
Unsymmetric Cisplatin-Based Pt(IV) Conjugates Containing a PARP-1 Inhibitor Pharmacophore Tested on Malignant Pleural Mesothelioma Cell Lines

 , ,
, ,
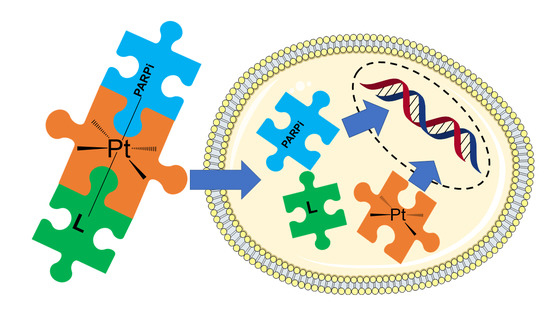
Abstract
:
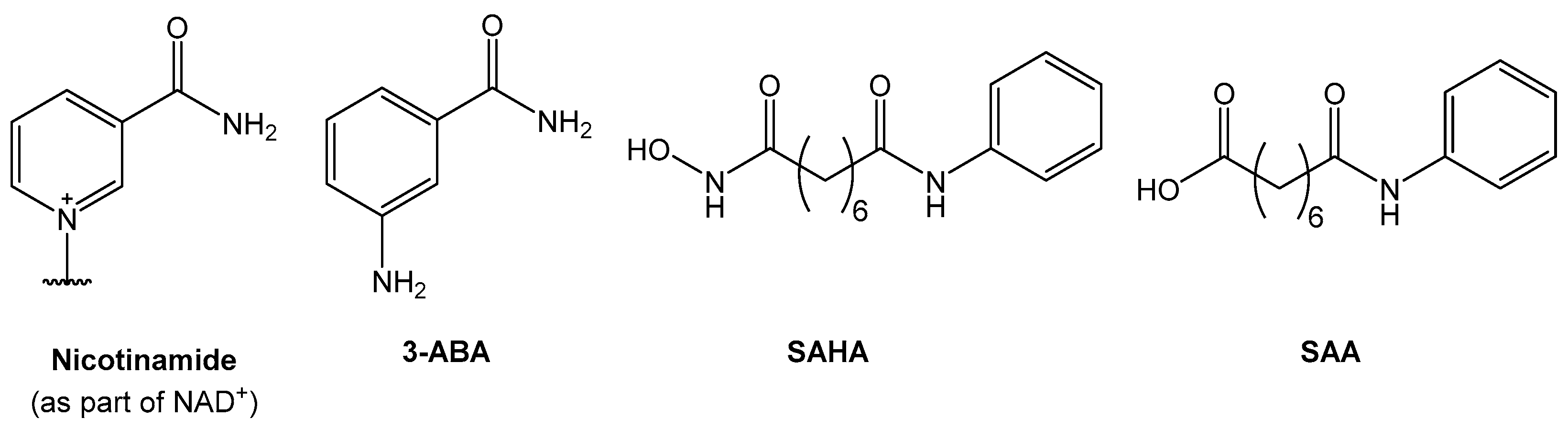
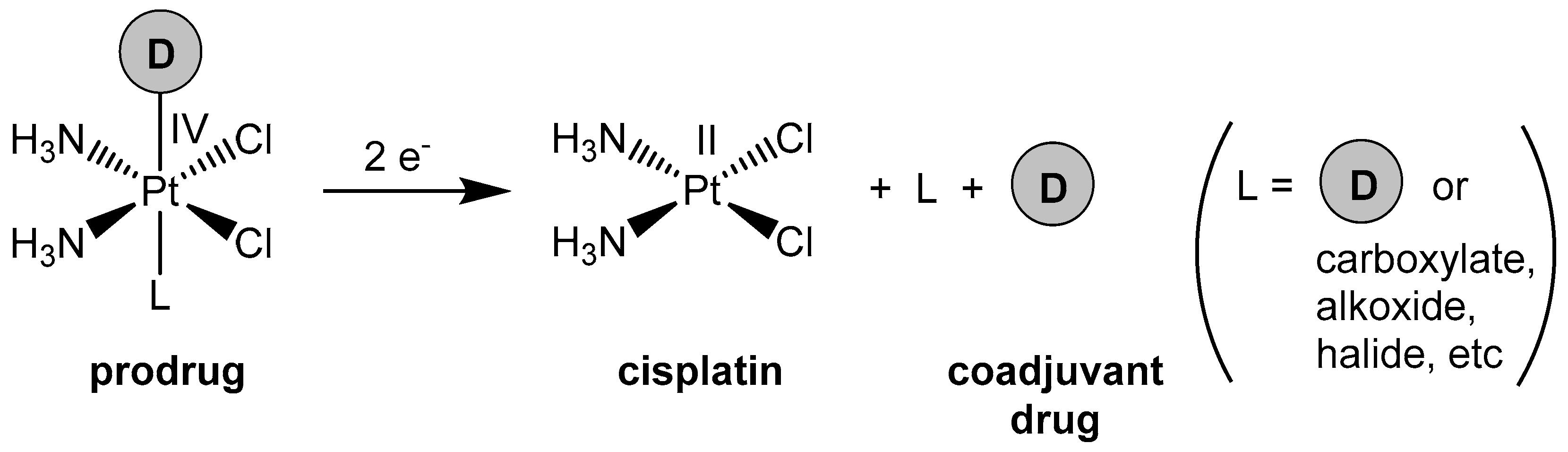
1. Introduction
2. Results and Discussion
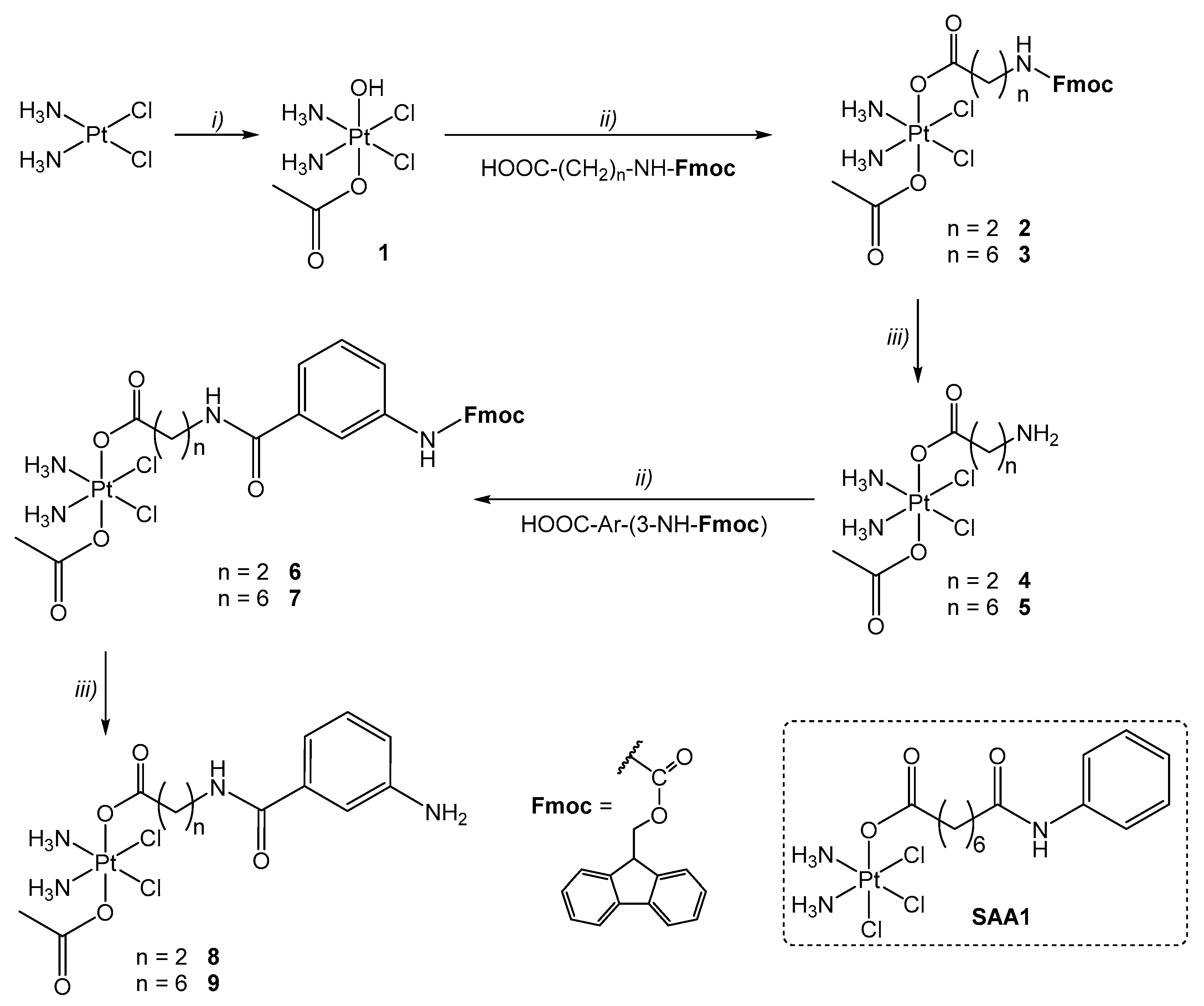
2.1. Synthesis of the Bifunctional Pt(IV) Prodrugs
2.2. Evaluation of the Antiproliferative Activity
2.3. Cell Uptake
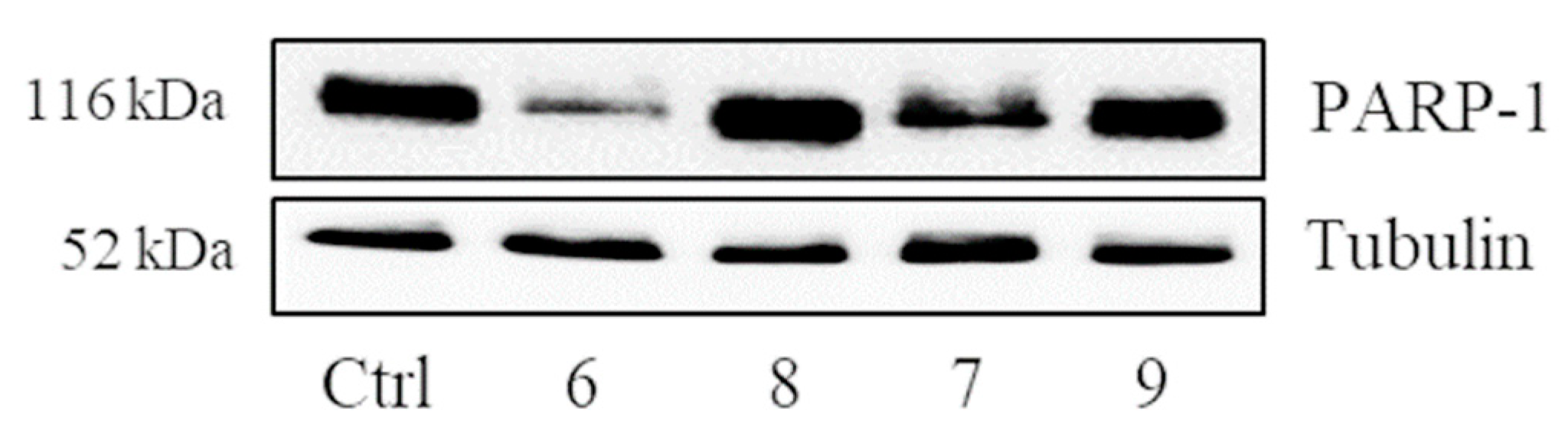
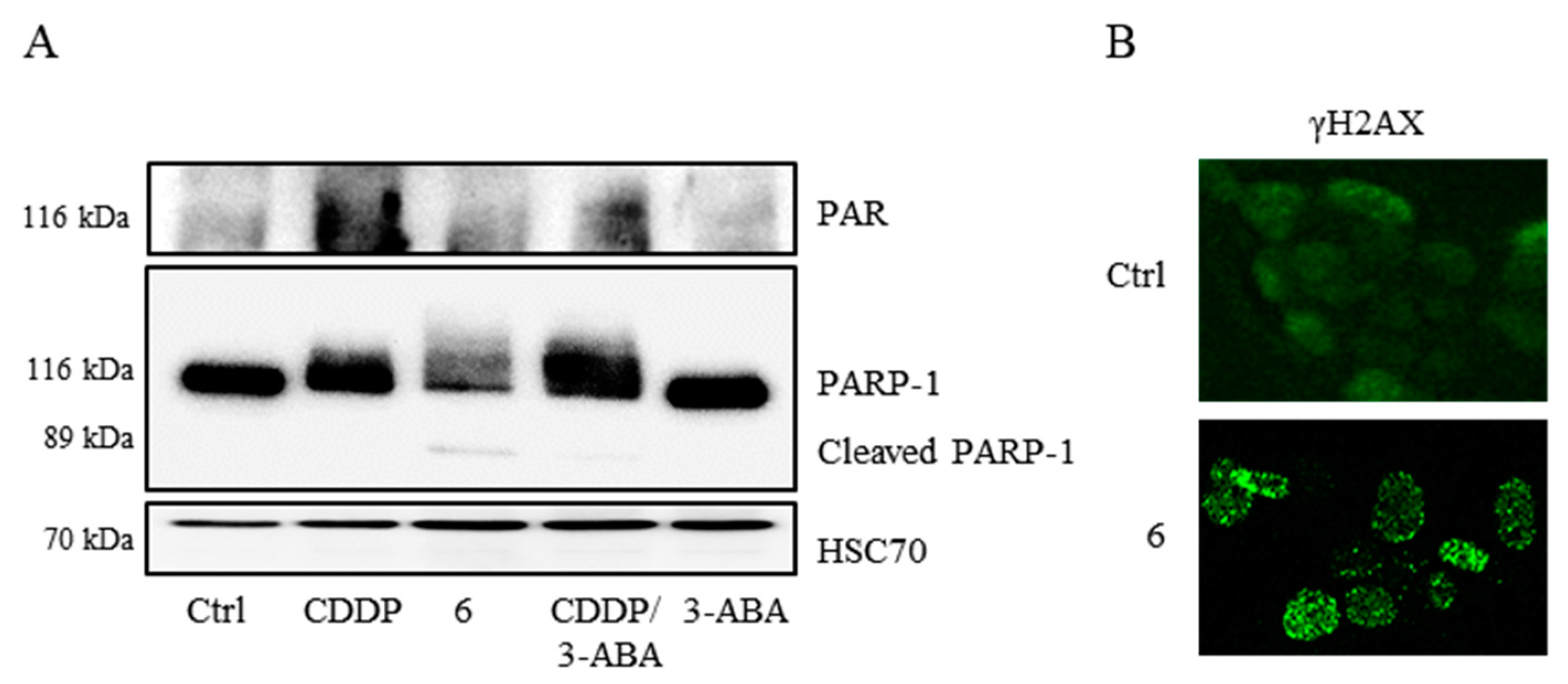
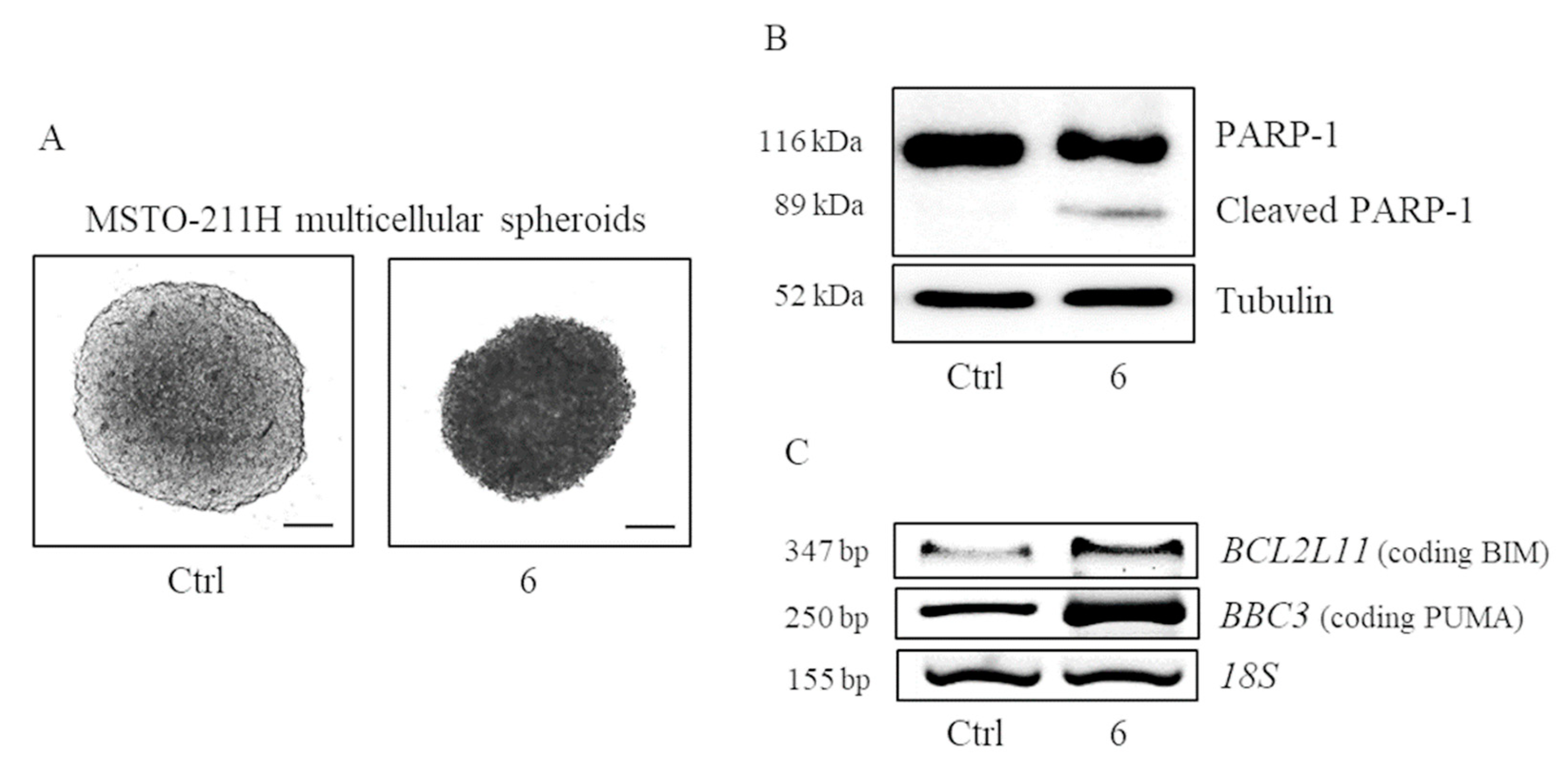
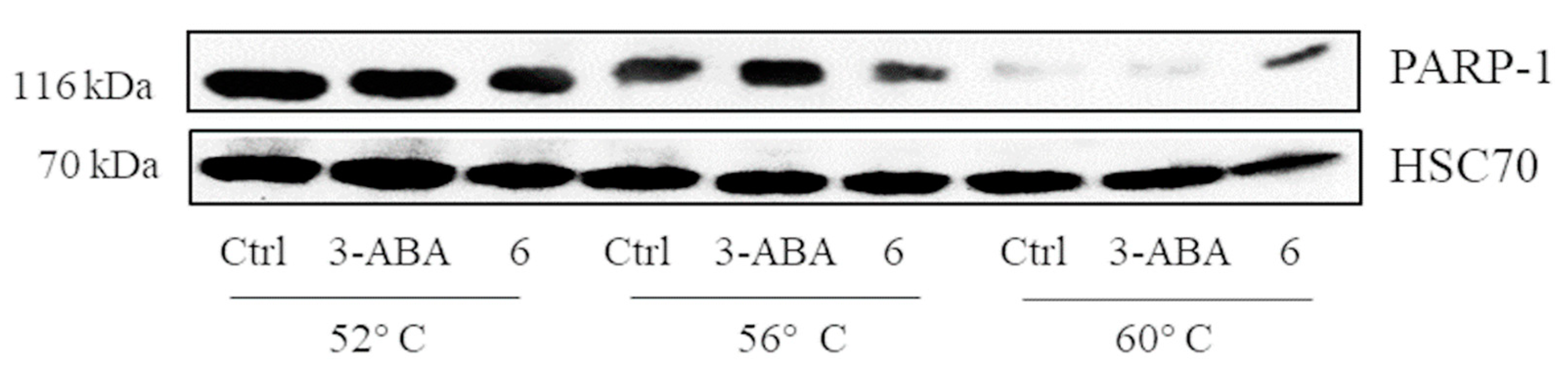
2.4. Verification of the Targets of 6
3. Materials and Methods
3.1. Chemical Procedures
3.2. Synthesis of the Pt(IV) Prodrugs
3.2.1. Synthesis of (OC-6-44)-Acetato-[7-(fmoc-amino)-heptanoato]-diammine-dichlorido-platinum(IV), 3
3.2.2. Synthesis of (OC-6-44)-Acetato-(7-aminoheptanoato)-diammine-dichlorido-platinum(IV), 5
3.2.3. Synthesis of (OC-6-44)-Acetato[3-(3-(fmoc-amino)-benzamido)-propanoato]-diammine-dichlorido-platinum(IV), 6
3.2.4. Synthesis of (OC-6-44)-Acetato-[7-(3-(fmoc-amino)-benzamido)-heptanoato]-diammine-dichlorido-platinum(IV), 7
3.2.5. Synthesis of (OC-6-44)-Acetato[3-(3-aminobenzamido)-propanoato]-diammine-dichlorido-platinum(IV), 8
3.2.6. Synthesis of (OC-6-44)-Acetato-[7-(3-aminobenzamido)-heptanoato]-diammine-dichlorido-platinum(IV), 9
3.3. Reduction Studies
3.4. Lipophilicity
3.5. Biological Procedures
3.5.1. Cell Viability Assay
3.5.2. Cell Uptake
3.5.3. Multicellular Spheroids
3.5.4. Cell Lysis and Immunoblot
3.5.5. γH2AX Foci Immunofluorescence
3.5.6. Cellular Thermal Shift Assay (CETSA®)
3.5.7. RNA Isolation and Real-Time PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Comba, P.; D’Angelo, M.; Fazzo, L.; Magnani, C.; Marinaccio, A.; Mirabelli, D.; Terracini, B. Mesothelioma in Italy: The Casale Monferrato model to a national epidemiological surveillance system. Ann. dell’Istituto Super. Sanità 2018, 54, 139–148. [Google Scholar] [CrossRef]
