
New Oxaliplatin-Pyrophosphato Analogs with Improved In Vitro Cytotoxicity

 , ,
, ,  , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
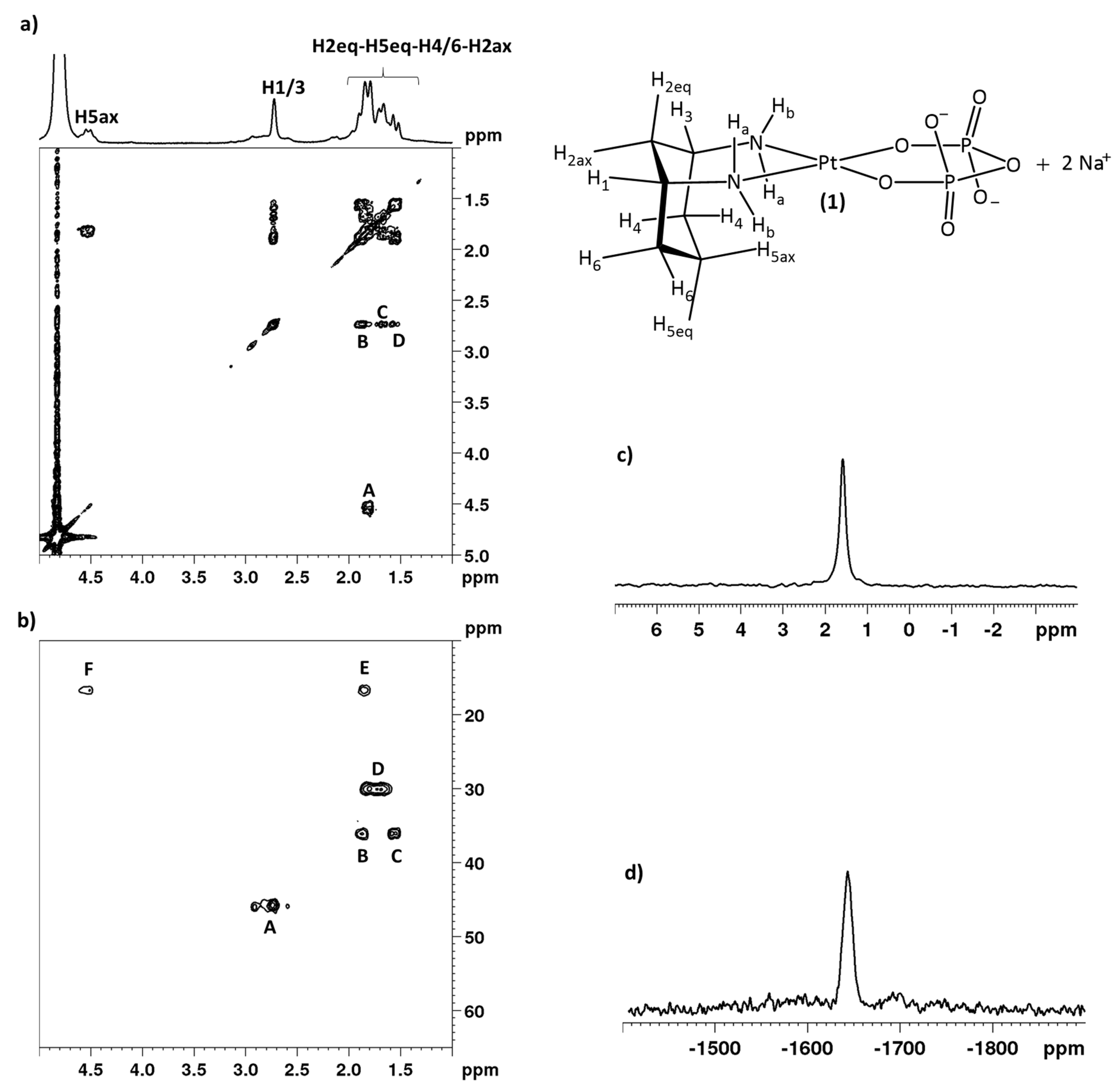
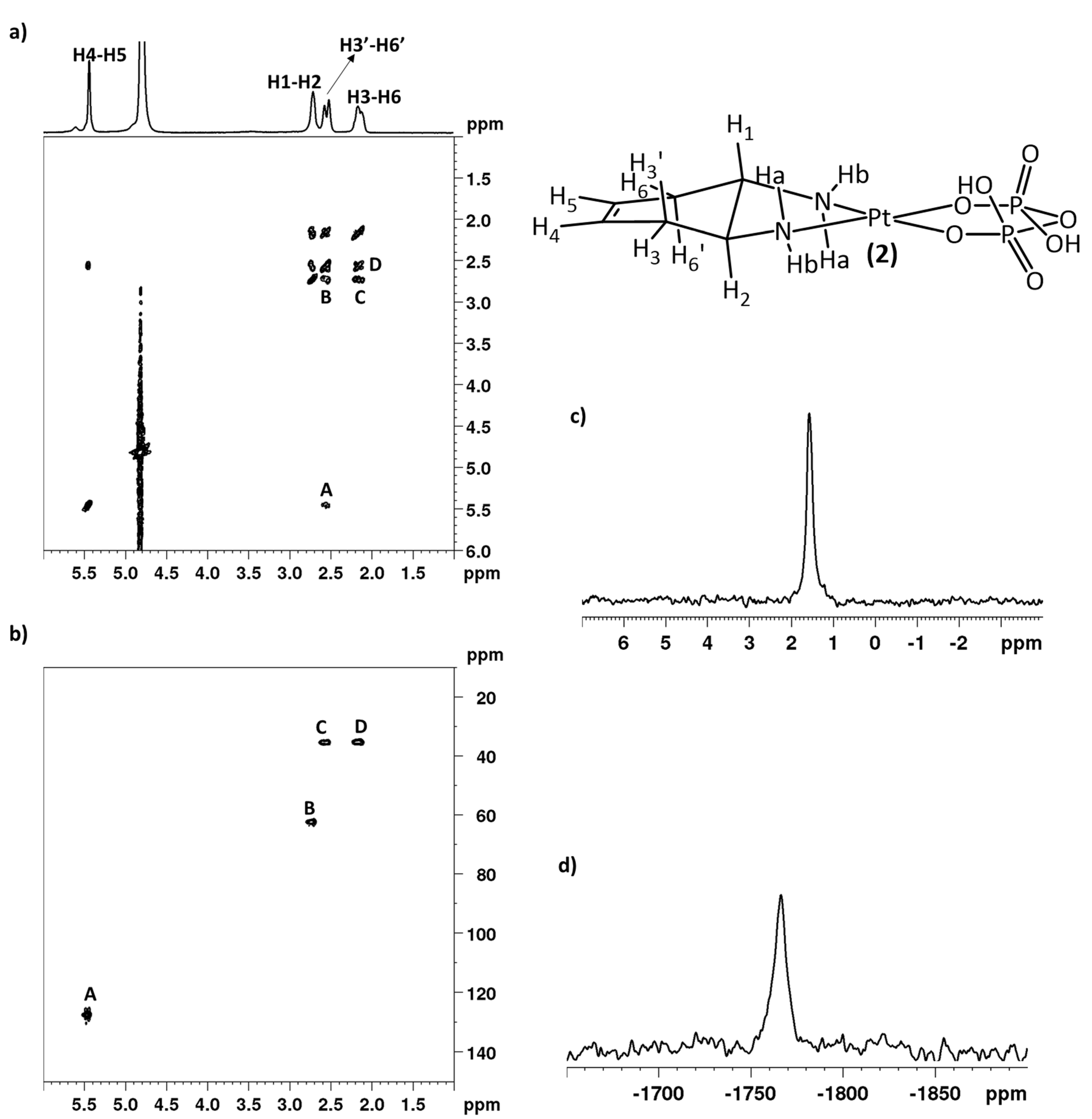
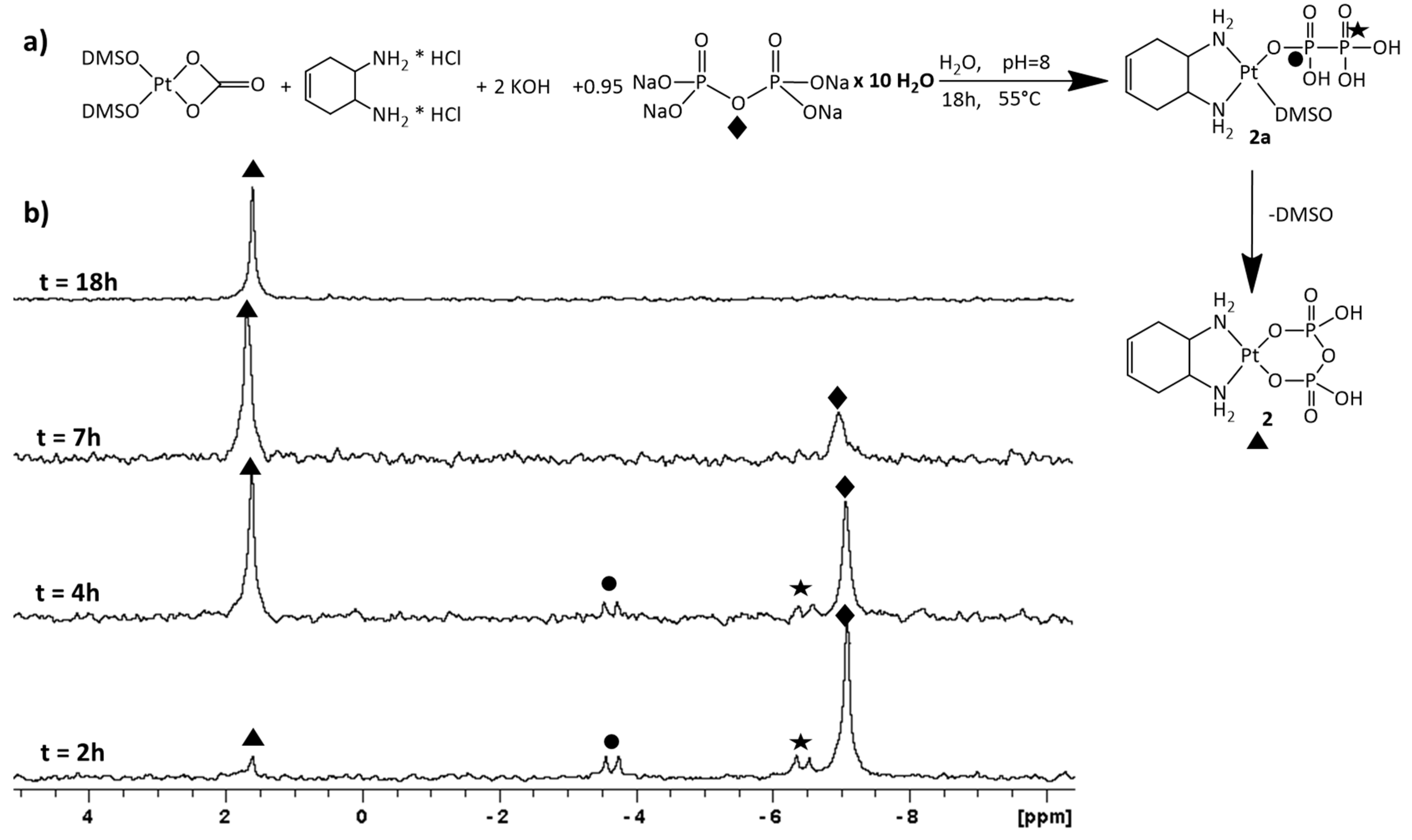
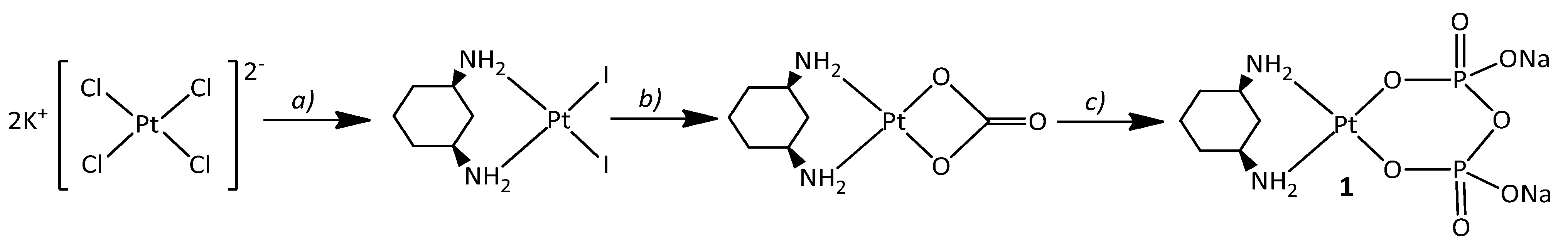
2.1. Synthesis and NMR Characterization
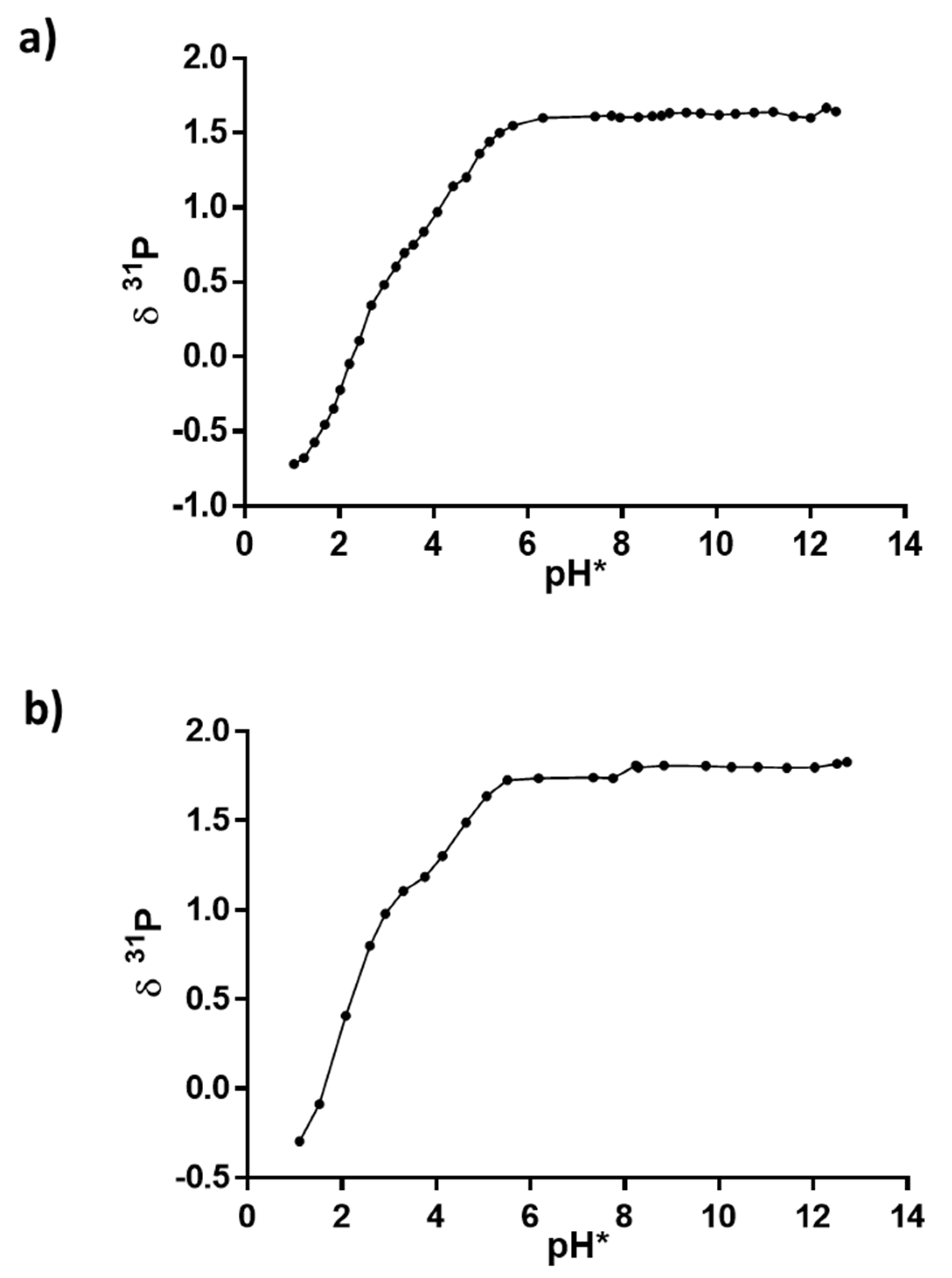
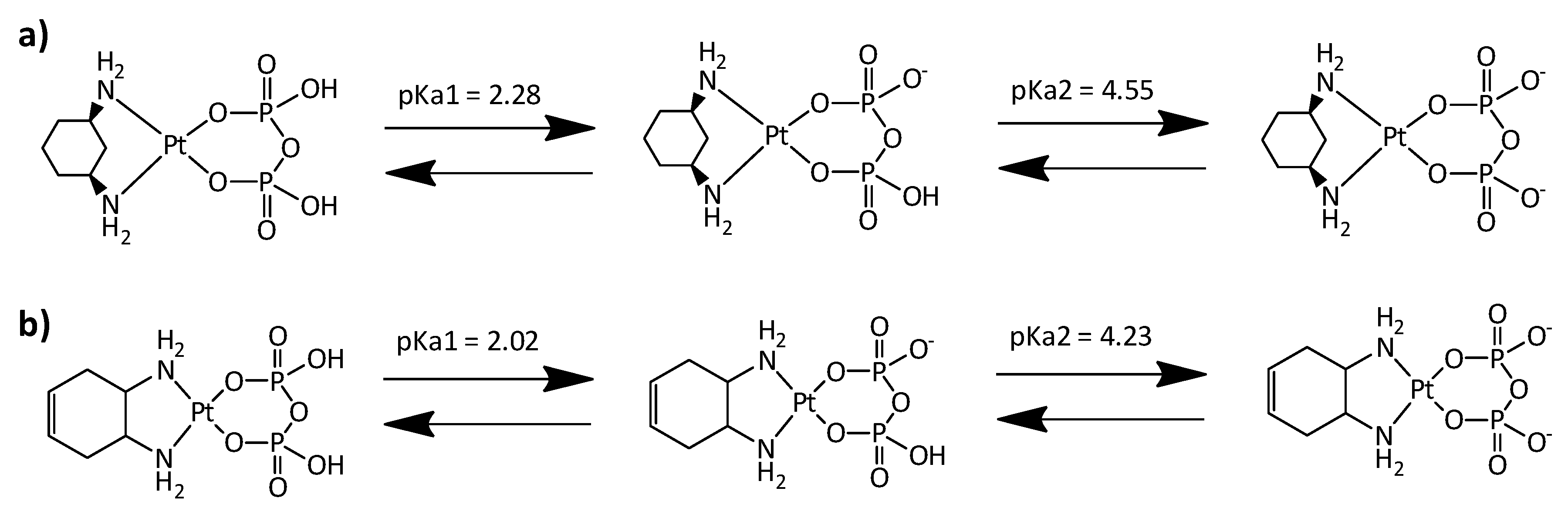
2.2. NMR Experiments at Different pH Values and Calculation of pKa Values
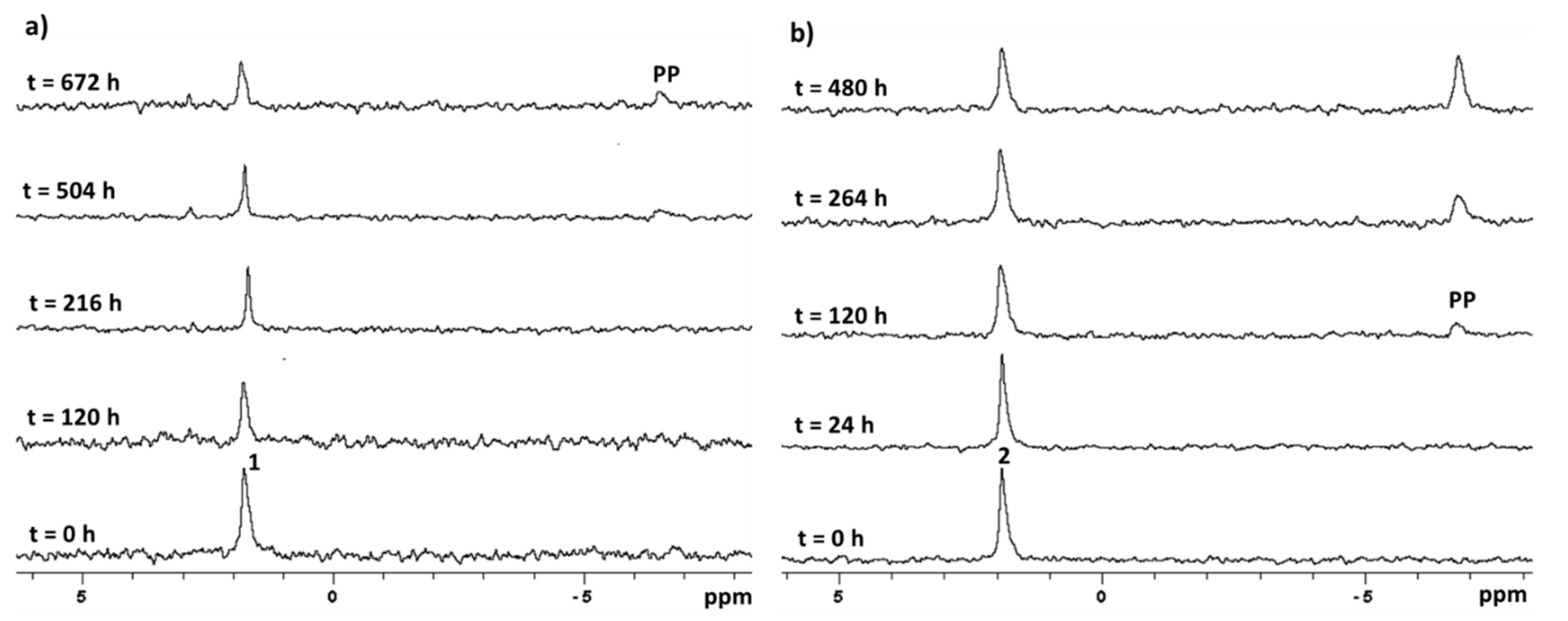
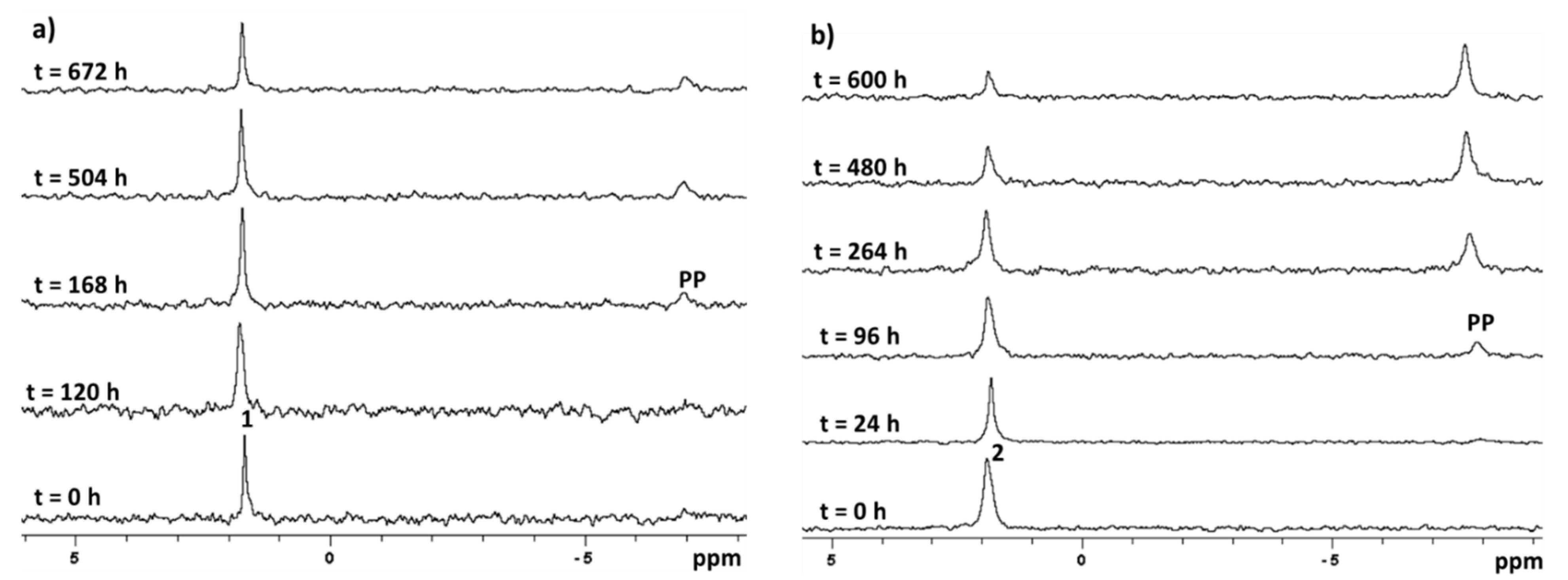
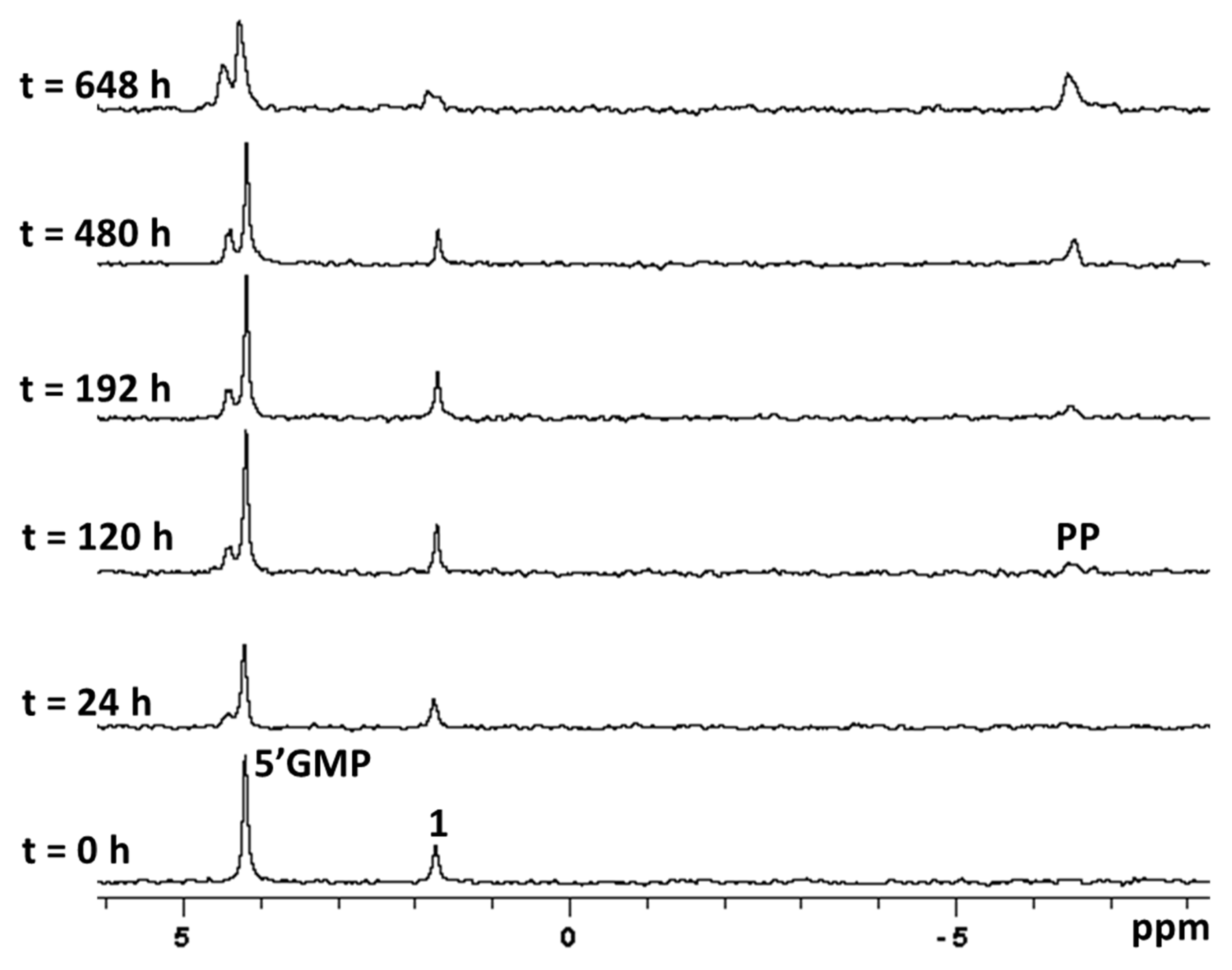
2.3. Stability in Physiological Conditions
2.4. Stability in Acidic Environment
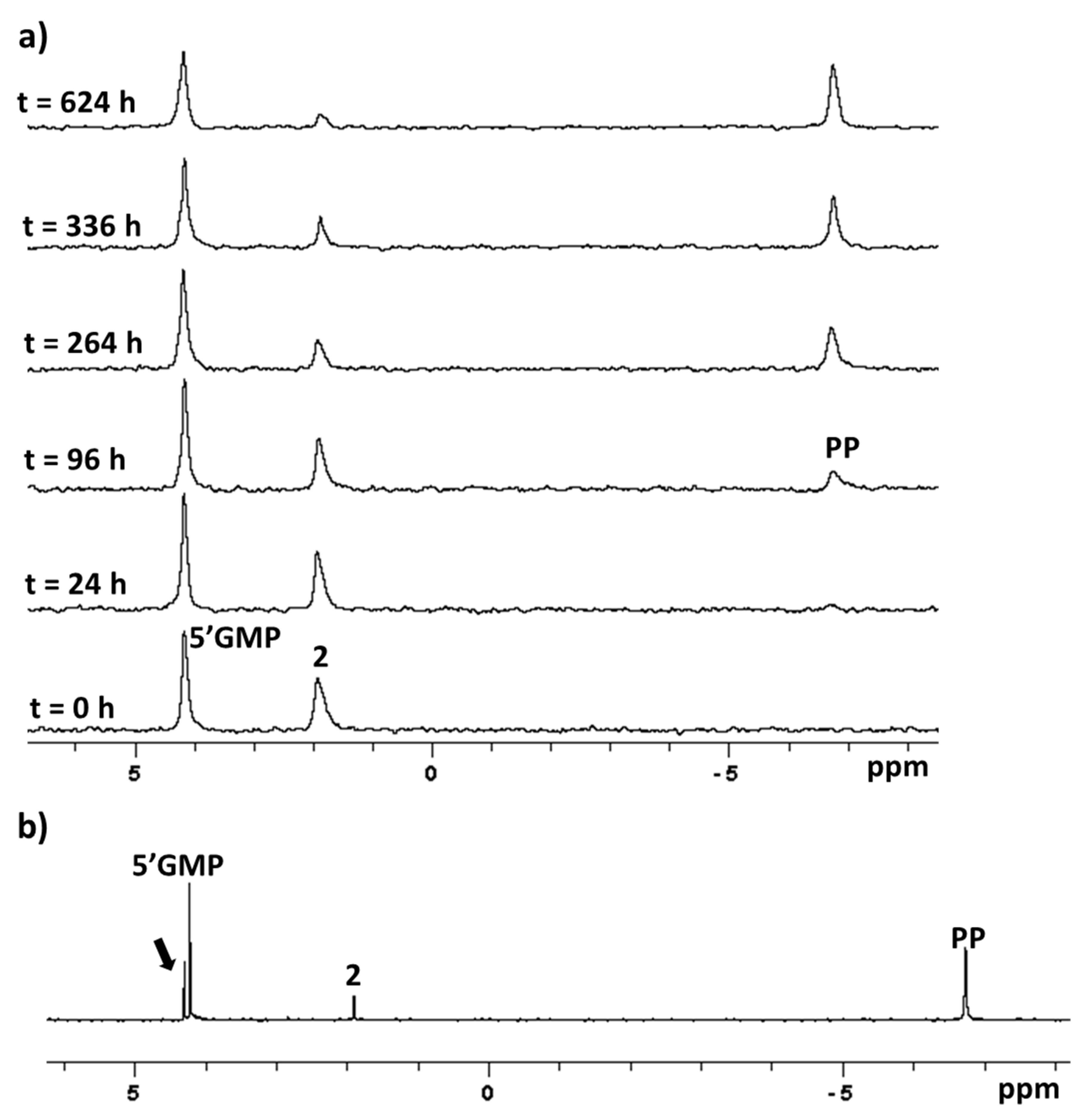
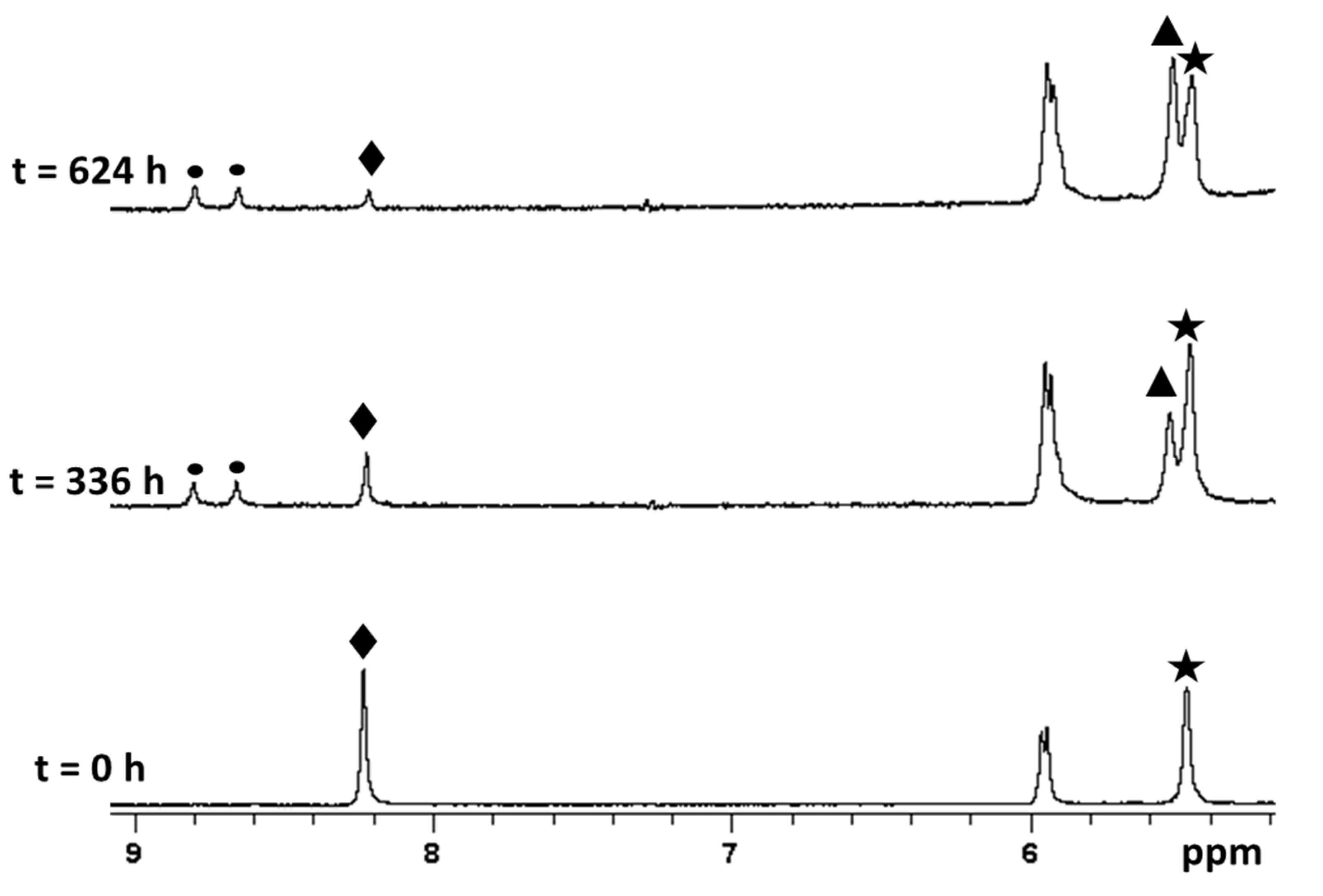
2.5. Reaction with 5’-GMP
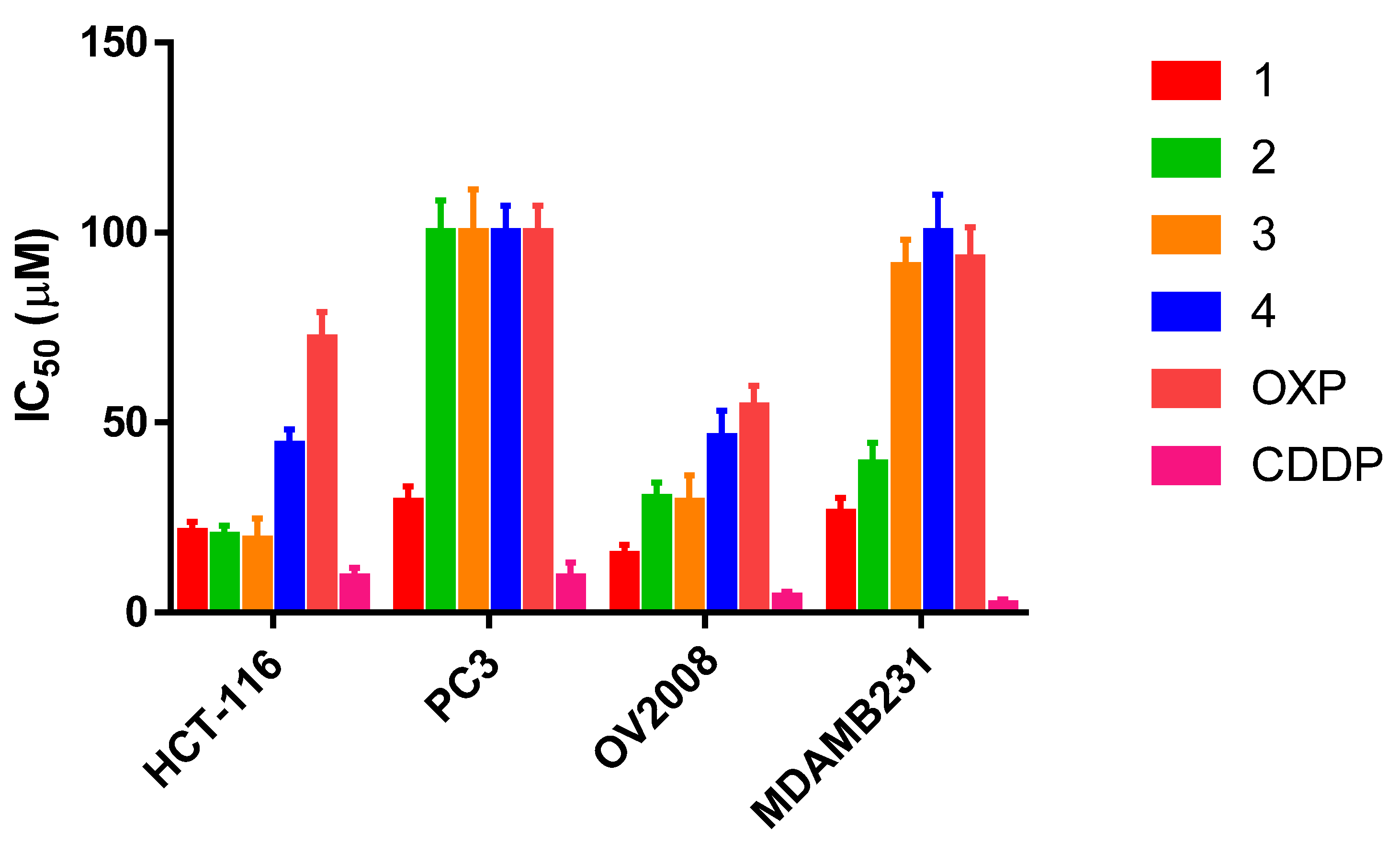
2.6. Cytotoxicity Assays
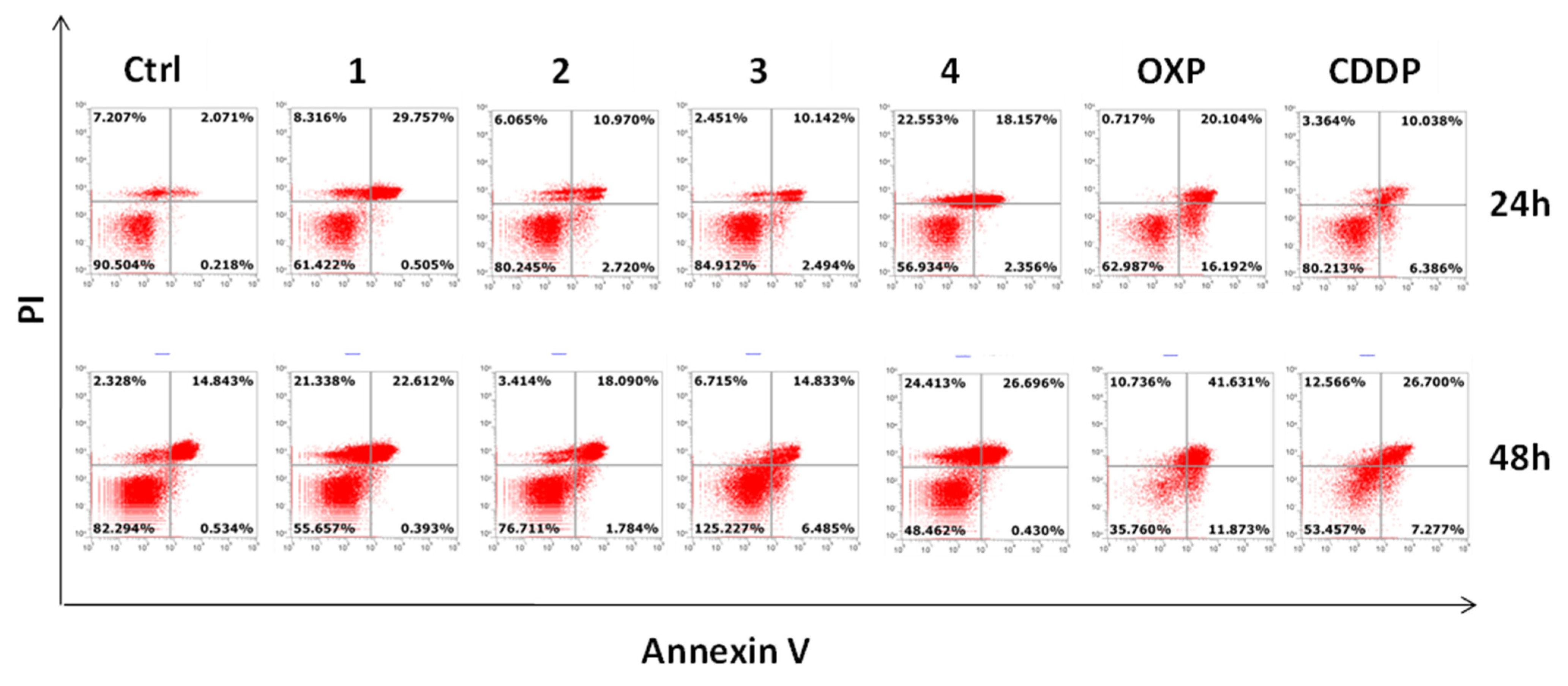
2.7. Apoptosis Assay
3. Materials and Methods
3.1. Reagents and Equipment
3.2. Preparation and Characterization of Platinum Complexes
3.2.1. [PtI2(cis-1,3-DACH)](DACH = diaminocyclohexane)
3.2.2. [Pt(CO3)(cis-1,3-DACH)]
3.2.3. [Pt(CO3)(DMSO)2] (DMSO = dimethylsulfoxide)
3.2.4. [PtI2(1R,2R-DACH)]
3.2.5. [Pt(CO3)(1R,2R-DACH)]
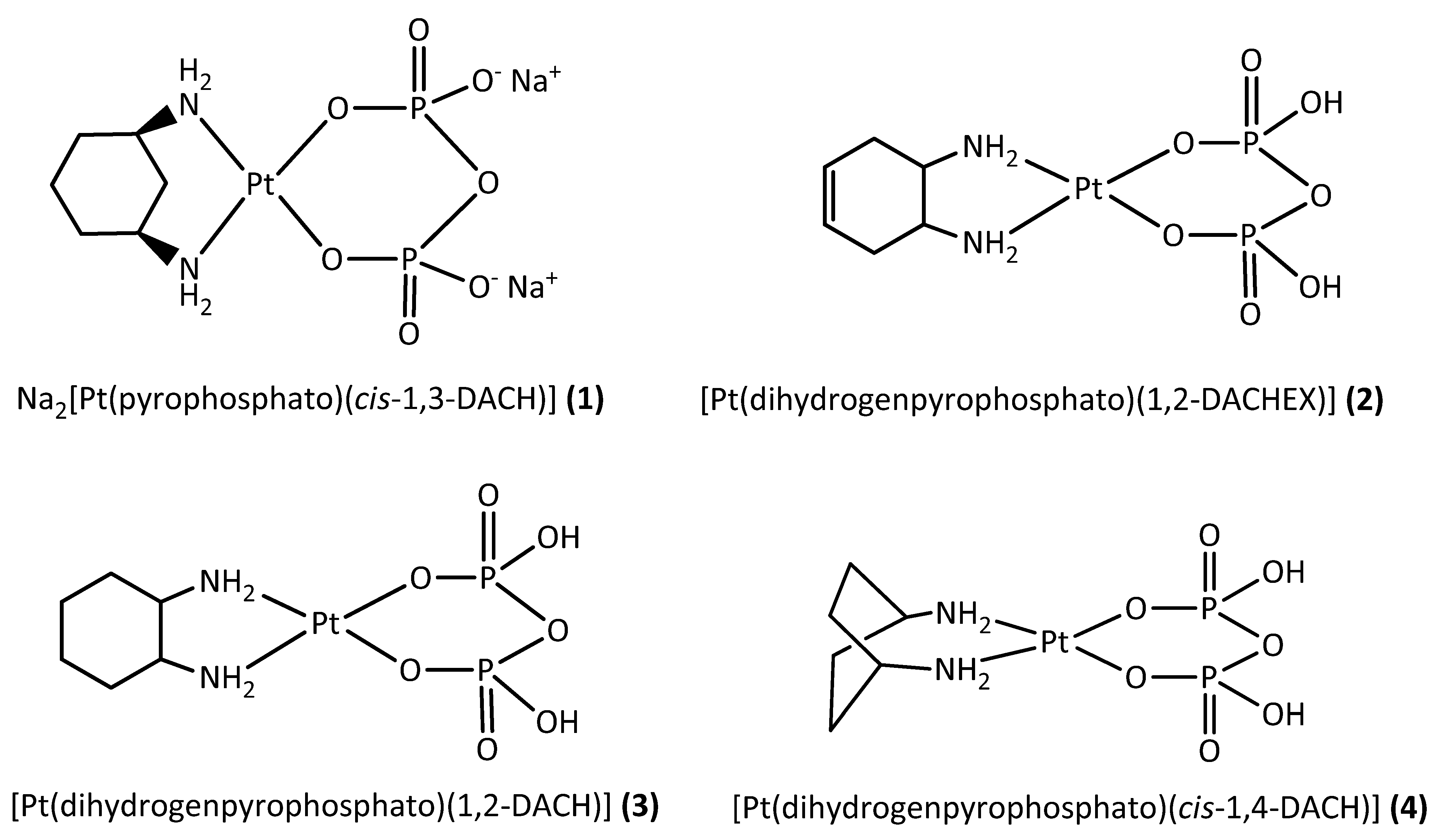
