global Cancer Data. Available online: https://www.who.int/cancer/PRGlobocanFinal.pdf (accessed on 12 September 2018).
- World Health Organisation. Global Cancer Observatory (GLOBOCAN). Available online: https://gco.iarc.fr/ (accessed on 12 September 2018).
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Das, S.K. Epigenetic regulations through DNA methylation and hydroxymethylation: Clues for early pregnancy in decidualization. Biomol. Concepts 2014, 5, 95–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Lin, K.; Luo, Y.; Perry, J.; Wang, Y.; Song, J. Crystal Structure of Human DNA Methyltransferase 1. HHS Public Access 2015, 35, 2520–2531. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.H.P.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [Green Version]
- Khandige, S.; Shanbhogue, V.V.; Chakrabarty, S.; Kapettu, S. Methylation Markers: A Potential Force Driving Cancer Diagnostics Forward. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2011, 19, 105–110. [Google Scholar] [CrossRef]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-methylated CpG islands associated with gene silencing in the adult body tissues of the ogye chicken using RNA-Seq and reduced representation bisulfite sequencing. Front. Genet. 2019. [Google Scholar] [CrossRef]
- Janitz, K.; Janitz, M. Assessing Epigenetic Information, 1st ed.; Elsevier Inc.: London, UK, 2011; ISBN 9780123757098. [Google Scholar]
- Oberley, M.J.; Farnham, P.J. Probing Chromatin Immunoprecipitates with CpG-Island Microarrays to Identify Genomic Sites Occupied by DNA-Binding Proteins. Methods Enzymol. 2003, 371, 577–596. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. 7 DNA methylation analysis of human cancer. In Handbook of Immunohistochemistry and in Situ Hybridization of Human Carcinomas; Elsevier Inc.: Burlington, NJ, USA, 2005; Volume 3, pp. 65–78. ISBN 9780120884049. [Google Scholar]
- el-Deiry, W.S.; Nelkin, B.D.; Celano, P.; Yen, R.W.; Falco, J.P.; Hamilton, S.R.; Baylin, S.B. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc. Natl. Acad. Sci. USA 1991, 88, 3470–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Bindseil, K.U.; Jakupovic, J.; Wolf, D.; Lavayre, J.; Leboul, J.; van der Pyl, D. Pure compound libraries; a new perspective for natural product based drug discovery. Drug Discov. Today 2001, 6, 840–847. [Google Scholar] [CrossRef]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [Green Version]
- Martinet, N.; Michel, B.Y.; Bertrand, P.; Benhida, R. Small molecules DNA methyltransferases inhibitors. Medchemcomm 2011, 3, 263. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development ( CADDD ): In silico -chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinforma. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- El-Saadi, M.W.; Williams-Hart, T.; Salvatore, B.A.; Mahdavian, E. Use of in-silico assays to characterize the ADMET profile and identify potential therapeutic targets of fusarochromanone, a novel anti-cancer agent. Silico Pharmacol. 2015, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, K.; Suetake, I.; Yamashita, E.; Suga, M.; Narita, H.; Nakagawa, A.; Tajima, S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011. [Google Scholar] [CrossRef] [Green Version]
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Contrera, J.F. Validation of Toxtree and SciQSAR in silico predictive software using a publicly available benchmark mutagenicity database and their applicability for the qualification of impurities in pharmaceuticals. Regul. Toxicol. Pharmacol. 2013, 67, 285–293. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of three” for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C.; Jeliazkova, N.; Netzeva, T.; Worth, A. The Benigni/Bossa Rulebase for Mutagenicity and Carcinogenicity—A Module of Toxtree; JRC Science Technology Reports: Luxembourg, 2008; pp. 1–70. [Google Scholar]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for evaluating chemical ADMET properties. J. Chem. Inf. Model. 2012, 11, 3099–3105. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided. Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef]
