
Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies

 ,
, 
 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Filtration of Natural Products
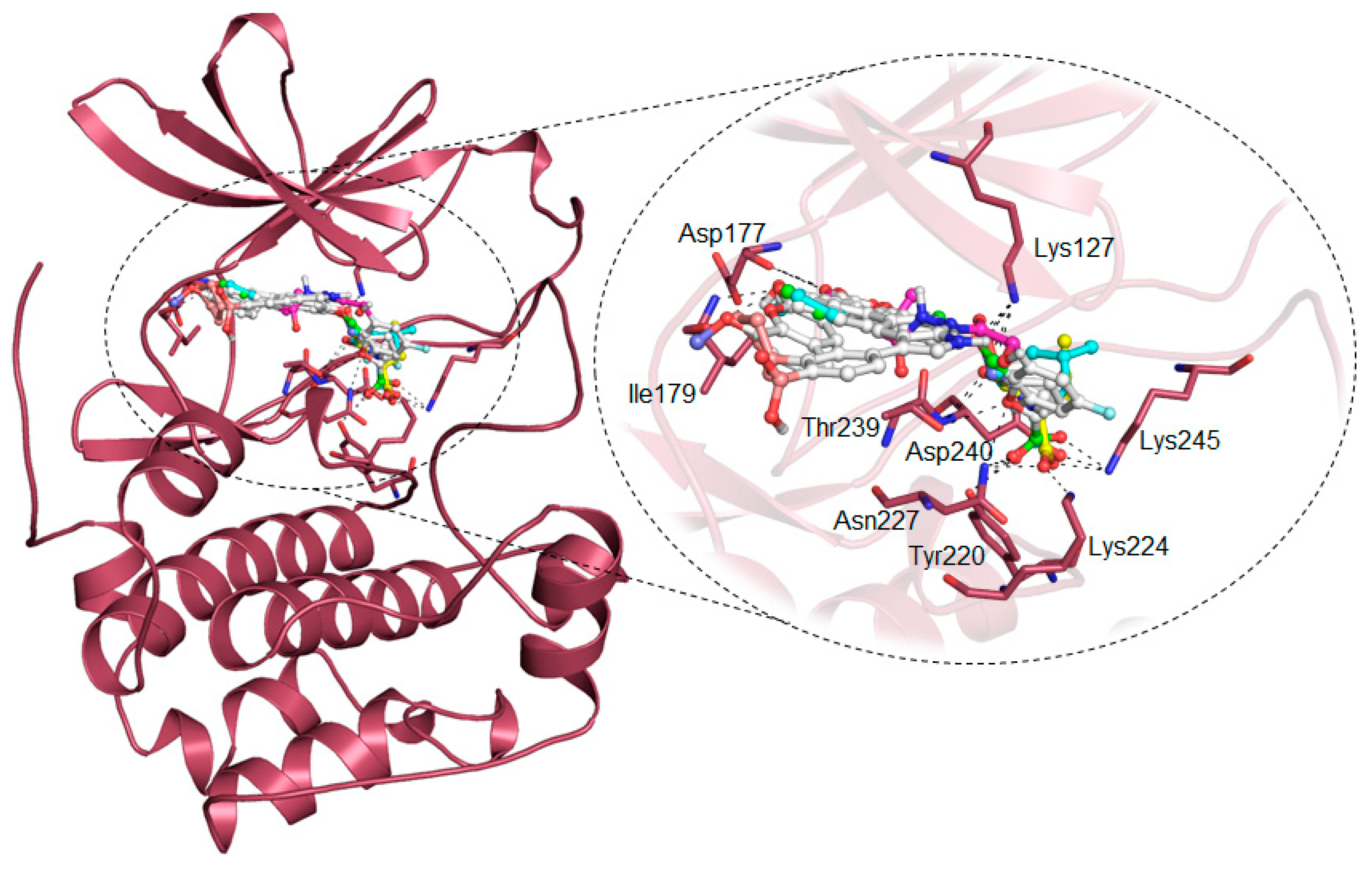
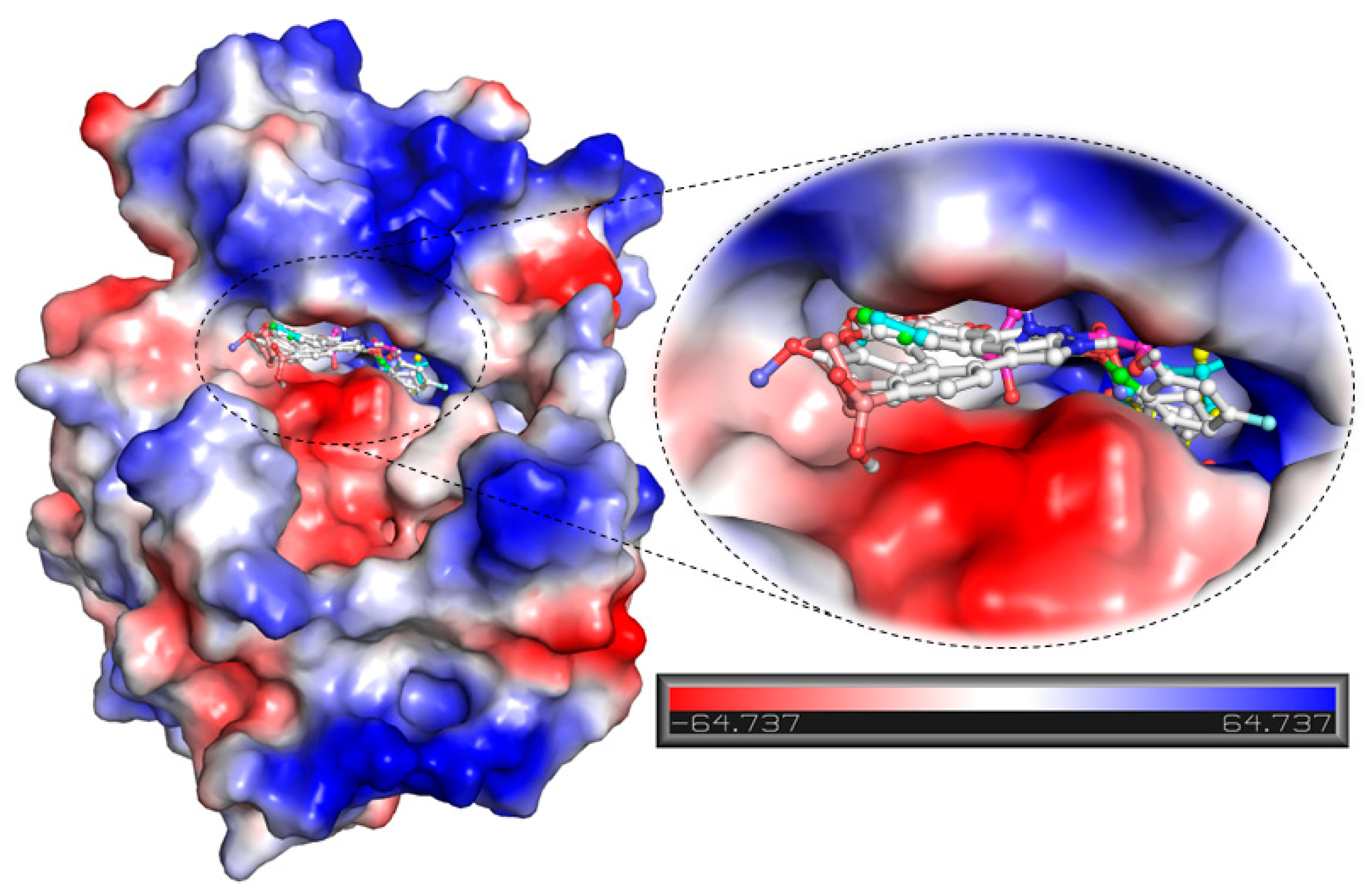
2.2. vHTS
2.3. Biological Activity Predictions
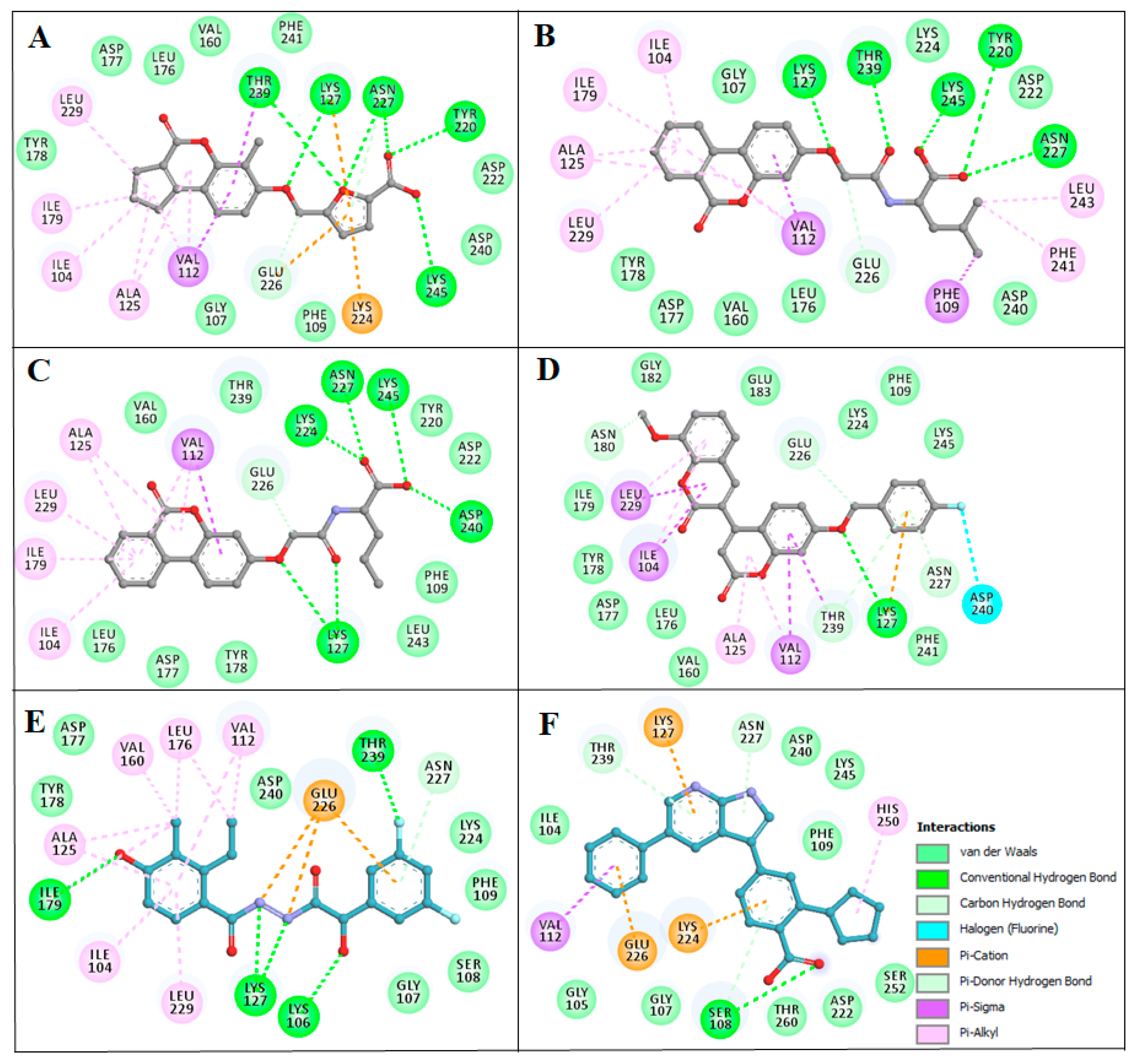
2.4. Structure-Activity Relationship of the Compounds
2.5. MD Simulations
2.6. Structural Deviations and Compactness
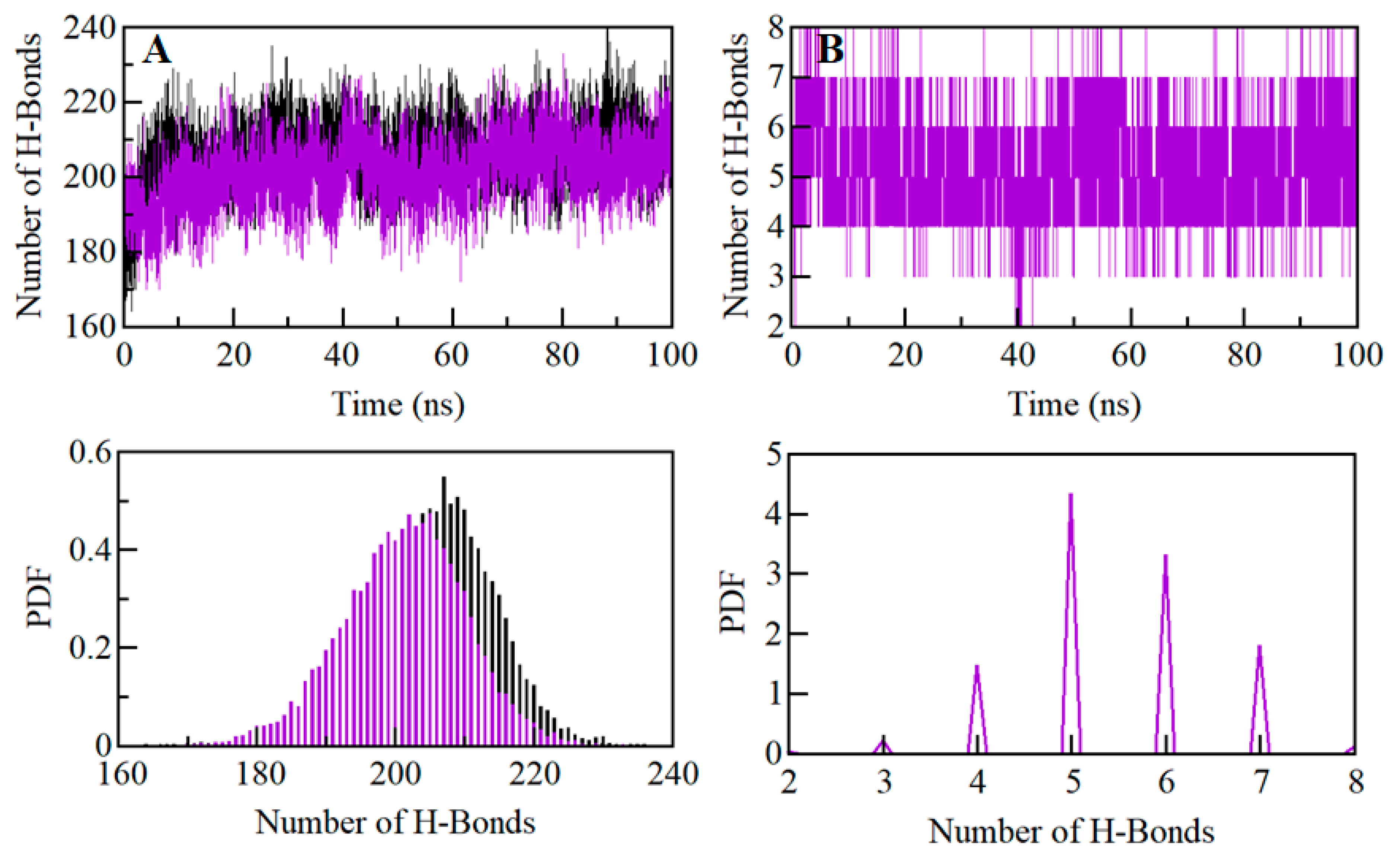
2.7. Dynamics of SGK1 Interactions: Hydrogen Bonds Analysis
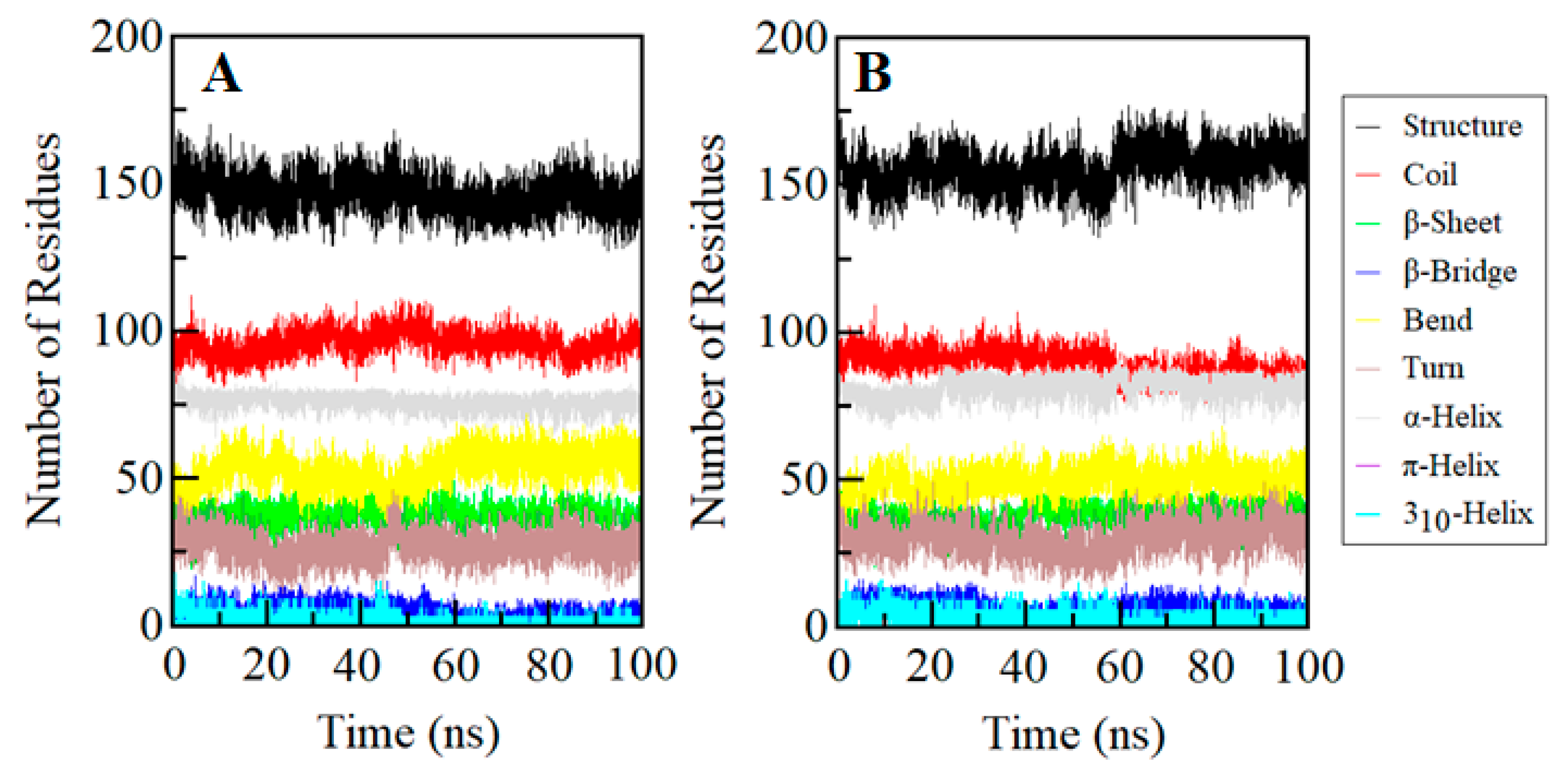
2.8. Secondary Structure Dynamics of SGK1
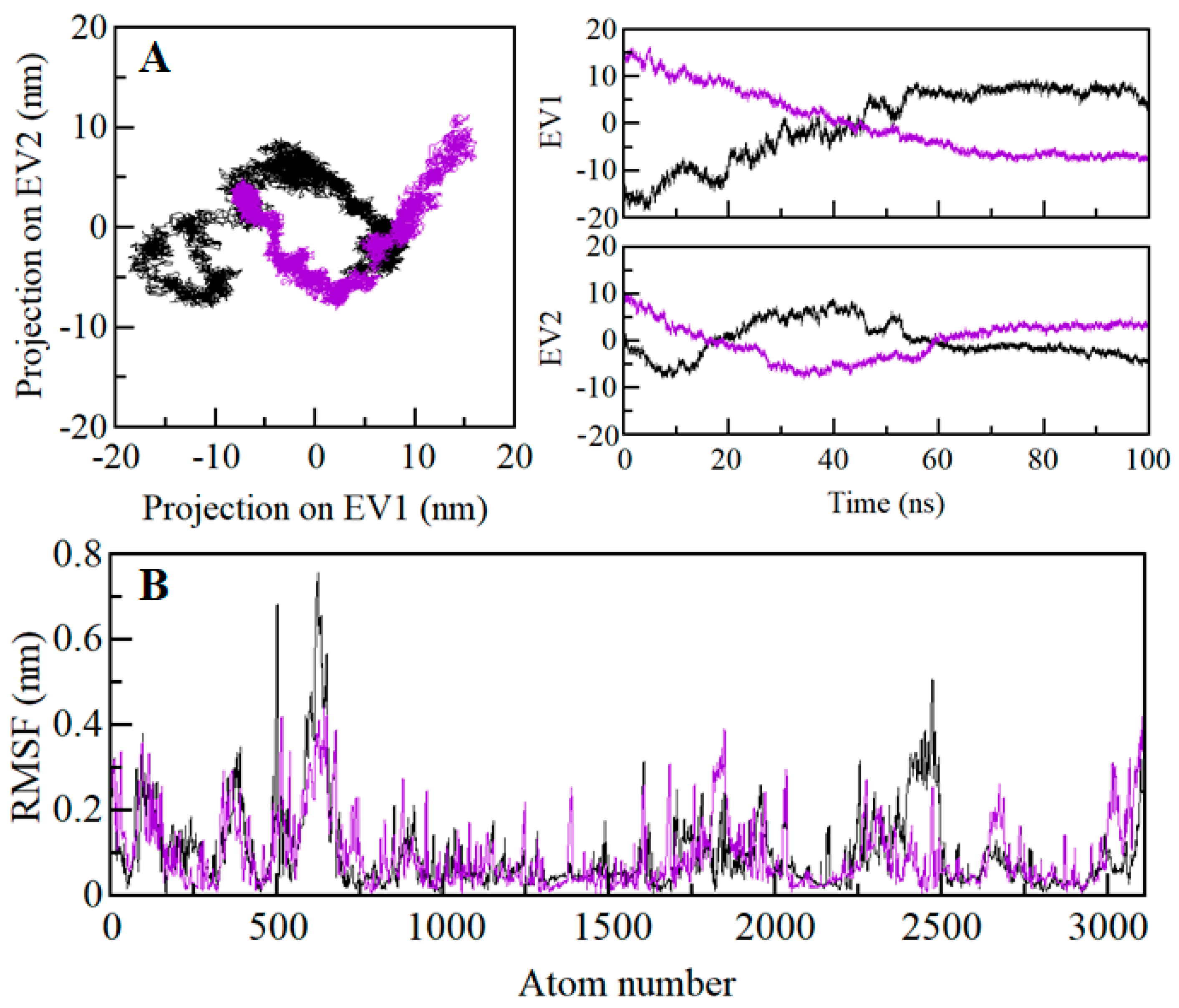
2.9. Principal Component and Free Energy Landscape Analysis
2.10. MPBSA Analysis
3. Materials and Methods
3.1. Computational Resources
3.2. Filtration of Natural Products
3.3. Structure-Based vHTS
3.4. Biological Activity Predictions of the Compounds
3.5. MD Simulations
3.6. MMPBSA Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: The rise of SGK in cancer signalling. Growth Factors 2010, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Strutz-Seebohm, N.; Seebohm, G.; Lang, U.E. Significance of SGK1 in the regulation of neuronal function. J. Physiol. 2010, 588, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Shumilina, E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. Faseb J. 2013, 27, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gamper, N.; Fillon, S.; Feng, Y.; Friedrich, B.; Lang, P.A.; Henke, G.; Huber, S.M.; Kobayashi, T.; Cohen, P.; Lang, F. K+ channel activation by all three isoforms of serum- and glucocorticoid-dependent protein kinase SGK. Pflug. Arch. 2002, 445, 60–66. [Google Scholar] [CrossRef]