3.2.6. Na2[Pt(Pyrophosphato)(cis-1,3-DACH)] (1; pyrophosphate = P2O74−)
3.2.7. [Pt(Dihydrogenpyrophosphato)(1,2-DACHEX)] (2; 1,2-DACHEX = racemic trans-1,2-diamino-4-cyclohexene)
3.2.8. [Pt(Dihydrogenpyrophosphato)(1R,2R-DACH)] (3)
3.2.9. [Pt(Dihydrogenpyrophosphato)(cis-1,4-DACH)] (4)
3.3. NMR Experiments at Different pHs and Calculation of pKa Values
3.4. Stability in Physiological or Acidic Conditions
3.5. Reactivity with 5′-GMP
3.6. Cell Viability Assay
3.7. Annexin V/PI Apoptosis Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalt. Trans. 2008, 3, 183–194. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D. Platinum(IV) anticancer prodrugs—Hypotheses and facts. Dalt. Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carozzi, V.; Marmiroli, P.; Cavaletti, G. The Role of Oxidative Stress and Anti-Oxidant Treatment in Platinum-Induced Peripheral Neurotoxicity. Curr. Cancer Drug Targets 2010, 10, 670–682. [Google Scholar] [CrossRef]
- Galanski, M. Recent Developments in the Field of Anticancer Platinum Complexes. Recent Pat. Anticancer. Drug Discov. 2006, 1, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Guo, Z. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 202–224. [Google Scholar] [CrossRef]