- López-Camacho, E.; García-Godoy, M.J.; García-Nieto, J.; Nebro, A.J.; Aldana-Montes, J.F. A new multi-objective approach for molecular docking based on rmsd and binding energy. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Spinger: Cham, Switzerland, 2016; Volume 9702, pp. 65–77. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoffin, R.; Slater, M. The Virtual Elaboration of Fragment Ideas: Growing, Merging and Linking Fragments with Realistic Chemistry. Drug Discov. Dev. Deliv. 2015, 7, 2–5. [Google Scholar]
- Patlewicz, G.; Rodford, R.; Walker, J.D. Quantitative structure-activity relationships for predicting mutagenicity and carcinogenicity. Environ. Toxicol. Chem. 2003, 22, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Cheng, F.; Chen, L.; Du, Z.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of chemical Ames mutagenicity. J. Chem. Inf. Model. 2012, 52, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Pillans, P. P-glycoprotein and its role in drug-drug interactions. Australianprescriber 2014, 37, 137–139. [Google Scholar] [CrossRef]
- He, J.; Peng, T.; Yang, X.; Liu, H. Development of QSAR models for predicting the binding affinity of endocrine disrupting chemicals to eight fish estrogen receptor. Ecotoxicol. Environ. Saf. 2018, 148, 211–219. [Google Scholar] [CrossRef]
- Levine, R.R. Factors affecting gastrointestinal absorption of drugs. Am. J. Dig. Dis. 1970, 15, 171–188. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J.; Egan, W.J.; Kenneth, M.; Merz, J.; Baldwin, J.J.; Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the assay: Chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.; Lee, Y.K.; Koo, J.S. Differential expression of the epigenetic methylation-related protein DNMT1 by breast cancer molecular subtype and stromal histology. J. Transl. Med. 2016, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Saldívar-González, F.I.; Gómez-García, A.; Chávez-Ponce De León, D.E.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Inhibitors of DNA methyltransferases from natural sources: A computational perspective. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Svedružić, Ž.M. Dnmt1: Structure and Function; Elsevier: Amsterdam, The Netherlands, 2011; Volume 101, ISBN 9780123876850. [Google Scholar]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzębski, Z.; Flis, S. DNA Methyltransferase inhibitor and their emerging role in epigenetic therapu of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar]
- Kumar, A.; Voet, A.; Zhang, K.Y.J. Fragment Based Drug Design: From Experimental to Computational Approaches. Curr. Med. Chem. 2012, 19, 5128–5147. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Giri, S.; Bader, A. A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov. Today 2015, 20, 37–49. [Google Scholar] [CrossRef]
- Myers, S.; Baker, A. Drug discovery—An operating model for a new era. Nat. Biotechnol. 2001. [Google Scholar] [CrossRef]
- Noori, H.R.; Spanagel, R. In silico pharmacology: Drug design and discovery’s gate to the future. Silico Pharmacol. 2013, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Front. Chem. 2018, 6. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Chapter 5—Analysis of the Content of SAR Databases BT—Ligand Efficiency Indices for Drug Discovery. In Expert Opinion on Drug Discovery; Taylor Francis Online: London, UK, 2013; ISBN 978-0-12-404635-1. [Google Scholar]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.E.; Jones, B.C. Human cytochromes P450 and their role in metabolism-based drug-drug interactions. In Drug-Drug Interactions; Marcel Dekker: New York, NY, USA, 2002; p. 2. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Classification | Natural Product Compounds Collected from the PubChem Database | Natural Product Compounds which Passed the Initial Pharmacological Filters |
|---|---|---|
| Alkaloids | 39,887 | 1949 |
| Flavonoids | 20,393 | 191 |
| Phenolics | 46,268 | 138 |
| Saponins | 5077 | 2 |
| Tannins | 1042 | 0 |
| Terpenes | 55,979 | 321 |
| Fragment | Number of Ligands Generated from DataWarrior Software | Ligands which Passed the Toxicological Screening |
|---|---|---|
| 3-[(2R)-4-propyl-2-morpholinyl]phenol | 12,800 | 163 |