- Novello, S.; Pinto, C.; Torri, V.; Porcu, L.; Di Maio, M.; Tiseo, M.; Ceresoli, G.; Magnani, C.; Silvestri, S.; Veltri, A.; et al. The Third Italian Consensus Conference for Malignant Pleural Mesothelioma: State of the art and recommendations. Crit. Rev. Oncol. Hematol. 2016, 104, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Wallyn, F.; Albelda, S.M.; Munck, C. Novel therapies for malignant pleural mesothelioma. Lancet Oncol. 2018, 19, E161–E172. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical Mechanisms of Cisplatin Cytotoxicity. Anti-Cancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, C. Update on PARP1 inhibitors in ovarian cancer. Ann. Oncol. 2011, 22, viii72–viii76. [Google Scholar] [CrossRef]
- Betti, M.; Casalone, E.; Ferrante, D.; Aspesi, A.; Morleo, G.; Biasi, A.; Sculco, M.; Mancuso, G.; Guarrera, S.; Righi, L.; et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. 2017, 405, 38–45. [Google Scholar] [CrossRef]
- Fennell, D.A.; King, A.; Mohammed, S.; Branson, A.; Brookes, C.; Darlison, L.; Dawson, A.G.; Gaba, A.; Hutka, M.; Morgan, B.; et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): An open-label, single-arm, phase 2a clinical trial. Lancet Respir. Med. 2021, 9, 593–600. [Google Scholar] [CrossRef]
- Michels, J.; Vitale, I.; Senovilla, L.; Enot, D.P.; Garcia, P.; Lissa, D.; Olaussen, K.A.; Brenner, C.; Soria, J.C.; Castedo, M.; et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013, 12, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, M.; Fujihara, H.; Fujimori, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yamamoto, N.; Kishi, Y.; Hamada, Y.; Masutani, M. Synergetic Effects of PARP Inhibitor AZD2281 and Cisplatin in Oral Squamous Cell Carcinoma in Vitro and in Vivo. Int. J. Mol. Sci. 2016, 17, 272. [Google Scholar] [CrossRef] [Green Version]