- Gabano, E.; Ravera, M.; Osella, D. The Drug Targeting and Delivery Approach Applied to Pt-Antitumour Complexes. A Coordination Point of View. Curr. Med. Chem. 2009, 16, 4544–4580. [Google Scholar] [CrossRef] [PubMed]
- Appleton, T.G.; Hall, J.R.; McMahon, I.J. Multinuclear NMR study of reactions of methylphosphonic acid, CH3PO3H2, and (aminoalkyl)phosphonic acids, NH2(CH2)nPO3H2 (n = 1–3), with the cis-diamminediaquaplatinum(II) cation and cis-diamminedihydroxoplatinum(II). Inorg. Chem. 1986, 25, 720–725. [Google Scholar] [CrossRef]
- Galanski, M.; Slaby, S.; Jakupec, M.A.; Keppler, B.K. Synthesis, characterization, and in vitro antitumor activity of osteotropic diam(m)ineplatinum(II) complexes bearing a N,N-bis(phosphonomethyl)glycine ligand. J. Med. Chem. 2003, 46, 4946–4951. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Lin, M.; Zhu, J.; Zhang, J.; Li, Y.; Guo, Z. Platinum(II) compounds bearing bone-targeting group: Synthesis, crystal structure and antitumor activity. Chem. Commun. 2010, 46, 1212–1214. [Google Scholar] [CrossRef]
- Nakatake, H.; Ekimoto, H.; Aso, M.; Ogawa, A.; Yamaguchi, A.; Suemune, H. Dialkyl Bisphosphonate Platinum(II) Complex as a Potential Drug for Metastatic Bone Tumor. Chem. Pharm. Bull. 2011, 59, 710–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadar, R.A.; Franssen, G.M.; Van Dijk, N.W.M.; Codee-van der Schilden, K.; de