| 3-(2-methyl-2-azabicyclo[3.2.1]oct-5-yl)phenol | 12,800 | 8041 |
| α-quinidine | 12,800 | 3143 |
| (R)-N-methylsalsolinol | 12,800 | 2216 |
| No | Compound Name | Virtual Screening | Rigid Docking | Flexible Docking | |||
|---|---|---|---|---|---|---|---|
| ΔGbinding | RMSD | ΔGbinding | RMSD | ΔGbinding | RMSD | ||
| 1 | C-7756 | −12.89 | 1.047 | −12.97 | 1.529 | −14.45 | 1.563 |
| 2 | C-5769 | −10.11 | 1.210 | −10.96 | 1.932 | −13.17 | 1.560 |
| 3 | C-1723 | −10.23 | 1.723 | −10.80 | 1.831 | −12.51 | 1.691 |
| 4 | C-2129 | −9.71 | 1.594 | −12.04 | 1.929 | −12.43 | 1.495 |
| 5 | C-2140 | −10.32 | 0.979 | −11.88 | 1.686 | −12.42 | 1.685 |
| S1 | SAH | −9.19 | 1.322 | −10.52 | 1.608 | −11.02 | 1.563 |
| S2 | SAM | −5.64 | 2.347 | −11.69 | 2.564 | −12.05 | 2.372 |
| S3 | SFG | −8.33 | 1.396 | −10.54 | 1.493 | −10.67 | 1.492 |
| No | Compound Name | Molecular Weight (Da) | log P | H- Don | H- Acc | TPSA (Å2) | Rotatable Bond | Druglikeness | Drug Score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C-7756 | 492.78 | 1.97 | 2 | 3 | 24.67 | 10 | 1.45 | 0.41 |
| 2 | C-5769 | 490.72 | 0.91 | 3 | 4 | 44.9 | 8 | −2.25 | 0.23 |
| 3 | C-1723 | 494.76 | 1.31 | 3 | 4 | 44.9 | 10 | −1.11 | 0.34 |
| 4 | C-2129 | 496.64 | 1.57 | 5 | 5 | 69.57 | 7 | −0.61 | 0.33 |
| 5 | C-2140 | 494.63 | 1.46 | 5 | 5 | 69.57 | 6 | −0.89 | 0.24 |
| S1 | SAH | 384.41 | −3.72 | 4 | 11 | 212.38 | 7 | −18.44 | 0.42 |
| S2 | SAM | 399.45 | −3.93 | 4 | 11 | 187.08 | 7 | −9.09 | 0.39 |
| S3 | SFG | 382.40 | −3.95 | 5 | 12 | 214.72 | 7 | −19.10 | 0.42 |
| No | Compound Name | Genotoxic Carcinogenicity | Non-Genotoxic Carcinogenicity | QSAR Carcinogenicity | Mutagenicity (Salmonella Typhimurium) |
|---|---|---|---|---|---|
| 1 | C-7756 | No | No | No | No |
| 2 | C-5769 | No | No | No | No |
| 3 | C-1723 | No | No | No | No |
| 4 | C-2129 | No | No | No | No |
| 5 | C-2140 | No | No | No | No |
| S1 | SAH | Yes | No | No | No |
| S2 | SAM | Yes | No | No | No |
| S3 | SFG | Yes | No | No | No |
| No | Compound Name | P-gp Substrate/Inhibi tor | CYP450 Substrate/Inhibi tor | CYP Inhibitory Promiscuity | Biodegradable | AMES Toxicity | Carcinogenicity |
|---|---|---|---|---|---|---|---|
| 1 | C-7756 | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| 2 | C-5769 | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| 3 | C-1723 | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| 4 | C-2129 | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| 5 | C-2140 | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| S1 | SAH | Substrate, non-inhibitor | Non-substrate, non-inhibitor | Low | Not Ready | None | None |
| S2 | SAM | Substrate, non-inhibitor | Substrate (CYP450 3A4), non-inhibitor | Low | Not Ready | None | None |
| S3 | SFG | Non-substrate, non-inhibitor | Non-substrate, non-inhibitor | Low | Not Ready | None | None |
| No | Compound Name | GI Absorption | Lipinski | Veber | Egan | Bioavailability Score | PAINS | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|---|
| 1 | C-7756 | High | 0 | 0 | 0 | 0.55 | 0 | 5.59 |
| 2 | C-5769 | High | 0 | 0 | 0 | 0.55 | 0 | 6.09 |
| 3 | C-1723 | High | 0 | 0 | 0 | 0.55 | 0 | 5.51 |
| 4 | C-2129 | High | 0 | 0 | 0 | 0.55 | 1 | 5.11 |
| 5 | C-2140 | High | 0 | 0 | 0 | 0.55 | 1 | 5.12 |
| S1 | SAH | Low | 1 | 1 | 1 | 0.55 | 0 | 4.69 |
| S2 | SAM | Low | 1 | 1 | 1 | 0.55 | 0 | 4.94 |
| S3 | SFG | Low | 1 | 1 | 1 | 0.55 | 0 | 4.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkaff, A.H.; Saragih, M.; Imana, S.N.; Nasution, M.A.F.; Tambunan, U.S.F. Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design. Molecules 2021, 26, 375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020375
Alkaff AH, Saragih M, Imana SN, Nasution MAF, Tambunan USF. Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design. Molecules. 2021; 26(2):375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020375
Chicago/Turabian StyleAlkaff, Ahmad Husein, Mutiara Saragih, Shabrina Noor Imana, Mochammad Arfin Fardiansyah Nasution, and Usman Sumo Friend Tambunan. 2021. "Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design" Molecules 26, no. 2: 375. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020375