- Oei, A.L.; van Leeuwen, C.M.; Ahire, V.R.; Rodermond, H.M.; ten Cate, R.; Westermann, A.M.; Stalpers, L.J.A.; Crezee, J.; Kok, H.P.; Krawczyk, P.M.; et al. Enhancing synthetic lethality of PARP-inhibitor and cisplatin in BRCA-proficient tumour cells with hyperthermia. Oncotarget 2017, 8, 28116–28124. [Google Scholar] [CrossRef] [Green Version]
- Novohradsky, V.; Zajac, J.; Vrana, O.; Kasparkova, J.; Brabec, V. Simultaneous delivery of olaparib and carboplatin in PEGylated liposomes imparts this drug combination hypersensitivity and selectivity for breast tumor cells. Oncotarget 2018, 9, 28456–28473. [Google Scholar] [CrossRef] [Green Version]
- Mensah, L.B.; Morton, S.W.; Li, J.H.; Xiao, H.H.; Ouadir, M.A.; Elias, K.M.; Penn, E.; Richson, A.K.; Ghoroghchian, P.P.; Liu, J.; et al. Layer-by-layer nanoparticles for novel delivery of cisplatin and PARP inhibitors for platinum-based drug resistance therapy in ovarian cancer. Bioeng. Transl. Med. 2019, 4, e10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusoh, N.A.; Ahmad, H.; Gill, M.R. Combining PARP Inhibition with Platinum, Ruthenium or Gold Complexes for Cancer Therapy. Chemmedchem 2020, 15, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Zhang, S.; Song, L.Y.; Qu, M.; Zou, Z.H. Synergistic lethality between PARP-trapping and alantolactone-induced oxidative DNA damage in homologous recombination-proficient cancer cells. Oncogene 2020, 39, 2905–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. PARP inhibition in platinum-based chemotherapy: Chemopotentiation and neuroprotection. Pharmacol. Res. 2018, 137, 104–113. [Google Scholar] [CrossRef]
- Gabano, E.; Ravera, M.; Osella, D. Pros and cons of bifunctional platinum(IV) antitumor prodrugs: Two are (not always) better than one. Dalton Trans. 2014, 43, 9813–9820. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(IV) anticancer prodrugs—Hypotheses and facts. Dalton Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef] [PubMed]