Weijert, M.; Oosterwijk, E.; Iafisco, M.; Margiotta, N.; Heskamp, S.; van den Beucken, J.J.J.P.; et al. Bone tumor–targeted delivery of theranostic 195mPt-bisphosphonate complexes promotes killing of metastatic tumor cells. Mater. Today Bio. 2021, 9, 100088. [Google Scholar] [CrossRef] [PubMed]
- Nadar, R.A.; Farbod, K.; der Schilden, K.C.; Schlatt, L.; Crone, B.; Asokan, N.; Curci, A.; Brand, M.; Bornhaeuser, M.; Iafisco, M.; et al. Targeting of radioactive platinum-bisphosphonate anticancer drugs to bone of high metabolic activity. Sci. Rep. 2020, 10, 5889. [Google Scholar] [CrossRef] [Green Version]
- Piccinonna, S.; Margiotta, N.; Pacifico, C.; Lopalco, A.; Denora, N.; Fedi, S.; Corsini, M.; Natile, G. Dinuclear Pt(ii)-bisphosphonate complexes: A scaffold for multinuclear or different oxidation state platinum drugs. Dalt. Trans. 2012, 41, 9689–9699. [Google Scholar] [CrossRef] [PubMed]
- Iafisco, M.; Margiotta, N. Silica xerogels and hydroxyapatite nanocrystals for the local delivery of platinum-bisphosphonate complexes in the treatment of bone tumors: A mini-review. J. Inorg. Biochem. 2012, 117, 237–247. [Google Scholar] [CrossRef]