- Zhao, B.; Lehr, R.; Smallwood, A.M.; Ho, T.F.; Maley, K.; Randall, T.; Head, M.S.; Koretke, K.K.; Schnackenberg, C.G. Crystal structure of the kinase domain of serum and glucocorticoid-regulated kinase 1 in complex with AMP–PNP. Protein Sci. 2007, 16, 2761–2769. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, B.A.; Cilia, S.; Maiyar, A.C.; Vaysberg, M.; Firestone, G.L. The serum-and glucocorticoid-induced protein kinase-1 (Sgk-1) mitochondria connection: Identification of the IF-1 inhibitor of the F1F0-ATPase as a mitochondria-specific binding target and the stress-induced mitochondrial localization of endogenous Sgk-1. Biochimie 2013, 95, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Valinsky, W.C.; Touyz, R.M.; Shrier, A. Aldosterone, SGK1, and ion channels in the kidney. Clin. Sci. 2018, 132, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Artunc, F.; Vallon, V. The physiological impact of the serum-and glucocorticoid-inducible kinase SGK1. Curr. Opin. Nephrol. Hypertens. 2009, 18, 439. [Google Scholar] [CrossRef]
- Lang, F.; Voelkl, J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin. Investig. Drugs 2013, 22, 701–714. [Google Scholar] [CrossRef]