- Kenny, R.G.; Chuah, S.W.; Crawford, A.; Marmion, C.J. Platinum(IV) Prodrugs—A Step Closer to Ehrlich’s Vision? Eur. J. Inorg. Chem. 2017, 12, 1596–1612. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D. Multi-action Pt(IV) anticancer agents; do we understand how they work? J. Inorg. Biochem. 2019, 191, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. A view on multi-action Pt(IV) antitumor prodrugs. Inorg. Chim. Acta 2019, 492, 32–47. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(IV) anticancer agents; are we en route to the holy grail or to a dead end? J. Inorg. Biochem. 2021, 217, 111353. [Google Scholar] [CrossRef] [PubMed]
- Benafif, S.; Hall, M. An update on PARP inhibitors for the treatment of cancer. Oncotargets Ther. 2015, 8, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Costantino, G.; Macchiarulo, A.; Camaioni, E.; Pellicciari, R. Modeling of poly(ADP-ribose)polymerase (PARP) inhibitors. Docking of ligands and quantitative structure-activity relationship analysis. J. Med. Chem. 2001, 44, 3786–3794. [Google Scholar] [CrossRef]
- Ferraris, D.V. Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.M.; Soto, M.; Quevedo, C.; Alonso, C.; Cepeda, V.; Fuertes, M.A.; Nguewa, P.A. Poly(ADP-ribose) Polymerase-1 Inhibitor 3-Aminobenzamide Enhances Apoptosis Induction by Platinum Complexes in Cisplatin-Resistant Tumor Cells. Med. Chem. 2006, 2, 47–53. [Google Scholar] [CrossRef]
- Zheng, Y.D.; Xu, X.Q.; Peng, F.; Yu, J.Z.; Wu, H. The poly(ADP-ribose) polymerase-1 inhibitor 3-aminobenzamide suppresses cell growth and migration, enhancing suppressive effects of cisplatin in osteosarcoma cells. Oncol. Rep. 2011, 25, 1399–1405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.J.; Kan, Y.Y.; Tian, Y.J.; Wang, Z.; Zhang, J. Effects of poly (ADP-ribosyl) polymerase (PARP) inhibitor on cisplatin resistance & proliferation of the ovarian cancer C13*cells. Indian J. Med. Res. 2013, 137, 527–532. [Google Scholar] [PubMed]