- Bose, R.N.; Maurmann, L.; Mishur, R.J.; Yasui, L.; Gupta, S.; Grayburn, W.S.; Hofstetter, H.; Salley, T.; Milton, T. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum-phosphato complexes towards human ovarian cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Mishur, R.J.; Zheng, C.; Gilbert, T.M.; Bose, R.N. Synthesis, X-ray crystallographic, and NMR characterizations of platinum(II) and platinum(IV) pyrophosphato complexes. Inorg. Chem. 2008, 47, 7972–7982. [Google Scholar] [CrossRef] [PubMed]
- Moghaddas, S.; Majmudar, P.; Marin, R.; Dezvareh, H.; Qi, C.; Soans, E.; Bose, R.N. Phosphaplatins, next generation platinum antitumor agents: A paradigm shift in designing and defining molecular targets. Inorganica Chim. Acta 2012, 393, 173–181. [Google Scholar] [CrossRef]
- Bose, R.N.; Moghaddas, S.; Belkacemi, L.; Tripathi, S.; Adams, N.R.; Majmudar, P.; McCall, K.; Dezvareh, H.; Nislow, C. Absence of Activation of DNA Repair Genes and Excellent Efficacy of Phosphaplatins against Human Ovarian Cancers: Implications To Treat Resistant Cancers. J. Med. Chem. 2015, 58, 8387–8401. [Google Scholar] [CrossRef]
- Curci, A.; Gandin, V.; Marzano, C.; Hoeschele, J.D.; Natile, G.; Margiotta, N. Novel Kiteplatin Pyrophosphate Derivatives with Improved Efficacy. Inorg. Chem. 2017, 56, 7482–7493. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Kostrhunova, H.; Novohradsky, V.; Pracharova, J.; Curci, A.; Margiotta, N.; Natile, G.; Brabec, V. Anticancer kiteplatin pyrophosphate derivatives show unexpected target selectivity for DNA. Dalt. Trans. 2017, 46, 14139–14148. [Google Scholar] [CrossRef] [PubMed]