- Sherk, A.B.; Frigo, D.E.; Schnackenberg, C.G.; Bray, J.D.; Laping, N.J.; Trizna, W.; Hammond, M.; Patterson, J.R.; Thompson, S.K.; Kazmin, D. Development of a small-molecule serum-and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008, 68, 7475–7483. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Lan, C.; Jiao, G.; Fu, W.; Long, X.; An, Y.; Wang, K.; Zhou, J.; Chen, T.; Li, Y. Therapeutic inhibition of SGK1 suppresses colorectal cancer. Exp. Mol. Med. 2017, 49, e399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbruzzese, C.; Mattarocci, S.; Pizzuti, L.; Mileo, A.M.; Visca, P.; Antoniani, B.; Alessandrini, G.; Facciolo, F.; Amato, R.; D’Antona, L. Determination of SGK1 mRNA in non-small cell lung cancer samples underlines high expression in squamous cell carcinomas. J. Exp. Clin. Cancer Res. 2012, 31, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, T.F.; Boini, K.M.; Beier, N.; Scholz, W.; Fuchß, T.; Lang, F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell. Physiol. Biochem. 2011, 28, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Towhid, S.T.; Liu, G.-L.; Ackermann, T.F.; Beier, N.; Scholz, W.; Fuchß, T.; Toulany, M.; Rodemann, H.-P.; Lang, F. Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cell. Physiol. Biochem. 2013, 32, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. High throughput screening, docking, and molecular dynamics studies to identify potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. J. Biomol. Struct. Dyn. 2019, 37, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, M.; Ali, S.; Khan, F.I.; Khan, P.; Taneja, P.; Hassan, M.I. Binding