- Sakogawa, K.; Aoki, Y.; Misumi, K.; Hamai, Y.; Emi, M.; Hihara, J.; Shi, L.; Kono, K.; Horikoshi, Y.; Sun, J.Y.; et al. Involvement of homologous recombination in the synergism between cisplatin and poly (ADP-ribose) polymerase inhibition. Cancer Sci. 2013, 104, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.C.; Hu, W.W.; Wang, Z.M.; Gou, S.H. Platinum(IV) prodrugs multiply targeting genomic DNA, histone deacetylases and PARP-1. Eur. J. Med. Chem. 2017, 141, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.F.; Li, C.; Zhou, Q.Y.; Deng, Z.Q.; Tong, Z.X.; Tse, M.K.; Zhu, G.Y. Synthesis, Cytotoxicity, and Mechanistic Investigation of Platinum(IV) Anticancer Complexes Conjugated with Poly(ADP-ribose) Polymerase Inhibitors. Inorg. Chem. 2019, 58, 16279–16291. [Google Scholar] [CrossRef]
- Babu, T.; Sarkar, A.; Karmakar, S.; Schmidt, C.; Gibson, D. Multiaction Pt(IV) Carbamate Complexes Can Codeliver Pt(II) Drugs and Amine Containing Bioactive Molecules. Inorg. Chem. 2020, 59, 5182–5193. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991. [Google Scholar] [CrossRef]
- Xu, Z.F.; Tang, W.K.; Zhou, Q.Y.; Chen, S.; Siu, C.K.; Zhu, G.Y. On the hydrolytic stability of unsymmetric platinum(iv) anticancer prodrugs containing axial halogens. Inorg. Chem. Front. 2021, 9. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; Zanellato, I.; Fregonese, F.; Pelosi, G.; Platts, J.A.; Osella, D. Antiproliferative activity of a series of cisplatin-based Pt(IV)-acetylamido/carboxylato prodrugs. Dalton Trans. 2016, 45, 5300–5309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravera, M.; Gabano, E.; Tinello, S.; Zanellato, I.; Osella, D. May glutamine addiction drive the delivery of antitumor cisplatin-based Pt(IV) prodrugs? J. Inorg. Biochem. 2017, 167, 27–35. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; Bonzani, D.; Zanellato, I.; Arrais, A.; Cantamessa, S.; Biggiogera, M.; Osella, D. Hybrid inorganic (nonporous silica)/organic (alginate) core-shell platform for targeting a cisplatin-based Pt(IV) anticancer prodrug. J. Inorg. Biochem. 2018, 189, 185–191. [Google Scholar] [CrossRef]