- Papadia, P.; Gandin, V.; Barbanente, A.; Ruello, A.G.; Marzano, C.; Micoli, K.; Hoeschele, J.D.; Natile, G.; Margiotta, N. A minimal structural variation can overcome tumour resistance of oxaliplatin: The case of 4,5-dehydrogenation of the cyclohexane ring. RSC Adv. 2019, 9, 32448–32452. [Google Scholar] [CrossRef] [Green Version]
- Hoeschele, J.D.; Kasparkova, J.; Kostrhunova, H.; Novakova, O.; Pracharova, J.; Pineau, P.; Brabec, V. Synthesis, antiproliferative activity in cancer cells and DNA interaction studies of [Pt(cis-1,3-diaminocycloalkane)Cl2] analogs. JBIC J. Biol. Inorg. Chem. 2020. [Google Scholar] [CrossRef]
- Yamazaki, T.; Buqué, A.; Ames, T.D.; Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. Oncoimmunology 2020, 9, 1721810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berners-Price, S.J.; Ronconi, L.; Sadler, P.J. Insights into the mechanism of action of platinum anticancer drugs from multinuclear NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 65–98. [Google Scholar] [CrossRef]
- Ronconi, L.; Sadler, P.J. Applications of heteronuclear NMR spectroscopy in biological and medicinal inorganic chemistry. Coord. Chem. Rev. 2008, 252, 2239–2277. [Google Scholar] [CrossRef]