mechanism of caffeic acid and simvastatin to the integrin linked kinase for therapeutic implications: A comparative docking and MD simulation studies. J. Biomol. Struct. Dyn. 2019, 37, 4327–4337. [Google Scholar] [CrossRef]
- Gupta, P.; Mohammad, T.; Dahiya, R.; Roy, S.; Noman, O.M.A.; Alajmi, M.F.; Hussain, A.; Hassan, M.I. Evaluation of binding and inhibition mechanism of dietary phytochemicals with sphingosine kinase 1: Towards targeted anticancer therapy. Sci Rep. 2019, 9, 18727. [Google Scholar] [CrossRef]
- Khan, P.; Queen, A.; Mohammad, T.; Smita; Khan, N.S.; Hafeez, Z.B.; Hassan, M.I.; Ali, S. Identification of α-Mangostin as a Potential Inhibitor of Microtubule Affinity Regulating Kinase 4. J. Nat. Prod. 2019, 82, 2252–2261. [Google Scholar] [CrossRef]
- Mohammad, T.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification and evaluation of bioactive natural products as potential inhibitors of human microtubule affinity-regulating kinase 4 (MARK4). J. Biomol. Struct. Dyn. 2019, 37, 1813–1829. [Google Scholar] [CrossRef]
- Mohammad, T.; Arif, K.; Alajmi, M.F.; Hussain, A.; Islam, A.; Rehman, M.T.; Hassan, I. Identification of high-affinity inhibitors of pyruvate dehydrogenase kinase-3: Towards therapeutic management of cancer. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Mohammad, T.; Batra, S.; Dahiya, R.; Baig, M.H.; Rather, I.A.; Dong, J.