- Zanellato, I.; Bonarrigo, I.; Gabano, E.; Ravera, M.; Margiotta, N.; Betta, P.-G.; Osella, D. Metallo-drugs in the treatment of malignant pleural mesothelioma. Inorg. Chim. Acta 2012, 393, 64–74. [Google Scholar] [CrossRef]
- Ravera, M.; Gabano, E.; Zanellato, I.; Bonarrigo, I.; Alessio, M.; Arnesano, F.; Galliani, A.; Natile, G.; Osella, D. Cellular trafficking, accumulation and DNA platination of a series of cisplatin-based dicarboxylato Pt(IV) prodrugs. J. Inorg. Biochem. 2015, 150, 1–8. [Google Scholar] [CrossRef]
- Göschl, S.; Varbanov, H.P.; Theiner, S.; Jakupec, M.A.; Galanski, M.; Keppler, B.K. The role of the equatorial ligands for the redox behavior, mode of cellular accumulation and cytotoxicity of platinum(IV) prodrugs. J. Inorg. Biochem. 2016, 160, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Caron, G.; Ravera, M.; Gabano, E.; Bianco, S.; Platts, J.A.; Osella, D. Molecular interaction fields vs. quantum-mechanical-based descriptors in the modelling of lipophilicity of platinum(IV) complexes. Dalton Trans. 2013, 42, 3482–3489. [Google Scholar] [CrossRef] [Green Version]
- Platts, J.A.; Ermondi, G.; Caron, G.; Ravera, M.; Gabano, E.; Gaviglio, L.; Pelosi, G.; Osella, D. Molecular and statistical modeling of reduction peak potential and lipophilicity of platinum(IV) complexes. J. Biol. Inorg. Chem. 2011, 16, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Jaroszewicz, I.; Platts, J.A.; Kuduk-Jaworska, J. Calculation of lipophilicity for Pt(II) complexes: Experimental comparison of several methods. J. Inorg. Biochem. 2008, 102, 1424–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnesano, F.; Natile, G. Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA. Coord. Chem. Rev. 2009, 253, 2070–2081. [Google Scholar] [CrossRef]