- Papadia, P.; Micoli, K.; Barbanente, A.; Ditaranto, N.; Hoeschele, J.D.; Natile, G.; Marzano, C.; Gandin, V.; Margiotta, N. Platinum(IV) complexes of trans-1,2-diamino-4-cyclohexene: Prodrugs affording an oxaliplatin analogue that overcomes cancer resistance. Int. J. Mol. Sci. 2020, 21, 2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresser, M.J.; Tracey, A.S.; Parkinson, K.M. Vanadium(V) oxyanions: The interaction of vanadate with pyrophosphate, phosphate, and arsenate. J. Am. Chem. Soc. 1986, 108, 6229–6234. [Google Scholar] [CrossRef]
- Iacobazzi, R.M.; Cutrignelli, A.; Stefanachi, A.; Porcelli, L.; Lopedota, A.A.; Di Fonte, R.; Lopalco, A.; Serratì, S.; Laquintana, V.; Silvestris, N.; et al. Hydroxy-Propil-β-Cyclodextrin Inclusion Complexes of two Biphenylnicotinamide Derivatives: Formulation and Anti-Proliferative Activity Evaluation in Pancreatic Cancer Cell Models. Int. J. Mol. Sci. 2020, 21, 6545. [Google Scholar] [CrossRef]
- Feltham, R.D.; Hayter, R.G. The electrolyte type of ionized complexes. J. Chem. Soc. 1964, 4587. [Google Scholar] [CrossRef]
- Bergamini, P.; Marvelli, L.; Ferretti, V.; Gemmo, C.; Gambari, R.; Hushcha, Y.; Lampronti, I. Bis(dimethylsulfoxide)carbonateplatinum(ii), a new synthon for a low-impact, versatile synthetic route to anticancer Pt carboxylates. Dalt. Trans. 2016, 45, 10752–10760. [Google Scholar] [CrossRef]
- S.C., D. A rapid method for the synthesis of cis-[Pt(NH3)2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Kaleida Graph 3.5; Synergy Software: Reading, PA, USA, 2000.














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbanente, A.; Iacobazzi, R.M.; Azzariti, A.; Hoeschele, J.D.; Denora, N.; Papadia, P.; Pacifico, C.; Natile, G.; Margiotta, N. New Oxaliplatin-Pyrophosphato Analogs with Improved In Vitro Cytotoxicity. Molecules 2021, 26, 3417. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113417
Barbanente A, Iacobazzi RM, Azzariti A, Hoeschele JD, Denora N, Papadia P, Pacifico C, Natile G, Margiotta N. New Oxaliplatin-Pyrophosphato Analogs with Improved In Vitro Cytotoxicity. Molecules. 2021; 26(11):3417. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113417
Chicago/Turabian StyleBarbanente, Alessandra, Rosa Maria Iacobazzi, Amalia Azzariti, James D. Hoeschele, Nunzio Denora, Paride Papadia, Concetta Pacifico, Giovanni Natile, and Nicola Margiotta. 2021. "New Oxaliplatin-Pyrophosphato Analogs with Improved In Vitro Cytotoxicity" Molecules 26, no. 11: 3417. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26113417