-J.; Hassan, I. Identification of High-Affinity Inhibitors of Cyclin-Dependent Kinase 2 Towards Anticancer Therapy. Molecules 2019, 24, 4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Khan, F.I.; Mohammad, T.; Khan, P.; Hasan, G.M.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Exploring molecular insights into the interaction mechanism of cholesterol derivatives with the Mce4A: A combined spectroscopic and molecular dynamic simulation studies. Int. J. Biol. Macromol. 2018, 111, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Naz, F.; Khan, F.I.; Mohammad, T.; Khan, P.; Manzoor, S.; Hasan, G.M.; Lobb, K.A.; Luqman, S.; Islam, A.; Ahmad, F. Investigation of molecular mechanism of recognition between citral and MARK4: A newer therapeutic approach to attenuate cancer cell progression. Int. J. Biol. Macromol. 2018, 107, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Khan, P.; Rahman, S.; Queen, A.; Manzoor, S.; Naz, F.; Hasan, G.M.; Luqman, S.; Kim, J.; Islam, A.; Ahmad, F.; et al. Elucidation of Dietary Polyphenolics as Potential Inhibitor of Microtubule Affinity Regulating Kinase 4: In silico and In vitro Studies. Sci. Rep. 2017, 7, 9470. [Google Scholar] [CrossRef]
- Dahiya, R.; Mohammad, T.; Gupta, P.; Haque, A.; Alajmi, M.F.; Hussain, A.; Hassan, M.I. Molecular interaction studies on ellagic acid for its anticancer potential targeting pyruvate dehydrogenase kinase 3. RSC Adv. 2019, 9, 23302–23315. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Mohammad, T.; Roy, S.; Anwar, S.; Gupta, P.; Haque, A.; Khan, P.; Kazim, S.N.; Islam, A.; Ahmad, F. Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy. Int. J. Biol. Macromol. 2019, 136, 1076–1085. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- V da Costa, G.; Ferreira, E.F.; da S Ramos, R.; B da Silva, L.; MF de Sá, E.; KP da Silva, A.; M Lobato, C.; NP Souto, R.; da Silva, T.d.P.; Henrique, C. Hierarchical Virtual Screening of Potential Insectides Inhibitors of Acetylcholinesterase and Juvenile Hormone from Temephos. Pharmaceuticals 2019, 12, 61. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Bahadur, R.P.; Chakrabarti, P.; Janin, J. Hydration of protein–protein interfaces. Proteins Struct. Funct. Bioinform. 2005, 60, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; Andersen, T.; McDougal, O.M. Accessible high-throughput virtual screening molecular docking software for students and educators. PLoS Comput. Biol. 2012, 8, e1002499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2006, 35, D5–D12. [Google Scholar] [CrossRef]
- Berman, H.M.; Bourne, P.E.; Westbrook, J.; Zardecki, C. The protein data bank. In Protein Structure; CRC Press: Boca Raton, FL, USA, 2003; pp. 394–410. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ai, H.; Chen, W.; Yin, Z.; Hu, H.; Zhu, J.; Zhao, J.; Zhao, Q.; Liu, H. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Turner, P. Grace-5.1. 22/qtGrace v 0.2. 4. 2018. Available online: http://plasma-gate.weizmann.ac.il/Grace/doc/UsersGuide.html (accessed on 10 February 2020).