- Arnesano, F.; Losacco, M.; Natile, G. An Updated View of Cisplatin Transport. Eur. J. Inorg. Chem. 2013, 2013, 2701–2711. [Google Scholar] [CrossRef]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef] [Green Version]
- Shamseddin, M.; Obacz, J.; Garnett, M.J.; Rintoul, R.C.; Francies, H.E.; Marciniak, S.J. Use of preclinical models for malignant pleural mesothelioma. Thorax 2021, thoraxjnl-2020-216602. [Google Scholar] [CrossRef]
- Notarstefano, V.; Sabbatini, S.; Sabbatini, M.; Arrais, A.; Belloni, A.; Pro, C.; Vaccari, L.; Osella, D.; Giorgini, E. Hyperspectral characterization of the MSTO-211H cell spheroid model: A FPA–FTIR imaging approach. Clin. Spectrosc. 2021, 3, 100011. [Google Scholar] [CrossRef]
- Molina, D.M.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.H.; Nordlund, P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Platts, J.A.; Oldfield, S.P.; Reif, M.M.; Palmucci, A.; Gabano, E.; Osella, D. The RP-HPLC measurement and QSPR analysis of log P-o/w values of several Pt(II) complexes. J. Inorg. Biochem. 2006, 100, 1199–1207. [Google Scholar] [CrossRef]
- Zanellato, I.; Boidi, C.D.; Lingua, G.; Betta, P.G.; Orecchia, S.; Monti, E.; Osella, D. In vitro anti-mesothelioma activity of cisplatin-gemcitabine combinations: Evidence for sequence-dependent effects. Cancer Chemother. Pharmacol. 2011, 67, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, A.; Aceto, M.; Cassino, C.; Gabano, E.; Osella, D. Uptake of antitumor platinum(II)-complexes by cancer cells, assayed by inductively coupled plasma mass spectrometry (ICP-MS). J. Inorg. Biochem. 2004, 98, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Manente, A.G.; Pinton, G.; Zonca, S.; Tavian, D.; Habib, T.; Jithesh, P.V.; Fennell, D.; Nilsson, S.; Moro, L. KDM6B histone demethylase is an epigenetic regulator of estrogen receptor beta expression in human pleural mesothelioma. Epigenomics 2016, 8, 1227–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Log k′ | IC50 MSTO-211H (μM) | IC50 REN (μM) | IC50 BR95 (μM) | IC50 MM98 (μM) | Pt Uptake (ng Pt per 106 Cells) |