- Ferreira, E.F.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.; Leão, R.P.; Ferreira, A.L.; Federico, L.B.; Silva, C.H.; Rosa, J. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.; Neto, M.; Silva, L.; da S Ramos, R.; da S Costa, J.; Brasil, D.; Lobato, C.; da Costa, G.; Bittencourt, J.; da Silva, C. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [Green Version]
- Ramos, R.S.; Macêdo, W.J.; Costa, J.S.; da Silva, C.H.d.P.; Rosa, J.M.; da Cruz, J.N.; de Oliveira, M.S.; de Aguiar Andrade, E.H.; e Silva, R.B.; Souto, R.N. Potential inhibitors of the enzyme acetylcholinesterase and juvenile hormone with insecticidal activity: Study of the binding mode via docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–23. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Ahamad, S.; Islam, A.; Ahmad, F.; Dwivedi, N.; Hassan, M.I. 2/3D-QSAR, molecular docking and MD simulation studies of FtsZ protein targeting benzimidazoles derivatives. Comput. Biol. Chem. 2019, 78, 398–413. [Google Scholar] [CrossRef]
- Naz, F.; Shahbaaz, M.; Bisetty, K.; Islam, A.; Ahmad, F.; Hassan, M.I. Designing New Kinase Inhibitor Derivatives as Therapeutics Against Common Complex Diseases: Structural Basis of Microtubule Affinity-Regulating Kinase 4 (MARK4) Inhibition. OMICS 2015, 19, 700–711. [Google Scholar] [CrossRef]
- Naz, H.; Jameel, E.; Hoda, N.; Shandilya, A.; Khan, P.; Islam, A.; Ahmad, F.; Jayaram, B.; Hassan, M.I. Structure guided design of potential inhibitors of human calcium-calmodulin dependent protein kinase IV containing pyrimidine scaffold. Bioorg. Med. Chem. Lett. 2016, 26, 782–788. [Google Scholar] [CrossRef]
- Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.I.; Wang, Y. Identification and Evaluation of Inhibitors of Lipase from Malassezia restricta using Virtual High-Throughput Screening and Molecular Dynamics Studies. Int. J. Mol. Sci. 2019, 20, 884. [Google Scholar] [CrossRef] [Green Version]
- Gulzar, M.; Syed, S.B.; Khan, F.I.; Khan, P.; Ali, S.; Hasan, G.M.; Taneja, P.; Hassan, M.I. Elucidation of interaction mechanism of ellagic acid to the integrin linked kinase. Int. J. Biol. Macromol. 2019, 122, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.K.; Singh, D.; Lagunin, A.; Poroikov, V. PASS-assisted exploration of new therapeutic potential of natural products. Med. Chem. Res. 2011, 20, 1509–1514. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Hoda, N.; Naz, H.; Jameel, E.; Shandilya, A.; Dey, S.; Hassan, M.I.; Ahmad, F.; Jayaram, B. Curcumin specifically binds to the human calcium-calmodulin-dependent protein kinase IV: Fluorescence and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2016, 34, 572–584. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID. | Molecular Weight | No. of Rotatable Bonds | No. of H-Bond Donor | No. of H-Bond Acceptor | logP | Lipinski Violation |
|---|---|---|---|---|---|---|
| ZINC00319000 | 340.33 | 4 | 1 | 5 | 3.10 | 0 |
| ZINC02106991 | 387.43 | 7 | 2 | 5 | 3.48 | 0 |
| ZINC02115387 | 369.37 | 7 | 2 | 5 | 3.23 | 0 |
| ZINC02121074 | 444.41 | 5 | 0 | 6 | 4.62 | 0 |
| EMD638683 | 364.34 | 4 | 4 | 6 | 2.03 | 0 |
| GSK650394 | 382.46 | 4 | 2 | 3 | 6.25 | 1 |
| Compound ID | Absorption | Distribution | Metabolism | Excretion | Toxicity | |
|---|---|---|---|---|---|---|
| GI Absorption (%) | Water Solubility | BBB/CNS Permeation | CYP2D6 Inh/Subs | OCT2 Substrate | AMES Toxicity | |
| ZINC00319000 | 95.20 | Soluble | No | No | No | No |
| ZINC02106991 | 59.18 | Soluble | No | No | No | No |
| ZINC02115387 | 60.62 | Soluble | No | No | No | No |
| ZINC02121074 | 99.84 | Soluble | No | No | No | No |
| EMD638683 | 60.54 | Soluble | No | No | No | No |
| GSK650394 | 98.96 | Poor | No | No | No | Yes |
| S. No. | Compound ID | Affinity (kcal/mol) |
|---|---|---|
| 1. | ZINC02092709 | −11.8 |
| 2. | ZINC02095133 | −11.7 |
| 3. | ZINC02119552 | −11.7 |
| 4. | ZINC02116493 | −11.4 |
| 5. | ZINC02129029 | −11.4 |
| 6. | ZINC02127993 | −11.3 |
| 7. | ZINC02131906 | −11.2 |
| 8. | ZINC02092851 | −11.1 |
| 9. | ZINC02114667 | −11.1 |
| 10. | ZINC02127995 | −11.1 |
| 11. | ZINC02136713 | −11.1 |
| 12. | ZINC00848446 | −11.0 |
| 13. | ZINC01105783 | −11.0 |
| 14. | ZINC02114669 | −11.0 |
| 15. | ZINC02117876 | −11.0 |
| 16. | ZINC02119756 | −11.0 |
| 17. | ZINC02120975 | −11.0 |
| 18. | ZINC02121074 | −11.0 |
| 19. | ZINC02130599 | −11.0 |
| 20. | ZINC02137640 | −11.0 |
| 21. | ZINC02149843 | −11.0 |
| 22. | ZINC00319000 | −10.9 |
| 23. | ZINC02096281 | −10.9 |
| 24. | ZINC02106991 | −10.9 |
| 25. | ZINC02112637 | −10.9 |
| 26. | ZINC02114126 | −10.9 |
| 27. | ZINC02115387 | −10.9 |
| 28. | ZINC02117188 | −10.9 |
| 29. | ZINC02117586 | −10.9 |
| 30. | ZINC02120692 | −10.9 |
| 31. | EMD638683 | −8.6 |
| 32. | GSK650394 | −9.1 |
| Compound ID. | Pa | Pi | Biological Activity |
|---|---|---|---|
| ZINC00319000 | 0.515 | 0.053 | Anti-inflammatory |
| ZINC02106991 | 0.787 | 0.009 | Antiarthritic |
| ZINC02115387 | 0.751 | 0.012 | Antiarthritic |
| ZINC02121074 | 0.375 | 0.080 | Kinase inhibitor |
| EMD638683 | 0.376 | 0.004 | SGK inhibitor |
| GSK650394 | 0.740 | 0.002 | SGK1 inhibitor |
| Compound ID | Chemical Name | Molecular Formula | Structure |
|---|---|---|---|
| ZINC00319000 | 5-[(methyl-oxo-BLAHyl) oxymethyl]furan-2-carboxylic | C19H16O6 |  |
| ZINC2106991 | (2S)-2-[[2-[(6-keto-7,8,9,10 tetrahydrobenzo[c]isochromen-3-yl)oxy]acetyl]amino]-4-methyl-valerate | C21H25NO6 |  |
| ZINC02115387 | (2S)-2-[[2-(6-ketobenzo[c]isochromen-3-yl)oxyacetyl]amino]valerate | C20H19NO6 |  |
| ZINC02121074 | 3-[7-(4-fluorobenzyl)oxy-2-keto-chromen-4-yl]-8-methoxy-coumarin | C26H17FO6 |  |
| EMD638683 | N’-[2-(3,5-Difluorophenyl)-2-hydroxyacetyl]-2-ethyl-4-hydroxy-3-methylbenzohydrazide | C18H18F2N2O4 |  |
| GSK650394 | 2-cyclopentyl-4-(5-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid | C25H22N2O2 |  |
| Molecule | No. of Atoms | Spatial Features | Aromatic | Hydrophobic | Donors | Acceptors | Negative Group | Positive Group |
|---|---|---|---|---|---|---|---|---|
| ZINC00319000 | 40 | 15 | 3 | 5 | 2 | 5 | 1 | 0 |
| ZINC2106991 | 52 | 19 | 2 | 9 | 1 | 5 | 1 | 0 |
| ZINC02115387 | 45 | 13 | 2 | 3 | 2 | 5 | 1 | 0 |
| ZINC02121074 | 50 | 12 | 4 | 2 | 0 | 6 | 0 | 0 |
| EMD638683 | 45 | 14 | 0 | 8 | 4 | 6 | 0 | 0 |
| GSK650394 | 50 | 15 | 4 | 6 | 2 | 3 | 1 | 1 |
| Complex | RMSD (nm) | RMSF (nm) | Rg (nm) | SASA (nm2) | Kinetic Energy | Enthalpy | Volume (nm3) | Density (g/L) |
|---|---|---|---|---|---|---|---|---|
| SGK1 | 0.45 | 0.14 | 1.88 | 135.94 | 157,875 | −817,080 | 642.38 | 1023.19 |
| SGK1-ZINC00319000 | 0.46 | 0.14 | 1.92 | 139.74 | 157,820 | −817,087 | 642.23 | 1023.56 |
| System | Secondary Structure Content (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Structure * | Coil | β-Sheet | β-Bridge | Bend | Turn | α-helix | Other # | |
| SGK1 | 50 | 32 | 13 | 2 | 17 | 10 | 25 | 1 |
| SGK1-ZINC00319000 | 52 | 30 | 13 | 2 | 16 | 10 | 27 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, T.; Siddiqui, S.; Shamsi, A.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M.I. Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies. Molecules 2020, 25, 823. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040823
Mohammad T, Siddiqui S, Shamsi A, Alajmi MF, Hussain A, Islam A, Ahmad F, Hassan MI. Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies. Molecules. 2020; 25(4):823. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040823
Chicago/Turabian StyleMohammad, Taj, Shiza Siddiqui, Anas Shamsi, Mohamed F. Alajmi, Afzal Hussain, Asimul Islam, Faizan Ahmad, and Md. Imtaiyaz Hassan. 2020. "Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies" Molecules 25, no. 4: 823. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040823