|---|---|---|---|---|---|---|
| Cisplatin, CDDP | 0.29 | 1.14 ± 0.07 | 3.07 ± 0.43 | 2.15 ± 0.29 | 1.75 ± 0.09 | 9.23 ± 0.96 |
| 6 | 4.22 | 0.69 ± 0.03 (1.6) | 1.59 ± 0.07 (1.9) | 0.83 ± 0.04 (2.6) | 0.98 ± 0.03 (1.8) | 10.64 ± 1.77 |
| 8 | 0.25 | 28.9 ± 2.9 (0.039) | 46.9 ± 3.2 (0.065) | 56.8 ± 4.3 (0.038) | 58.4 ± 2.7 (0.030) | 2.50 ± 1.16 |
| 7 | 5.84 | 1.48 ± 0.02 (0.77) | 2.22 ± 0.08 (1.4) | 2.19 ± 0.13 (0.98) | 0.45 ± 0.08 (3.9) | 10.86 ± 1.17 |
| 9 | 0.34 | 54.9 ± 3.1 (0.021) | 38.9 ± 2.2 (0.079) | 39.7 ± 2.7 (0.054) | 44.1 ± 3.5 (0.049) | 3.12 ± 0.78 |
| SAA1 | 1.36 | 2.13 ± 0.05 (0.53) | 5.73 ± 0.11 (0.53) | 3.53 ± 0.06 (0.61) | 1.96 ± 0.04 (0.89) | 5.38 ± 1.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabano, E.; Pinton, G.; Balzano, C.; Boumya, S.; Osella, D.; Moro, L.; Ravera, M. Unsymmetric Cisplatin-Based Pt(IV) Conjugates Containing a PARP-1 Inhibitor Pharmacophore Tested on Malignant Pleural Mesothelioma Cell Lines. Molecules 2021, 26, 4740. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164740
Gabano E, Pinton G, Balzano C, Boumya S, Osella D, Moro L, Ravera M. Unsymmetric Cisplatin-Based Pt(IV) Conjugates Containing a PARP-1 Inhibitor Pharmacophore Tested on Malignant Pleural Mesothelioma Cell Lines. Molecules. 2021; 26(16):4740. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164740
Chicago/Turabian StyleGabano, Elisabetta, Giulia Pinton, Cecilia Balzano, Sara Boumya, Domenico Osella, Laura Moro, and Mauro Ravera. 2021. "Unsymmetric Cisplatin-Based Pt(IV) Conjugates Containing a PARP-1 Inhibitor Pharmacophore Tested on Malignant Pleural Mesothelioma Cell Lines" Molecules 26, no. 16: 4740. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164740